
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Caesium fluoride-mediated hydrocarboxylation of alkenes and allenes: scope and mechanistic insights†
Ashot
Gevorgyan‡
 a,
Marc F.
Obst‡
b,
Yngve
Guttormsen
a,
Feliu
Maseras
c,
Kathrin H.
Hopmann
*b and
Annette
Bayer
*a
a,
Marc F.
Obst‡
b,
Yngve
Guttormsen
a,
Feliu
Maseras
c,
Kathrin H.
Hopmann
*b and
Annette
Bayer
*a
aDepartment of Chemistry, UiT The Arctic University of Norway, Norway. E-mail: annette.bayer@uit.no
bHylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, UiT The Arctic University of Norway, Norway. E-mail: kathrin.hopmann@uit.no
cInstitute of Chemical Research of Catalonia (ICIQ), Spain
First published on 11th September 2019
Abstract
A caesium fluoride-mediated hydrocarboxylation of olefins is disclosed that does not rely on precious transition metal catalysts and ligands. The reaction occurs at atmospheric pressures of CO2 in the presence of 9-BBN as a stoichiometric reductant. Stilbenes, β-substituted styrenes and allenes could be carboxylated in good yields. The developed methodology can be used for preparation of commercial drugs as well as for gram scale hydrocarboxylation. Computational studies indicate that the reaction occurs via formation of an organocaesium intermediate.
Introduction
CO2 provides a sustainable source of carbon that increasingly is being used in chemical synthesis.1 Construction of anthropogenic chemical carbon cycles2 by valorisation of CO2 into chemicals, materials, and fuels, is a promising strategy for replacing fossil carbon in the chemical industry.1,3 Various studies have shown that transition metal-based catalysts are able to selectively reduce CO2 into simple chemicals, such as formic acid, methanol, alkanes, and CO.3a CO2 can also be incorporated into carbonates, which are valuable starting materials for polymer science.3b Use of CO2 in C–C bond forming reactions opens new pathways towards value-added products and pharmaceuticals from CO2.4As part of our research interest to develop C–CO2 bond forming reactions,5 we became interested in the copper-catalysed hydrocarboxylation reactions reported by Hou,6a Sawamura6b and Skrydstrup6c (Scheme 1A). In these formal hydrocarboxylations, an initial hydroboration with 9-borabicyclo[3.3.1]nonane (9-BBN) transforms an alkene to an organoborane, which in a subsequent copper-catalysed step is carboxylated with CO2. In order to elucidate the mechanistic details of the carboxylation step, we embarked on a computational study of the reaction. Surprisingly, our computational analysis indicated the existence of a feasible carboxylation pathway that does not involve the copper complex. Our subsequent experiments confirmed that it is possible to carboxylate in situ formed organoboranes in absence of copper. Related reports of carboxylations with CO2 in absence of transition metals include fluoride-mediated carboxylations of organosilanes7b–f and KOtBu-mediated carboxylations of benzylboronic esters (Scheme 1B).7a However, none of these reports addressed a possible role of the counterion for the observed reactivity. To the best of our knowledge, a CsF-mediated hydrocarboxylation with in situ generated organoboranes has not been reported. In the following, we detail our findings of the CsF-mediated hydrocarboxylation of alkenes with CO2 (Scheme 1C). A detailed computational analysis indicates that the reaction proceeds via formation of organocaesium intermediates. The described transformation expands the repertoire of carboxylation reactions that can be performed without the use of transition metal catalysts.
