
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical labeling for fine mapping of IgG N-glycosylation by ETD-MS†
Lijun
Yang
a,
Zhenyu
Sun
b,
Lei
Zhang
b,
Yan
Cai
b,
Ye
Peng
b,
Ting
Cao
a,
Ying
Zhang
 *ab and
Haojie
Lu
*ab
*ab and
Haojie
Lu
*ab
aShanghai Cancer Center, Department of Chemistry, Fudan University, Shanghai 200032, China. E-mail: ying@fudan.edu.cn; luhaojie@fudan.edu.cn
bInstitutes of Biomedical Sciences, NHC Key Laboratory of Glycoconjugates Research, Fudan University, Shanghai 200032, China
First published on 23rd August 2019
Abstract
Immunoglobulin G (IgG), which contains four subclasses (IgG1–4), is one of the most important classes of glycoproteins in the immune system. Because of its importance in the immune system, a steady increase of interest in developing IgG as the biomarker or biotherapeutic agent for the treatment of diseases has been seen, as most therapeutic mAbs were IgG-based. N-Glycosylation of IgG is crucial for its effector function and makes IgG highly heterogeneous both in structure and function, although all four subclasses of IgG contain only a single N-glycosylation site in the Fc region with a highly similar amino acid sequence. Therefore, fine mapping of IgG glycosylation is necessary for understanding the IgG function and avoiding aberrant glycosylation in mAbs. However, site-specific and comprehensive N-glycosylation analysis of IgG subclasses still cannot be achieved by MS alone due to the partial sequence coverage and loss of connections among glycosylation of the protein sequence. We report here a chemical labeling strategy to improve the electron transfer dissociation efficiency in mass spectrometry analysis, which enables a 100% peptide sequence coverage of N-glycopeptides in all subclasses of IgG. Combined with high-energy collisional dissociation for the fragmentation of glycans, fine mapping of the N-glycosylation profile of IgG is achieved. This comprehensive glycosylation analysis strategy for the first time allows the discrimination of IgG3 and IgG4 intact N-glycopeptides with high similarity in sequence without the antibody-based pre-separation. Using this strategy, aberrant serum IgG N-glycosylation for four IgG subclasses associated with cirrhosis and hepatocellular carcinoma was revealed. Moreover, this method identifies 5 times more intact glycopeptides from human serum than the native-ETD method, implying that the approach can also accommodate large-scale site-specific profiling of glycoproteomes.
Introduction
Immunoglobulin G (IgG), which constitutes 10–20% of plasma proteins, plays an important role in the immune system to recognize and clear foreign antigens and pathogens.1 IgG can be broadly divided into four different subclasses (IgG1, IgG2, IgG3, and IgG4) with unique properties of their immune responses.1a,2 The N-glycosylation of IgG plays a key role in these immune reactions and it is closely associated with the pathogenesis and progression of diseases.3 For example, the degree of IgG Fc galactosylation varies in autoimmune diseases, infectious diseases and cancers.3a,4 Moreover, most current mAbs are IgG-based and glycosylation of IgG can significantly affect the potency and safety of biopharmaceuticals.5 Therefore, precise identification and quantitation of site-specific glycosylation is essential for understanding the biological functions of IgG, development of biomarkers for diseases and evaluating the quality of recombinantly expressed mAbs.1bMass spectrometry has shown great promise in characterization and quantitation of glycoproteomes.6 The site-specific glycosylation analysis in N-glycoproteomics involves the determination of the amino acid sequence, glycosylation site and glycan structure, to identify the glycopeptides with high confidence. However, due to the diversity of glycan structures, the relatively labile nature of glycosidic bond (compared with the peptide backbone), and the high sequence similarity of IgG glycopeptides (IgG1, EEQYN#STYR; IgG2, EEQFN#STFR; IgG3, EEQYN#STFR and IgG4, EEQFN#STYR, “#” representative glycosylation site), fine mapping glycosylation of IgG with high sequence coverage as well as glycan composition remains a challenging task. For the intact glycopeptide analysis, in order to solve the inconsistency in fragmentation energies between the glycoside bond and peptide bond, different fragmentation methods that provide complementary information were used. Higher-energy collision dissociation (HCD) and electron transfer dissociation (ETD) are two most common fragmentation methods in glycosylation analysis.7 HCD can break the peptide backbone and glycoside bond together, facilitating both the sequencing analysis of the peptide part and glycan part, though the fragmentation information is sometimes inadequate, while ETD predominantly cleaves the N–Cα bond along the peptide backbone to form c/z ions and preserves labile glycosidic bond, allowing for peptide sequence elucidation and site-specific analysis of the glycan modification.7,8 However, one of the most prominent technical problems in ETD is that the fragmentation is particularly ineffective for the precursor ions with low charge density (z < 3 or m/z > 850). These precursor ions with low-charge density are more compact and often undergo nondissociative electron transfer (ETnoD).9 The molecular weight of intact N-glycopeptides is generally larger than 2000 Da as the minimum molecular weight of the pentosaccharide core is 910.32 Da. Additionally, hydrophilic glycans are usually difficult to charge, further decreasing the charge density and thereby making ETnoD more prominent. Therefore, the development of an efficient ETD approach is essential in enabling comprehensive profiling of intact glycopeptides.10
In this work, for the first time, we reported a simple and robust method to increase the charge of N-glycopeptides through chemical labeling, thereby improving the ETD efficiency of N-glycopeptides. After optimization, a tertiary amine, N,N-dimethylethylenediamine (DMEN), was employed to label N-glycopeptides to improve the ETD efficiency of intact glycopeptides for mass spectrometric analysis, thereby allowing the discrimination of N-glycopeptides from different IgG subclasses. Moreover, we show the superiority of this approach in analyzing complex glycoproteomes and believe that it will further promote the use of ETD-MS for intact glycopeptide analysis.