Results and discussion
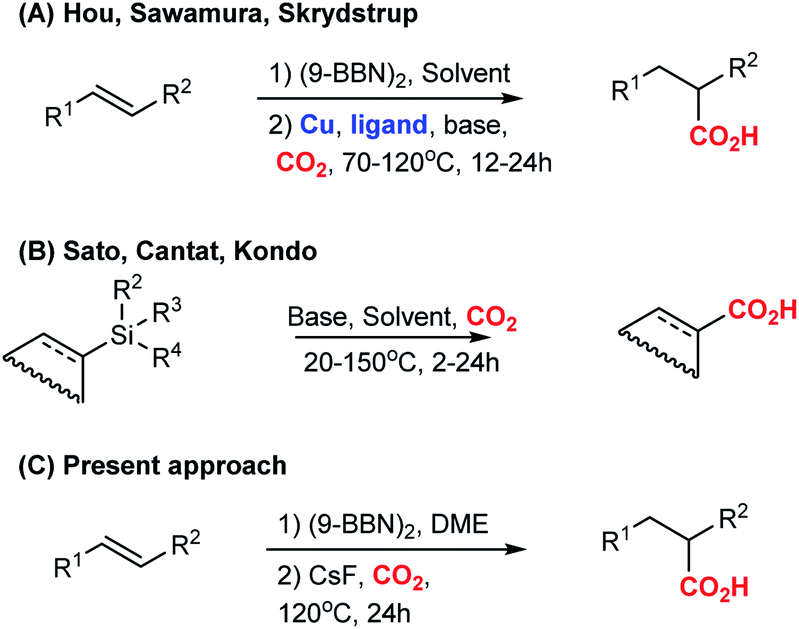
On basis of a preliminary computational investigation of the hydrocarboxylation of alkenes, we speculated that trans-stilbene 1a can be hydrocarboxylated via an organoborane intermediate in the absence of a transition metal catalyst, which is in contrast to previous reports.6 To test our hypothesis, we used 9-BBN in dioxane to convert 1a into an organoborane intermediate, which we attempted to carboxylate with CO2 in a CsF-mediated transformation (Table 1). Gratifyingly, the corresponding carboxylic acid 2a was obtained in 83% yield (Table 1, entry 2). In comparison, the previously reported copper-catalysed reaction6c gave the carboxylation product 2a in 78% yield (Table 1, entry 1). The higher yield in absence of copper was observed for several substrates (ESI, Scheme S1 and S2†). This phenomenon may be explained by a copper-promoted decarboxylation reaction slowly consuming the product 2a.8 To support this hypothesis, we mixed 2-phenylpropionic acid with the Cu complex under reaction conditions, which lead us to recover only 95% of the starting acid, while in the absence of Cu, the recovery of acid was 99% (ESI, Scheme S1†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Base (equiv.) | Solvent | °C/h | Yield % |
| a Reaction conditions: (1) 1a (0.444 mmol), (9-BBN)2 (1 equiv.), solvent (3 mL), 70 °C, 24 h. (2) (IPrCuI (5 mol%)), base (2–3 equiv.), CO2 120 mL, 80–120 °C, 24–28 h. b Isolated yields. c The active catalyst was prepared in situ (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). d The reaction mixture was run at 20 °C for 30 min before addition of CO2. | |||||
| 1 | IPrCuI(5)c | CsF(3) | Dioxane | 120/24 | 78 |
| 2 | — | CsF(3) | Dioxane | 120/24 | 83 |
| 3 | — | CsF(3) | THF | 120/24 | 61 |
| 4 | — | CsF(3) | Diglyme | 120/24 | 67 |
| 5 | — | CsF(3) | DME | 120/24 | 87 |
| 6 | — | CsF(3) | DMA | 120/24 | 0 |
| 7 | — | CsF(3) | Toluene | 120/24 | 70 |
| 8 | — | CsF(3) | MeCN | 120/24 | 0 |
| 9 | — | KF(3) | DME | 120/24 | 50 |
| 10 | — | NaF(3) | DME | 120/24 | 0 |
| 11 | — | Cs2CO3(3) | DME | 120/24 | 71 |
| 12 | — | K2CO3(3) | DME | 120/24 | 67 |
| 13 | — | KOtBu(3)d | DME | 120/24 | 47 |
| 14 | — | CsF(2) | DME | 120/24 | 57 |
| 15 | — | CsF(3) | DME | 80/24 | 59 |
| 16 | — | CsF(3) | DME | 120/28 | 85 |
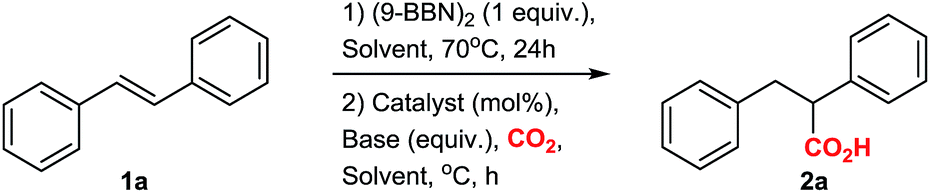
We proceeded to establish the optimum reaction conditions of the base-mediated carboxylation reaction. Screening of different solvents revealed that the reaction works well in ethers. The best yield was observed in dimethoxyethane (DME, 87%, Table 1, entry 5). The screening of different bases indicated that the optimal base is CsF (87% yield; Table 1, entry 5), while other fluoride containing bases like KF and NaF gave inferior results (50% and 0% yield, entry 9 and 10). Interestingly, also Cs2CO3 and K2CO3 gave good results (71% and 67% yield, entry 11 and 12), showing that not only the fluoride anion is important for the outcome of the reaction. On basis of the reports by Hou,6a Sawamura6b and Schomaker,7a we also attempted to employ alkoxides as base, but observed difficulties in our system. If mixed simultaneously, the reaction between alkoxide and CO2 lead to the corresponding carbonates, and no carboxylation product was formed. Alkoxide bases were effective only if the second reaction step was run without CO2 for minimum 30 minutes at 20 °C, followed by addition of CO2, which provided a yield of 47% (Table 1, entry 13). Further screening related to the stoichiometry of reagents, duration of the reaction, and temperature showed that the best conditions are 1 equiv. olefin and (9-BBN)2 and 3 equiv. CsF in DME at 120 °C for 24 h (Table 1, entry 5; for further details see ESI, Table S1†).