Results
The principle of chemical labeling to improve ETD efficiency for fine mapping of IgG N-glycopeptides is shown in Scheme 1. To achieve this goal, we chose tertiary and quaternary amines as the labeling reagents, which have been reported to improve the ETD efficiency of peptides, but there have been no reports about them in glycoproteomics.9a First, we systematically investigated several types of small molecules including tertiary and quaternary amines (Fig. 1a) and different reaction conditions to screen for the most optimum condition with both high derivatization efficiency and ETD efficiency. N-Glycopeptides from standard IgG were used to investigate both efficiencies. Preliminary data indicated that N,N-dimethylethylenediamine (DMEN) outperformed the other two and yielded the best derivatization efficiency and ETD efficiency (Fig. 2 and S1†). Therefore, DMEN was chosen for further studies, as shown in Fig. 1b.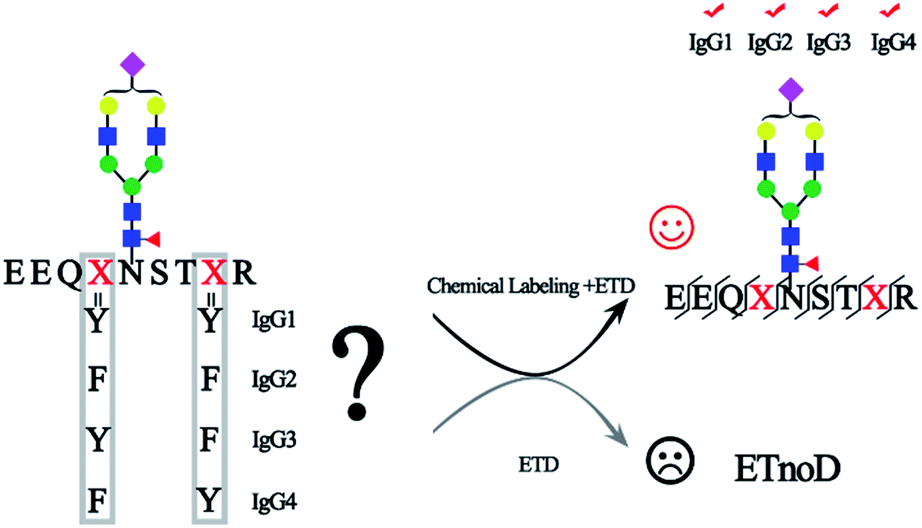 | ||
| Scheme 1 Chemical labeling to improve ETD efficiency for fine mapping of IgG N-glycopeptides. | ||
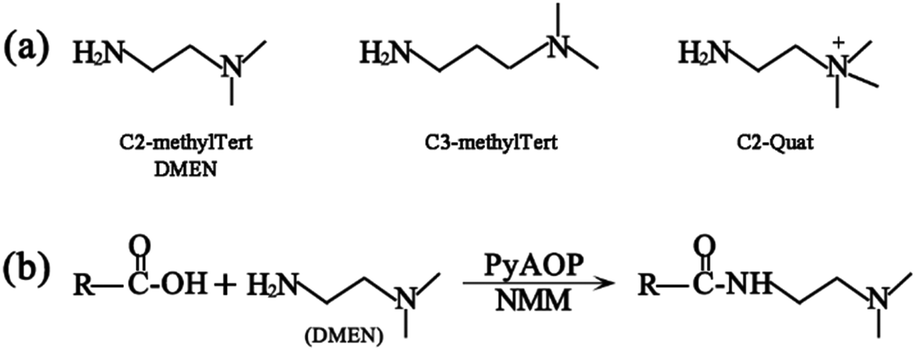 | ||
| Fig. 1 (a) Structure of the derivatization reagents and (b) illustration of the carboxylate amidation reaction between the carboxyl group of glycopeptides and the amine group of DMEN. | ||
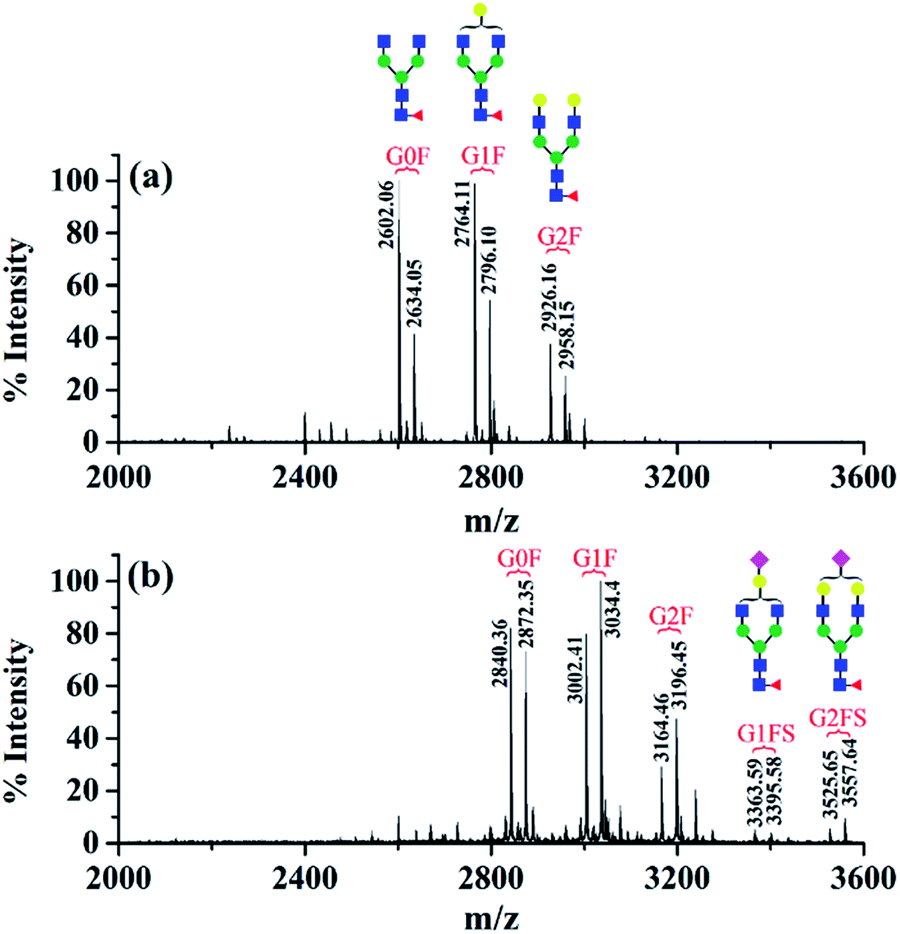 | ||
| Fig. 2 MALDI-TOF mass spectra of N-glycopeptides from standard IgG. (a) Before derivatization and (b) after dimethylation and DMEN-amidation. G0F indicates that the glycan composition is HexNAc4Hex3Fuc1, and G1F, G2F, G1FS, and G2FS are for HexNAc4Hex4Fuc1, HexNAc4Hex5Fuc1, HexNAc4Hex4Fuc1NeuAc1 and HexNAc4Hex5Fuc1NeuAc1, respectively. | ||
The first advantage of this approach is high labeling efficiency and the ionization efficiency of N-glycopeptides is improved (Fig. 2). To prevent the reaction of primary amines (Lys and N-termini) with the glycopeptide carboxyl groups, dimethylation (+28.03 Da, per reactive site) was performed prior to amidation. Subsequently, all the carboxyl groups including sialic acid, C-termini and the side chain carboxyl groups (Asp and Glu) were labeled with DMEN through the amidation reaction (+70.09 Da, per reactive site). For example, native glycopeptide IgG1–G0F (from IgG1 with the glycan composition of HexNAc4Hex3Fuc1) was detected as [M + H]+ at m/z 2634.05 and at m/z 2872.35 [M + H]+ after DMEN-amidation, respectively. More importantly, the sialylated glycopeptides IgG1/2–G1FS and IgG1/2–G2FS (S means sialic acid) were observed only after DMEN-amidation, indicating that the labeling was also applicable to sialic acid along with the benefit of protecting the sialic acid and improving the ionization efficiency significantly. Overall, the ionization efficiency of glycopeptides was improved in both MALDI-MS and LC-ESI-MS, primarily due to the introduction of a tertiary amine (Fig. S2†). Therefore, improved sensitivity of both neutral and sialylated N-glycopeptides was achieved through the DMEN labeling.