With the optimized conditions at hand, we explored the substrate scope of the reaction (Scheme 2; ESI Scheme S3†). Screening of different substrates showed that the CsF-mediated hydrocarboxylation works only on systems where the in situ hydroboration step (mediated by 9-BBN) generates benzylic or allylic borane intermediates. Indeed, styrene and cyclohexene were not reactive under optimal conditions (ESI Table S1†). On the other hand, stilbenes, β-substituted styrenes and allenes were successful substrates. Neither the pinacol ester of benzylboronic acid nor in situ-generated benzylic catechol esters (instead of the organoborane intermediate) were reactive in the CsF-mediated carboxylation (ESI Scheme S4†).
 | ||
| Scheme 2 Substrate scope of the CsF-mediated hydrocarboxylation. | ||
The CsF-mediated hydrocarboxylation of stilbene derivatives (1a–e) produced the corresponding carboxylic acids 2a–e with moderate to excellent yields (Scheme 2A). The conversion of (E)-α-methyl stilbenes (1d, 1e) was regioselective, providing exclusive carboxylation at the sterically less hindered β-position and resulting in formation of 2d and 2e, each as a mixture of diastereomers. The observed regioselectivity is assumed to be controlled by steric effects.6,9,10
The CsF-mediated hydrocarboxylation of β-substituted styrenes (1f–l) gave the α-carboxylated products 3a–g as the sole product in moderate to good yields (Scheme 2B). Interestingly, whereas the selectivity of the 9-BBN-initiated hydroboration of β-substituted styrenes is substrate-dependent and generally gives a non-regioselective mixture of boranes,6,9,10 our base-initiated carboxylation appears to convert only the benzylic boranes, providing a single carboxylation product with excellent regioselectivity for 3a–g (Scheme 2B). In contrast, the Cu-catalysed hydrocarboxylation does not differentiate between the regioisomeric borane intermediates, giving a mixture of carboxylic acids.6c For example, in the copper-catalysed hydrocarboxylation of indene (1k), we observed a mixture of α- and β-regioisomers with a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI, Scheme S2†).
:1 (ESI, Scheme S2†).
Allenes also proved to be suitable substrates for CsF-mediated hydrocarboxylation (Scheme 2C). Both aliphatic and aromatic allenes could be transformed to carboxylic acids 5a–f with good yields. The regioselectivity of the reaction was strongly dependent on the nature of the allene substituents. Allenes with aliphatic substituents gave the internal allylic carboxylic acid as a single product 5a–c (Scheme 2C). In contrast, allenes possessing aromatic substituents yielded the carboxylic acids 5d–f as isomeric mixtures, with the terminal carboxylic acids as the major product (Scheme 2C). Although borane-mediated hydroboration of allenes has been described,11 the selectivity is not well understood, and equilibria of internal and terminal allylic boranes have been proposed. Recently, Chida and Sato showed that the hydroboration of allenes in deuterated THF occurs predominantly at the terminal double bond.11f Carboxylation of alkyl allenes may then proceed from the terminal allylic borane with an allyl shift, or involve the internal allylic borane generated through equilibration. For aryl allenes, the direct carboxylation of the terminal allylic borane is preferred as the system is less likely to rearrange due to conjugation.
We further tested the possibility of asymmetric hydrocarboxylation using the (−)-isopinocampheylborane TMEDA complex – a chiral analogue of 9-BBN – in the initial hydroboration step (ESI Scheme S5†).12 Even though the hydroboration–oxidation of trans-β-methylstyrene gave the corresponding alcohol with 36% ee (ESI, Scheme S5A†), the hydroboration–carboxylation using our conditions led to racemic product (ESI, Scheme S5B†). The observed racemisation may be explained by the structural instability of intermediate organometallic compounds, such as the organoborane or an organocaesium (vide infra) at elevated temperatures.13
In order to show the versatility of the developed CsF-mediated hydrocarboxylation reaction, we applied our strategy in the synthesis of the commercial drugs butetamate 6a and butibufen 6b from β-substituted styrenes (Scheme 2D and E). Although in case of butetamate, four steps are required (hydroboration, carboxylation, preparation of acid anhydride, and esterification), only two isolations were needed, providing almost quantitative yields. Similarly, butibufen was obtained in 64% yield using the direct hydrocarboxylation of β-substituted styrene 1m (Scheme 2E).
Importantly, the hydrocarboxylation reaction can be scaled up (Scheme 2F). For this we changed the solvent from DME to diglyme (2-methoxyethyl ether), which has a higher boiling point, allowing the reaction to be performed in simple flasks using a CO2 balloon. Starting from 1.5 g of stilbene, we could prepare 1.427 g of the corresponding acid 2a (Scheme 2F). The yield at gram scale (76%, Scheme 2) is slightly larger compared to the small scale (67%, Table 1, entry 4), probably due to better recovery of material during work-up at larger scale.