Second, as expected, the charge states of N-glycopeptides were increased during LC-ESI-MS analysis after DMEN-amidation (Fig. S3† and 3). For neutral N-glycopeptides IgG1/2–G0F, G1F and G2F, the charge states of the native glycopeptides were mainly +2 and +3 (Fig. S3a†), which shifted to +3, +4 and +5 after DMEN-amidation (Fig. S3e†). This strategy is especially beneficial to the sialylated N-glycopeptides, because the chemical labeling brings more positive charges to the negatively charged sialylated N-glycopeptides. Before DMEN-amidation, the charge states of the N-glycopeptides bearing G1FS and G2FS were primarily +2 and +3 (Fig. S3b†), while after DMEN-amidation, the charge states changed to +3, +4, +5, and +6 (Fig. S3f†), which was also outperformed using m-NBA as the additive in the mobile phase to increase the charge states of N-glycopeptides.11 The above results confirmed that the DMEN-amidation increased the charge states of N-glycopeptides universally for both neutral glycopeptides and negatively charged sialylated glycopeptides (Fig. 3).
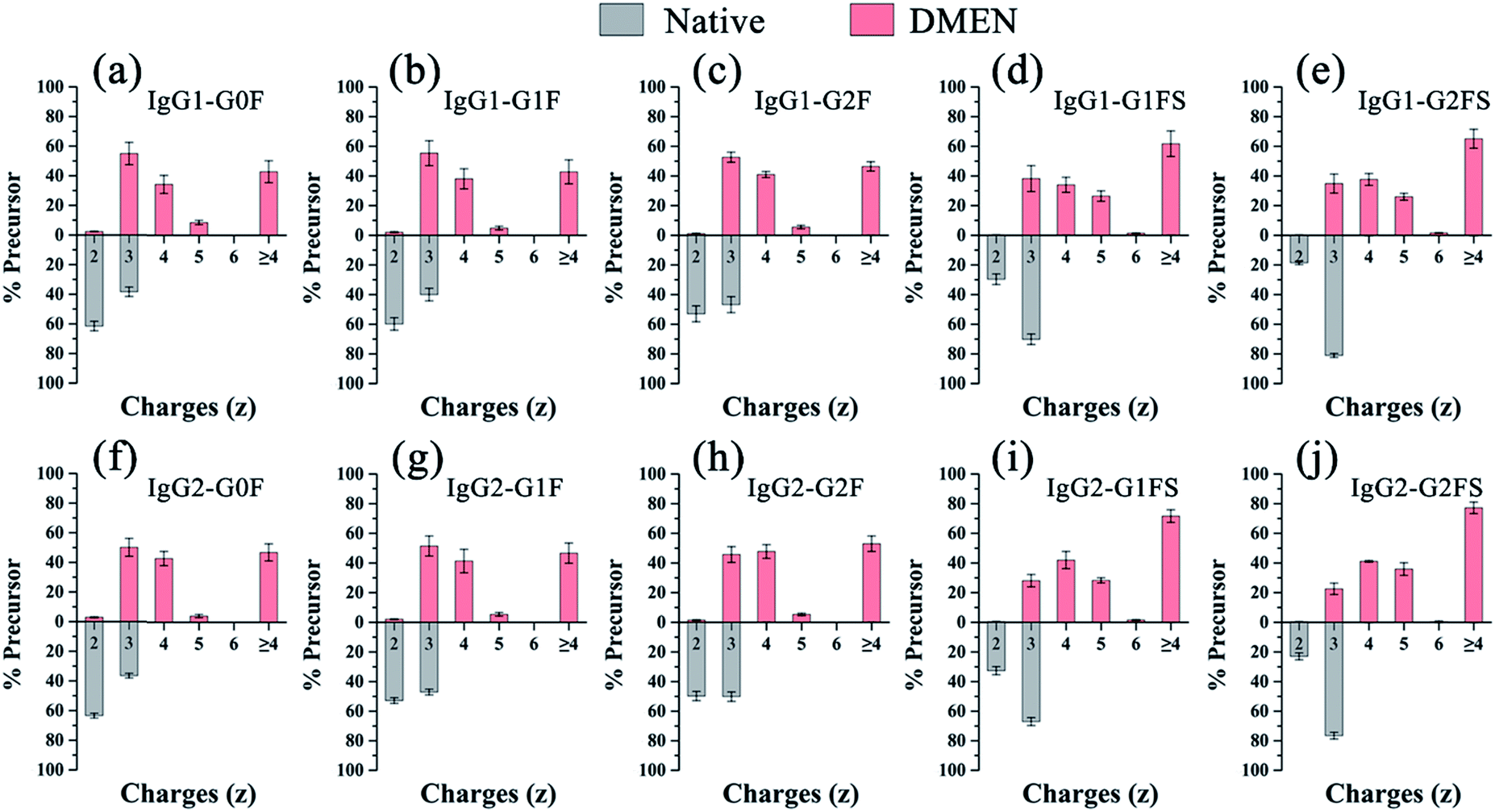 | ||
| Fig. 3 Charge state profile of N-glycopeptides from IgG. (a) IgG1-G0F, (b) IgG1-G1F, (c) IgG1-G2F, (d) IgG1-G1FS, (e) IgG1-G2FS, (f) IgG2-G0F, (g) IgG2-G1F, (h) IgG2-G2F, (i) IgG2-G1FS and (j) IgG2-G2FS. | ||
Encouraged by the charge state enhancement, we then investigated whether the ETD fragmentation efficiency of N-glycopeptides could be improved. The ETD efficiencies of native, dimethylated and DMEN-amidated N-glycopeptides were compared. As shown in Fig. 4, a few fragments were formed with the native and dimethylated N-glycopeptide, with the highest charge state of +3. However, after labeling, the ETD efficiency was significantly improved, and extensive fragmentation along the peptide backbone with consecutive c and z fragments was observed. Furthermore, as the charge state of precursor ions increased, more fragment ions were detected, providing 100% sequence coverage of the peptide (Fig. 4c and d). This further indicates the improvement in ETD efficiency. Additionally, HCD analysis was performed to further confirm the composition of glycans (Fig. S4†).
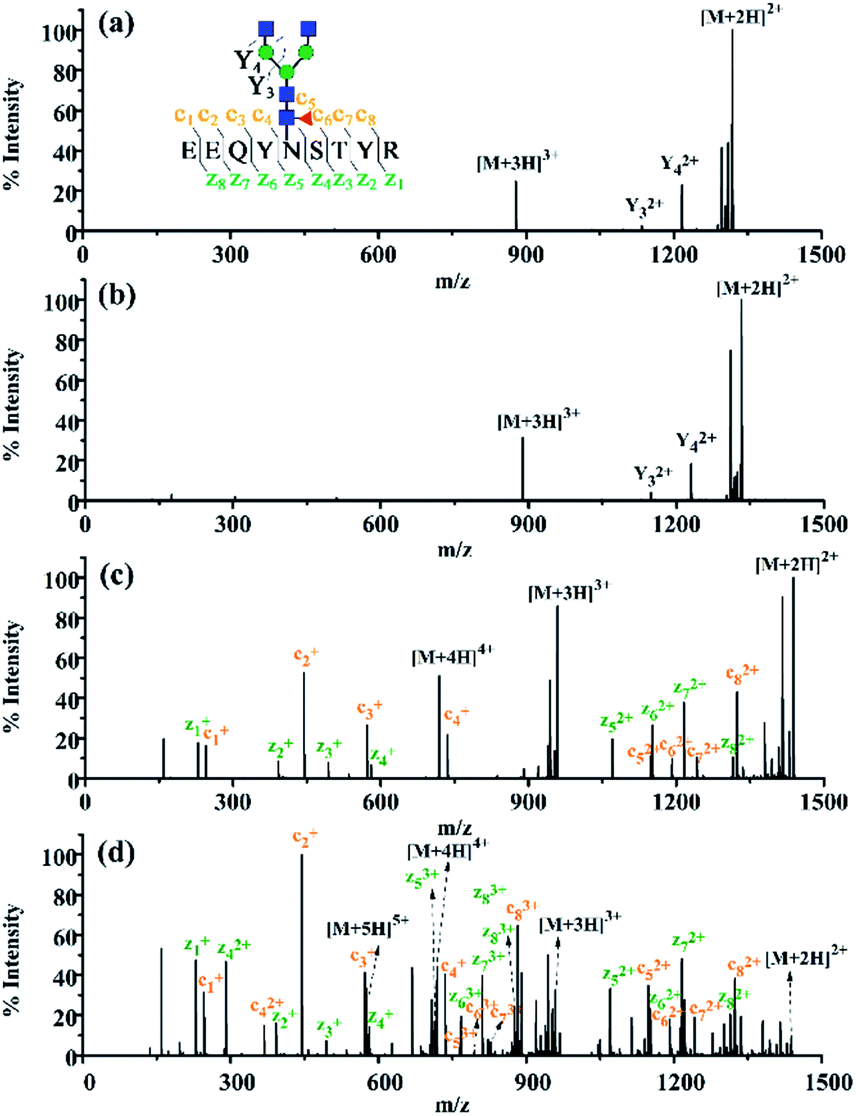 | ||
| Fig. 4 The ETD MS/MS spectra of the N-glycopeptide IgG1–G0F. (a) Native N-glycopeptide with the precursor [M + 3H]3+ at m/z 878.69, (b) dimethylated N-glycopeptide with the precursor [M + 3H]3+ at m/z 888.03, (c) DMEN-amidated N-glycopeptide with the precursor [M + 4H]4+ at m/z 718.84 and (d) DMEN-amidated N-glycopeptide with the precursor [M + 5H]5+ at m/z 575.28. | ||
Apart from N-glycopeptides of IgG1, great ETD efficiency was also observed for the N-glycopeptides of IgG2, IgG3, and IgG4 (Fig. S5†). N-glycopeptides of IgG3 and IgG4 (IgG3, EEQYN#STFR; IgG4, EEQFN#STYR), which share the same amino acid composition and are only different in the sequence of Y and F, usually neither can be well separated by chromatographic separation and nor can be distinguished in MS analysis according to the m/z. Therefore, the N-glycopeptides of IgG3 and IgG4 were always grouped together only when IgG3 was pre-separated using antibodies.12 Thanks to the 100% peptide sequence coverage in ETD MS/MS of this strategy, IgG3 and IgG4 N-glycopeptides could be unambiguously recognized with the product ions of c4–c7 and z2–z5 (Fig. S5c†), while N-glycopeptides of IgG3 and IgG4 were indistinguishable without labeling (data not shown). Overall, after DMEN derivatization, 115 IgG glycopeptides were identified. However, using the native-ETD method, only 2 IgG glycopeptides were identified. Moreover, taking a single run as an example, after DMEN derivatization, the average coverage of c and z ions was 79.17% and 84.72%, respectively. In comparison, the average coverage of both c and z ions of the native-ETD method was close to 0. Therefore, it is obvious that the ETD efficiency of glycopeptides is significantly improved after DMEN derivatization, which contributes to a significant improvement of the identification of IgG glycopeptides.
Then, we analyzed the aberrant N-glycosylation of IgG in different disease states. Herein, we combined DMEN-amidation with isotopic dimethylation (+0, +4, and +8) to quantify serum IgG N-glycopeptides in healthy, cirrhosis (CIR) and hepatocellular carcinoma (HCC) patients (Fig. S6†). Two N-glycopeptides (IgG1–G0F and IgG3–G0F) from standard IgG samples were first used to assess the accuracy and reproducibility of this relative