The computational analysis of the CsF-mediated carboxylation of in situ generated organoboranes provided insights into the mechanistic steps. Three boranes were included in the theoretical study (Fig. 1): b1 and b2, derived from the experimentally reactive alkenes trans-stilbene (1a) and trans-β-methylstyrene (1f), and b3, corresponding to the non-reactive alkene cyclohexene (1o). Three possible reaction mechanisms (referred to as A, B and C) were found by an automated search of the potential energy surface with the AFIR method.14 Mechanism A (ESI, Fig. S6†) is characterized by a nucleophilic attack of the reactive carbon of the borane on a CO2 molecule, followed by a transmetalation with CsF. This mechanism is considered not viable, as all the evaluated boranes show a computed Gibbs free activation energy of >50 kcal mol−1 for the first step (ESI, Table S2†).
 | ||
| Fig. 1 Computationally investigated boranes. | ||
Reaction mechanism B (Scheme 3) occurs through two steps: First, the formation of a B–F bond between the borane i0 and a CsF molecule yielding intermediate i1, and second, the nucleophilic attack of intermediate i1 on CO2. The latter step is characterized by a concerted formation of the C–CO2 bond and the cleavage of the B–C bond, releasing F-(9-BBN) and forming the product p1. The overall barrier computed for the different boranes with mechanism B (ESI, Table S3†) is significantly lower than with mechanism A (Table S2†). However, with values of 44.4 kcal mol−1 (cyclohexane-derived borane b3) to 52.3 kcal mol−1 (trans-β-methylstyrene-derived borane b2), the barriers are too high to be overcome at the reaction temperature of 120 °C.15
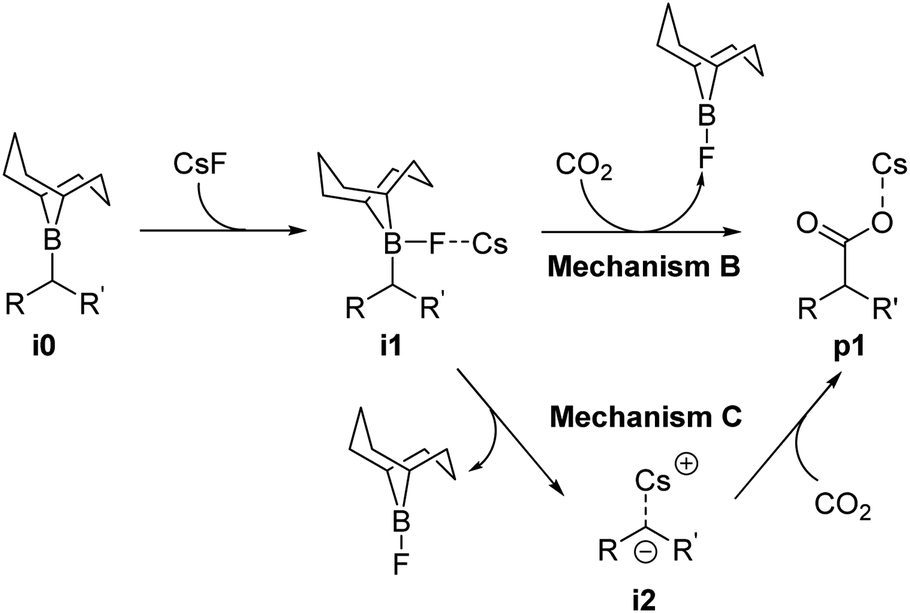 | ||
| Scheme 3 Computed reaction mechanisms B and C. | ||
The first step of mechanism C (Scheme 3) is the same as for B, the formation of intermediate i1. In the next step, the boron–carbon bond is cleaved, releasing a F-(9-BBN) molecule and forming the organocaesium intermediate i2 (Fig. 2). In the final step, i2 undergoes a nucleophilic attack on a CO2 molecule. Interestingly, at the insertion TS for substrate b1, CO2 shows no clear preference to interact with the cesium centre (Fig. 2; see also ESI, Fig. S9†), in contrast to other computational studies predicting CO2–Cs interactions.16 However, for b2, a preference for a weak CO2–Cs interaction is seen (ESI, Fig. S10†). The reason may be that the Cs atom experiences stronger interactions with the two phenyl rings of b1 than with the single aromatic ring in b2, making additional CO2–Cs interactions preferable for b2.
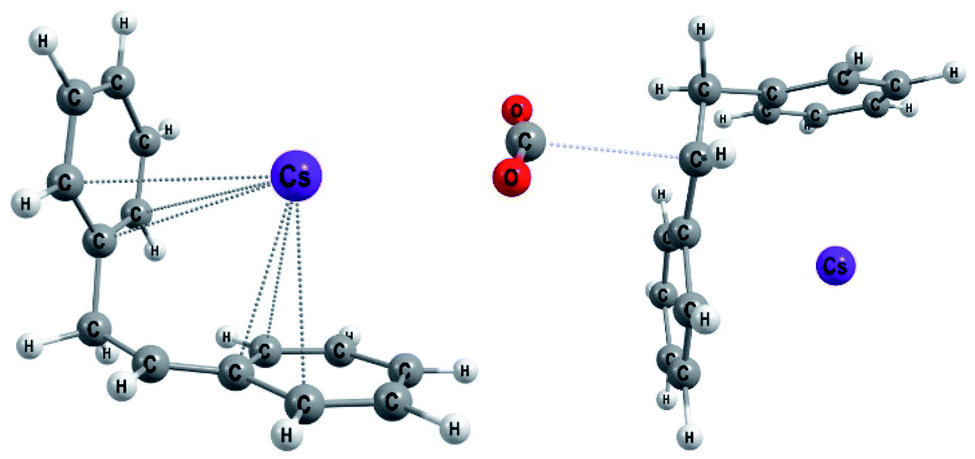 | ||
| Fig. 2 Optimized geometries for b1 (Mechanism C): the organocaesium intermediate i2 (left) and the C–CO2 bond formation TS (TSi2–p1, right). | ||
For boranes b1 and b2, the rate-limiting step of mechanism C is the cleavage of the boron–carbon bond with overall barriers of 34.0 kcal mol−1 for borane b1 (derived from trans-stilbene) and 36.7 kcal mol−1 for b2 (derived from trans-β-methylstyrene). Mechanism C is thus the preferred pathway for boranes b1 and b2. The full energy profile for carboxylation of b1via mechanism C is shown in Fig. 3.
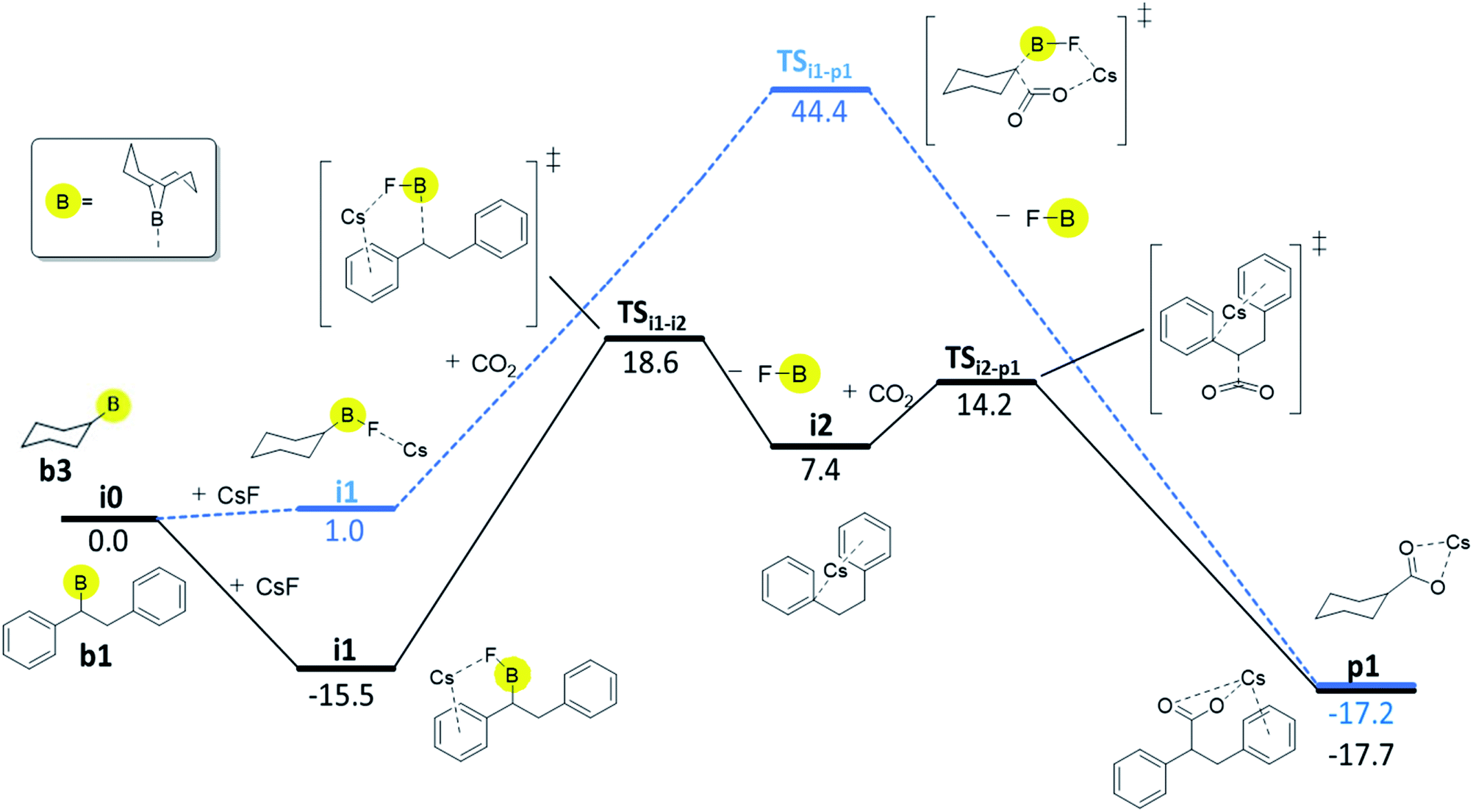 | ||
| Fig. 3 Computed Gibbs free energy profile (kcal/mol; DLPNO-CCSD(T)//ωB97XD) of the preferred reaction pathways, mechanism C for b1 (black solid line) and mechanism B for b3 (blue dashed line). | ||
For borane b3 (derived from cyclohexene), the rate-limiting step of mechanism C is the C–CO2 bond formation with an overall barrier of 51.5 kcal mol−1, which is not feasible. The lowest computed barrier for borane b3 is thus observed with mechanism B (Fig. 3), which at 44.4 kcal mol−1 is not feasible at the experimental temperature, in line with the experimentally observed lack of reactivity of cyclohexene.
Our computational and experimental results are in good agreement, indicating that the carboxylation of benzylic boranes occurs via reaction mechanism C, which features an organocaesium intermediate i2. The benzylic boranes b1 and b2 are able to stabilize the organocaesium intermediate i2via delocalization of the negative charge, and via cation–π interactions between caesium and the aromatic substituents on the organoborane. Similar Cs–π interactions have been observed in related computational studies.17 The cost of forming i2 is only 7.4 kcal mol−1 for b1 and 12.7 kcal mol−1 for b2. The cyclohexyl borane b3 lacks these stabilizing effects, resulting in a relative energy of 37.2 kcal mol−1 for the i2 intermediate. We therefore suggest that the stability of the organocaesium intermediate i2 is the factor determining the reactivity of olefins in the CsF-mediated hydrocarboxylation.
Conclusions
We report a CsF-mediated hydrocarboxylation of alkenes and allenes proceeding via a hydroboration with 9-BBN followed by a CsF-mediated carboxylation of the resulting organoboranes. The caesium fluoride-mediated carboxylation was effective for in situ generated benzylic and allylic organoboranes derived from stilbenes, β-substituted styrenes and allenes, providing the corresponding carboxylic acids with good yields and excellent regioselectivities. The developed methodology was demonstrated at gram-scale and was used for the production of commercial drugs. Computational studies indicate that benzylic organoboranes are transformed to organocaesium intermediates, which then undergo a nucleophilic attack on CO2. Stabilisation of the organocaesium intermediate by the aromatic substituent account for the observed selectivity towards benzylic organoboranes.Methods