quantitation method. The results demonstrated that this quantitation method has good reproducibility and accuracy (R2 > 0.99, CV < 20%, n = 3) (Fig. S7 and Table S1†). In addition, IgG was purified from human serum with high reproducibility (Fig. S8†). Finally, 109 site-specific IgG N-glycopeptides were identified in total, including 41 from IgG1, 32 from IgG2, 18 from IgG3 and 18 from IgG4 (Table S2†). This is the first time that N-glycopeptides of four IgG subclasses were well distinguished and quantified by MS alone. Our quantitative proteomics results confirmed previously reported that an increase in galactose-deficient core fucosylated N-glycopeptides (G0F and G0FN) and a decrease in agalactosylated core fucosylated N-glycopeptides (G1F and G2F) of four IgG subclasses in CIR and HCC patients (Fig. 5, Tables S3 and S4†).3a,12c,13 We also found that G1, G2, G1FS, and G2FS glycoforms were decreased in IgG1 and IgG2, which has not been reported. Besides, for the N-glycopeptides that contain the same glycoforms but belong to different IgG subclasses, the variation degree among IgG subclasses was different. The individual differences in IgG3 were more obvious, suggesting that the changes in glycosylation may be due to glycopeptide abundance instead of the glycan abundance and indicating the necessity of quantifying intact glycopeptides.12a
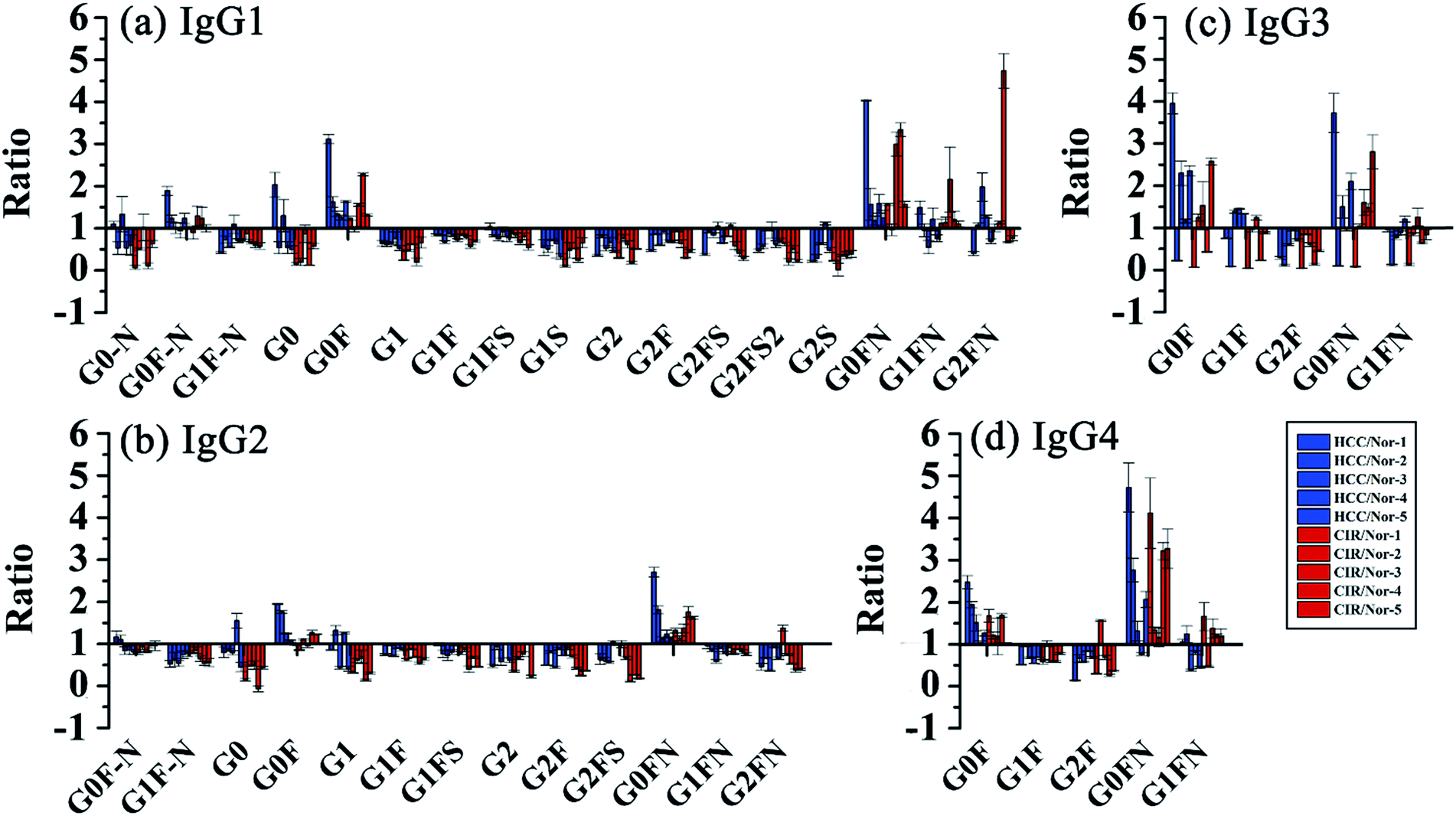 | ||
| Fig. 5 Changes of site specific IgG N-glycopeptides in CIR and HCC. Glycopeptides from (a) IgG1, (b) IgG2, (c) IgG3 and (d) IgG4. | ||
Moreover, given that the labeling method was simple and easily adopted, we further applied this strategy to study other glycoproteins and complex glycoproteomes to verify its superiority. After labeling, charges of N-glycopeptides from fetuins greatly increased and showed outstanding ETD efficiency, even for the molecular weight of glycopeptides over 7000 Da (Fig. S9 and S10†). For human serum N-glycoproteome (chromatographic separation is shown in Fig. S11†), the percentage of precursor charges over 3 (z > +3) in the ETD MS/MS spectra increased from 36.56% to 78.91% (Fig. S12a†). Moreover, most of the GPSMs (glycopeptide-spectrum matches) had charges over 3, indicating that the low MS/MS success rate (number of MS/MS scans successfully converted to GPSMs) of native-ETD