Experimental and computational details are given in the ESI.† The ESI includes experimental procedures and analytical data, an example input for DLPNO-CCSD(T) calculations, computed energies for the full reaction pathways for b1, b2 and b3, and a comparison of computed C–CO2 TS structures. A separate xyz file contains all optimized coordinates in a format that allows easy visualization with Mercury.General procedure for metal–free hydrocarboxylation of stilbenes, β-substituted styrenes and allenes
Inside the glove box, a 45 mL pressure tube was charged with the corresponding olefin or allene (1.5 mmol), (9-BBN)2 (1 equiv. for olefins or 0.7 equiv. for allenes) and dry DME (7 mL). The flask was closed with a suitable cap, removed from the glove box and heated to 70 °C (olefin) or 50 °C (allene) for 24 h. Afterwards, the pressure tube was transferred back to the glove box. To the reaction mixture at 20 °C was added CsF (3 equiv.). The pressure tube was closed with the cap and removed from the glove box. Afterwards CO2 (120 mL) was added via a syringe, which was followed by stirring of the reaction mixture at 120 °C for 24 h. Next, the reaction mixture was diluted with 30 mL Et2O and transferred into a 500 mL separating funnel. The resulting mixture was extracted with 30 mL saturated basic (NaHCO3, 1 M KOH) solution (3 times). The resulting basic solution was washed with 15 mL Et2O (once), acidified (50–55 mL 6 M HCl) and extracted with 30 mL Et2O (3 times). The resulting solution of Et2O was distilled to dryness to give the corresponding acid (in case of 5c the final solution of Et2O was dried using Na2SO4, which was followed by careful evaporation of solvents).Computational methods
Density functional theory (DFT) calculations were performed with the ωB97XD hybrid functional,18 as implemented in Gaussian 16, Revision B.01.19 Geometries were optimized with the SDD ECP and basis set for Cs and the 6-31+G* basis set for all other elements. Initial guess structures for the transition states were obtained through linear transit calculations and through artificial force induced reaction modelling (AFIR) as implemented in GRRM.14 Solvation effects were included in the final geometry optimizations via the IEFPCM model (1,4-dioxane). Explicit solvent molecules may bind to specific points in the system, but we do not expect them to affect the overall mechanistic picture,17 and because of this they were omitted from the calculation. Vibrational, entropic, and temperature corrections were computed at 393.15 Kelvin, with the same level of theory as geometry optimizations. Electronic energies were obtained with DLPNO-CCSD(T)20 using ORCA 4.1.1.21 The ZORA operator as well as the basis sets SARC-ZORA-TZVPP (for Cs) and ZORA-def2-QZVPP (all other elements) were employed. The final Gibbs free energies (ΔGDPLNO-CCSD(T)//ωB97XD) in the main text correspond to the DLPNO-CCSD(T) electronic energies combined with the DFT-based vibrational, entropic