methods was due to the low charge states of native glycopeptides (Fig. S12b†). This also proves our hypothesis that an increase in the charges of intact glycopeptides can greatly improve their ETD efficiency and thereafter can improve the identification of intact glycopeptides. On comparing native-ETD with DMEN-ETD, the number of GPSMs greatly increased from 67 to 1100 (average value, n = 3), providing over 15-fold improvement. Finally, a total of 553 N-glycopeptides were identified after DMEN-labeling (Table S5†). In comparison, only 98 N-glycopeptides were identified without labeling (Fig. S13†). The molecular weight distribution further shows that DMEN-labeling could help to identify glycopeptides with higher molecular weight compared to that without derivatization (Fig. S14†). Furthermore, we compared this strategy with EThcD fragmentation, through which only 334 N-glycopeptides were identified, indicating that the chemical labeling-assisted ETD was also superior to the EThcD method. Overall, this strategy improved the ETD efficiency of N-glycopeptides universally and allowed the identification of more N-glycopeptides with high confidence. In addition, the number of glycopeptides identified from serum using native-HCD and DMEN-ETD method were compared, 604 glycopeptides were identified by native-HCD method, and 553 glycopeptides were identified by DMEN-ETD method. The number of glycopeptides identified were close but complementary (Fig. S15†). We also analyzed the glycopeptides identified from serum using DMEN-HCD method, 613 glycopeptides were identified and there was also a good complementary between DMEN-HCD and DMEN-ETD methods. Therefore, DMEN derivatization didn’t affect the HCD fragmentation. The two fragmentation method could be used together to improve the identification of glycopeptides. Moreover, from the serum N-glycoproteome data, we observed another advantage of this method. The DMEN-amidation greatly improved the enrichment selectivity of HILIC toward glycopeptides. For native serum N-glycopeptide samples, only 66.15% MS/MS spectra contained the glycan specific oxonium ions, while for DMEN-amidated samples 86.22% MS/MS spectra contained the glycan specific ions, showing that the DMEN-amidation greatly improved the HILIC enrichment selectivity of N-glycopeptides.
Conclusions
We provide here a novel approach to address the challenge in ETD analysis of intact glycopeptides through chemical labeling of glycopeptides. Through labeling, the charges and ionization efficiency of intact glycopeptides were significantly increased, and ETD efficiency and HILIC enrichment selectivity of intact N-glycopeptides were greatly improved. Thereby, fine-mapping of intact glycopeptides was achieved and new insights into the site-specific microheterogeneity of proteins were provided. As far as we know, this is the first time that IgG3 and IgG4 N-glycopeptides have been identified and distinguished by mass spectrometry in the same MS run without antibody pre-separation. Combining this strategy with isotopic dimethylation labeling, serum IgG N-glycopeptides from different pathological states, including CIR, HCC and healthy people, were quantified, revealing the decrease of IgG Fc galactosylation and the differences between IgG subclasses. Moreover, as most mAbs are IgG based, we anticipate that this approach may facilitate the quality control of the glycosylation of IgG to ensure clinical safety and efficiency of mAb drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program (2016YFA0501303 and 2018YFC0910301) and NSF (Grants 21974025 and 21974026).References
-
(a) G. Vidarsson, G. Dekkers and T. Rispens, Front. Immunol., 2014, 5, 520 CrossRef PubMed
; (b) K. Hansen, A. M. Lau, K. Giles, J. M. McDonnell, W. B. Struwe, B. J. Sutton and A. Politis, Angew. Chem., Int. Ed., 2018, 57, 17194–17199 CrossRef CAS PubMed
- S. Engelhart, R. J. Glynn and P. H. Schur, Semin. Arthritis Rheum., 2017, 47, 276–280 CrossRef CAS PubMed
-
(a) S. F. Ren, Z. J. Zhang, C. J. Xu, L. Guo, R. Q. Lu, Y. H. Sun, J. M. Guo, R. H. Qin, W. J. Qin and J. X. Gu, Cell Res., 2016, 26, 963–966 CrossRef PubMed
- I. Gudelj, G. Lauc and M. Pezer, Cell. Immunol., 2018, 333, 65–79 CrossRef CAS PubMed
-
(a) M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 1235681 CrossRef PubMed
-
(a) J. N. Zheng, H. P. Xiao and R. H. Wu, Angew. Chem., Int. Ed., 2017, 56, 7107–7111 CrossRef CAS PubMed
-
(a) M. Q. Liu, W. F. Zeng, P. Fang, W. Q. Cao, C. Liu, G. Q. Yan, Y. Zhang, C. Peng, J. Q. Wu, X. J. Zhang, H. J. Tu, H. Chi, R. X. Sun, Y. Cao, M. Q. Dong, B. Y. Jiang, J. M. Huang, H. L. Shen, C. C. L. Wong, S. M. He and P. Y. Yang, Nat. Commun., 2017, 8, 343 CrossRef PubMed
- F. F. Ma, R. X. Sun, D. M. Tremmel, S. D. Sacket, J. Odorico and L. J. Li, Anal. Chem., 2018, 90, 5857–5864 CrossRef CAS PubMed
-
(a) B. L. Frey, D. T. Ladror, S. B. Sondalle, C. J. Krusemark, A. L. Jue, J. J. Coon and L. M. Smith, J. Am. Soc. Mass Spectrom., 2013, 24, 1710–1721 CrossRef CAS PubMed
-
(a) N. M. Riley and J. J. Coon, Anal. Chem., 2018, 90, 40–64 CrossRef CAS PubMed
- C. W. Lin, M. A. Haeuptle and M. Aebi, Anal. Chem., 2016, 88, 8484–8494 CrossRef CAS PubMed
-
(a) Q. T. Hong, C. B. Lebrilla, S. Miyamoto and L. R. Ruhaak, Anal. Chem., 2013, 85, 8585–8593 CrossRef CAS PubMed
- M. Sanda and R. Goldman, Anal. Chem., 2016, 88, 10118–10125 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and experimental procedures, eleven supplementary figures and six supplementary tables. See DOI: 10.1039/c9sc02491c |
| This journal is © The Royal Society of Chemistry 2019 |