and temperature corrections, and the standard state (SS, 393.15 K) conversion in case of a change in the number of moles:22 ΔGDPLNO-CCSD(T)//ωB97XD = ΔGωB97XD/IEFPCM − ΔEωB97XD/IEFPCM + ΔEDPLNO-CCSD(T) + SS. All ORCA and Gaussian calculations were performed on the Norwegian supercomputer Stallo at UiT, whereas GRRM calculations were performed on the computer cluster at ICIQ. More information on the computational details and example inputs as well as additional DFT energies are given in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been performed with support from NordForsk (Grant No. 85378), the Research Council of Norway (Centre of Excellence Grant No. 262695, KHH), the Tromsø Research Foundation (Grant No. TFS2016KHH to KHH), and Notur - The Norwegian Metacenter for Computational Science (CPU Grant No. nn9330k to KHH) and the Artic Centre for Sustainable Energy (ARC) at UiT (Grant No. 310059). We gratefully acknowledge the Faculty of Science and Technology at UiT for a travel grant to MFO. We thank M. K. Langer for HPLC analysis.Notes and references
- (a) M. Aresta, A. Dibenedetto and I. Tommasi, Energy Fuels, 2001, 15, 269–273 CrossRef CAS; (b) M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC; (c) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933, DOI:10.1038/ncomms6933.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS.
- For selected reviews on valorisation of CO2, see: (a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; (b) R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- For a review on transition metal-catalysed C–C bond forming reactions involving CO2, see: A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed.
- (a) J. Vaitla, Y. Guttormsen, J. K. Mannisto, A. Nova, T. Repo, A. Bayer and K. H. Hopmann, ACS Catal., 2017, 7, 7231–7244 CrossRef CAS; (b) M. Obst, L. Pavlovic and K. H. Hopmann, J. Organomet. Chem., 2018, 864, 115–127 CrossRef CAS; (c) L. Pavlovic, J. Vaitla, A. Bayer and K. H. Hopmann, Organometallics, 2018, 37, 941–948 CrossRef CAS.
- (a) T. Ohishi, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 8114–8117 CrossRef CAS PubMed; (b) H. Ohmiya, M. Tanabe and M. Sawamura, Org. Lett., 2011, 13, 1086–1088 CrossRef CAS PubMed; (c) M. Juhl, S. L. R. Laursen, Y. Huang, D. U. Nielsen, K. Daasbjerg and T. Skrydstrup, ACS Catal., 2017, 7, 1392–1396 CrossRef CAS.
- (a) R. D. Grigg, J. W. Rigoli, R. V. Hoveln, S. Neale and J. M. Schomaker, Chem.–Eur. J., 2012, 18, 9391–9396 CrossRef CAS PubMed; (b) T. Mita, K. Michigami and Y. Sato, Org. Lett., 2012, 14, 3462–3465 CrossRef CAS PubMed; (c) M. Yonemoto-Kobayashi, K. Inamoto, Y. Tanaka and Y. Kondo, Org. Biomol. Chem., 2013, 11, 3773–3775 RSC; (d) T. Mita, M. Sugawara, K. Saito and Y. Sato, Org. Lett., 2014, 16, 3028–3031 CrossRef CAS; (e) M. Yonemoto-Kobayashi, K. Inamoto and Y. Kondo, Chem. Lett., 2014, 43, 477–479 CrossRef CAS; (f) X. Frogneux, N. von Wolff, P. Thuery, G. Lefevre and T. Cantat, Chem. - Eur. J., 2016, 22, 2930–2934 CrossRef CAS.
- For selected examples on Cu-catalysed/initiated decarboxylation, see: (a) T. Cohen and R. A. Schambach, J. Am. Chem. Soc., 1970, 92, 3189–3190 CrossRef CAS; (b) O. Toussaini, P. Capdevielle and M. Maumy, Tetrahedron, 1984, 17, 3229–3233 CrossRef; (c) L. J. Gooßen, W. R. Thiel, N. Rodriguez, C. Linder and B. Melzer, Adv. Synth. Catal., 2007, 349, 2241–2246 CrossRef; (d) L. J. Gooßen, N. Rodriguez, C. Linder, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430–442 CrossRef.
- For a selected review on borane-mediated hydroboration of olefins, see: Selective Hydroboration and Synthetic Utility of Organoboranes Thus Obtained, A. Suzuki and R. S. Dhillon, Top. Curr. Chem., 1986, 130, 23–88 CrossRef CAS.
- The selectivity of hydroboration is substrate dependent. For selected examples on borane-mediated hydroboration of β-substituted styrenes, see: (a) H. C. Brown, P. K. Jadhav and A. K. Mandal, J. Org. Chem., 1982, 47, 5074–5083 CrossRef CAS; (b) H. C. Brown, D. J. Nelson and C. G. Scouten, J. Org. Chem., 1983, 48, 641–643 CrossRef CAS; (c) H. C. Brown and J. Chandrasekharan, J. Org. Chem., 1983, 48, 644–648 CrossRef CAS; (d) H. C. Brown, J. V. N. V. Prasad and S. H. Zee, J. Org. Chem., 1986, 51, 439–445 CrossRef CAS; (e) G. Erker and R. Aul, Chem. Ber., 1991, 124, 1301–1310 CrossRef CAS; (f) J. M. Clay and E. Vedejs, J. Am. Chem. Soc., 2005, 127, 5766–5767 CrossRef CAS; (g) P. Moquist, G.-Q. Chen, C. Muck-Lichtenfeld, K. Bussmann, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Sci., 2015, 6, 816–825 RSC.
- For selected examples on borane-mediated hydroboration of allenes, see: (a) H. C. Brown, R. Liotta and G. W. Kramer, J. Am. Chem. Soc., 1979, 101, 2966–2970 CrossRef CAS; (b) K. K. Wang, Y. G. Gu and C. Liu, J. Am. Chem. Soc., 1990, 112, 4424–4431 CrossRef CAS; (c) S.-C. Hung, Y.-F. Wen, J.-W. Chang, C.-C. Liao and B.-J. Uang, J. Org. Chem., 2002, 67, 1308–1313 CrossRef CAS PubMed; (d) L. Yang, Z. Lin, S.-H. Huang and R. Hong, Angew. Chem., Int. Ed., 2016, 55, 6280–6284 CrossRef CAS PubMed; (e) T. Suto, Y. Yanagita, Y. Nagashima, S. Takikawa, Y. Kurosu, N. Matsuo, T. Sato and N. Chida, J. Am. Chem. Soc., 2017, 139, 2952–2955 CrossRef CAS PubMed; (f) Y. Nagashima, K. Sasaki, T. Suto, T. Sato and N. Chida, Chem.–Asian J., 2018, 13, 1024–1028 CrossRef CAS PubMed.
- For a review on asymmetric hydroborations using (−)-isopinocampheylborane, see: M. Srebnik and P. V. Ramachandran, Aldrichimica Acta, 1987, 20, 9–24 CAS.
- Related chiral organolithium reagents racemize above -78 °C, see: (a) Stereochemical Aspects of Organolithium Compounds, ed. R. E. Gawley, Verlag Helvetica Chimica Acta, Postfach, CH-8042 Zürich, Switzerland, 2010, ISBN: 978-3-906-39061-1 Search PubMed; (b) A. Basu and S. Thayumanavan, Angew. Chem., Int. Ed., 2002, 41, 716–738 CrossRef CAS; (c) H. J. Reich, J. Org. Chem., 2012, 77, 5471–5491 CrossRef CAS PubMed.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC.
- For a discussion of feasible barriers at different temperatures, see : H. Ryu, J. Park, H. K. Kim, J. Y. Park, S.-T. Kim and M.-H. Baik, Organometallics, 2018, 37, 3228–3239 CrossRef CAS.
- H. D Velázquez, Z.-H. Wu, M. Vandichel and F. Verpoort, Catal. Lett., 2017, 147, 463–471 CrossRef.
- R. Kuniyil and F. Maseras, Theor. Chem. Acc., 2017, 136, 65 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Gaussian 16, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Guo, C. Riplinger, U. Becker, D. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- (a) F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 73–78 CAS; (b) F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8(1), 1327 Search PubMed.
- (a) K. H. Hopmann, Organometallics, 2016, 35, 3795–3807 CrossRef CAS; (b) C. J. Cramer, Essentials of Computational Chemistry: Theories and models, Wiley, 2004, p. 379 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02467k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |