
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePore-size dominated electrochemical properties of covalent triazine frameworks as anode materials for K-ion batteries†
Shu-Ying
Li‡
a,
Wen-Hao
Li‡
b,
Xing-Long
Wu
 ab,
Yuyang
Tian
a,
Jieyu
Yue
c and
Guangshan
Zhu
*a
ab,
Yuyang
Tian
a,
Jieyu
Yue
c and
Guangshan
Zhu
*a
aKey Laboratory of Polyoxometalate Science of the Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: zhugs100@nenu.edu.cn
bKey Laboratory for UV Light-Emitting Materials and Technology of Ministry of Education, Northeast Normal University, Changchun, Jilin 130024, P. R. China
cCollege of Chemistry, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Normal University, Jinan 250014, P. R. China
First published on 18th June 2019
Abstract
Two homologous covalent triazine frameworks (CTFs) have been developed for the first time as anode materials for high performance K-ion batteries (KIBs). The two-dimensional sheet-like structure as well as the regular channels in CTFs enable the process of intercalation/deintercalation of K-ions into/from the CTF interlayers reversibly. Particularly, a size effect of the porous structure is found to dominate the K-ion storage behavior. CTF-0 with a smaller pore size displays a higher K-ion storage capacity than CTF-1. Molecular simulations reveal the operation mechanism, showing that the depotassiation process in CTF-0 is exothermic while the depotassiation in CTF-1 is endothermic, which makes the deintercalation of K-ions from CTF-0 more feasible than from CTF-1 and contributes to the higher reversible capacity of CTF-0. This work provides a promising strategy for rational design of high-performance organic anode materials by structural modulation at the molecular scale.
Introduction
The huge demand for rechargeable Li-ion batteries (LIBs) in a variety of application scenarios such as powering electronic devices and vehicles leads to a heavy reliance on LIBs.1,2 However, the scarcity and uneven global distribution of commercially viable lithium resources have seriously hindered the long-term and widespread development of LIBs, which calls for alternative abundant energy storage devices, such as Na-ion (NIBs) and K-ion batteries (KIBs).3–6 In comparison to lithium resources which occur at a concentration of 0.0017 wt% in the Earth's crust, the abundance of sodium and potassium elements is 2.36 wt% and 2.09 wt%, respectively. Unlike NIBs which have gained widespread attention, KIBs have been less explored over a long-term period.7,8 It is noteworthy that in addition to the natural substantial reserves of element K, KIBs deliver a high energy density attributed to the lower negative redox potential of K/K+ (−2.93 V vs. the standard hydrogen electrode (SHE)) compared with Na/Na+ (−2.71 V vs. SHE).9 Thus, KIBs are promising next-generation batteries that will provide clean energy with high capacity and at low cost.10–13 Nevertheless, the larger ionic radius of K+ than Li+ and Na+ leads to a significant volume change for the intercalation-type electrode materials. The conversion electrodes will be subjected to structural deterioration as well as poor cycling stability during the potassiation/depotassiation process, which is especially severe in KIBs. Therefore, exploring novel anode materials suitable for K-ion storage is urgently needed for KIBs.14–17Owing to their merits of being inexpensive and sustainable, organic compounds are perceived to be ideal electrode materials for high-energy density and low-cost energy storage devices.18,19 Furthermore, in contrast to inorganic anode materials, the supramolecular assembly of organic materials is mainly sustained by van der Waals interactions rather than covalent or ionic bonds, which is beneficial for a larger interlayer spacing to accommodate the bigger sized K+ with insignificant lattice deformation. Thus, considerable efforts have been made to explore highly efficient organic electrodes applicable in KIBs.20–24 Among organic materials, two-dimensional (2D) covalent organic frameworks (COFs) represent a special class of layered materials with high crystallinity and porosity.25–28 Due to the diversity of building blocks, dimensional adjustability and designability of extended structures, 2D COFs are becoming an appealing option as electrode materials.29–34 Sakaushi and co-workers have reported a bipolar covalent triazine framework (CTF) electrode composed of aromatic rings in a honeycomb structure for high-performance sodium–organic energy storage devices.35 When exfoliated 2D CTFs were explored as anodes for NIBs, high capacities and excellent rate capabilities were achieved, owing to their robust conjugated porous structure.36 Furthermore, the well-defined covalent frameworks offer a uniform electrochemical environment which is critical for the investigation of the intercalation/deintercalation mechanism in K-ion storage. Nevertheless, K-storage anode materials based on COFs have rarely been investigated. It remains a challenge to exploit COFs as anode materials for KIBs with high capacity and long-term cycle life, and the intrinsic rules regulating the electrochemical performance of COFs are still elusive.
Herein, CTFs,37,38 a distinct and emerging class of COFs, have been exploited as anode materials for KIBs, and they store K-ions through the intercalation/deintercalation mechanism with high rate capability and long cycle life. Since CTF-0 and CTF-1 possess similar chemical compositions and topological structures (Fig. 1), the K-ion storage performance of CTF-0 and CTF-1 has been evaluated and compared simultaneously, not only to fabricate a high-performance rechargeable battery, but also to gain an insight into the mechanism of K-ion storage. Notably, the electrochemical performance is closely related to the pore size of the frameworks. CTF-0 with a smaller pore size displays better energy storage performance than CTF-1. The higher percentage of the diffusion-controlled process of K-ion storage and the larger apparent diffusion coefficient of CTF-0 according to the kinetic analysis both contribute to the better electrochemical performance. Furthermore, the dramatic size effect of the porous structure on the process of intercalation/deintercalation of K ions is revealed by molecular mechanistic simulations. It is exothermic when K+ is extracted from the interlayers of CTF-0 in the charging state, which facilitates the depotassiation process and is beneficial for a higher reversible capacity.
 | ||
| Fig. 1 Illustration for the formation of CTFs based on trimerization reaction of carbonitriles and the corresponding potassiation/depotassiation process in CTFs with different pore sizes. (a) CTF-0; (b) CTF-1. | ||
Results and discussion
Physical characterization of the CTFs
The preparation of the CTFs was performed in molten ZnCl2 at 400 °C for 40 h using either 1,3,5-tricyanobenzene (CTF-0 networks) or p-dicyanobenzene (CTF-1 networks) as the precursor according to a previously reported protocol.39,40 In order to gain an insight into the long-range order in CTFs, powder X-ray diffraction (PXRD) and Fourier transform infrared (FT-IR) measurements were carried out. The PXRD pattern of CTF-0 in Fig. 2a displays a diffraction peak at 2θ near 14°, arising from reflections from the (100) plane. According to the d spacing between the (001) diffraction planes (26°), the π–π stacking interlayer space of CTF-0 is calculated to be 3.4 Å. Possible two-dimensional structures were fabricated, where the layers of CTF-0 could stack either in an eclipsed or staggered manner. The simulated PXRD pattern according to the eclipsed AA stacking structure matches well with the experimental result. Thus, a framework belonging to the hexagonal space group (P6/m) with unit cell parameters of a = b = 7.3 Å and c = 3.4 Å is proposed for CTF-0 (inset in Fig. 2a). Additionally, the trimerization reaction is further confirmed by FT-IR spectroscopy (Fig. 2b).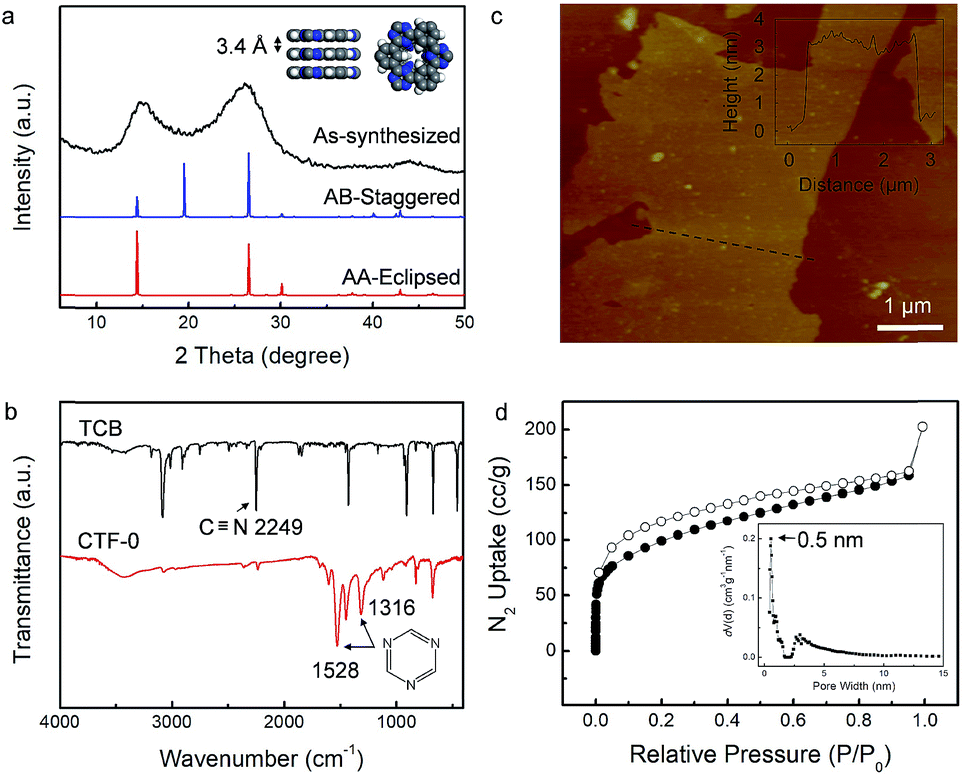 | ||
| Fig. 2 Structural and morphological characterization of CTF-0. (a) PXRD patterns; (b) FT-IR spectra (TCB stands for the monomer 1,3,5-tricyanobenzene); (c) AFM image and height profile along the black line; and (d) N2 adsorption isotherms collected at 77 K and (inset) the pore size distribution. The relative pressure (P/P0) range for determination of the CTF-0 BET surface area is from 2.0124 × 10−2 to 2.0034 × 10−1. | ||
The absence of the characteristic absorption peak of the carbonitrile band at 2249 cm−1 provides direct evidence for a successful polymerization reaction, in combination with the appearance of strong peaks at 1316 cm−1 and 1528 cm−1 corresponding to triazine rings. The FT-IR measurements confirm an almost complete conversion under the present conditions. Atomic force microscopy (AFM) investigations reveal that CTF-0 displays clear lamellar features (Fig. 2c), indicative of layer-by-layer stacked two-dimensional sheets within the CTF. The thickness of the exfoliated CTF-0 nanosheets is about 3 nm.
Meanwhile, the porosity of CTF-0 was determined by N2 adsorption/desorption isotherm measurement at 77 K (Fig. 2d). The sample exhibits a type I isotherm, suggesting the presence of microporous materials with a BET surface area of 446 m2 g−1. According to the pore size distribution plot, a narrow peak located at 0.5 nm is found, which accords with the pore width evaluated from the simulated model of CTF-0. The trimerization of 1,3,5-tricyanobenzene monomers generates a relatively dense organic framework with ultramicropores (<7 Å).41 Some mesoporous structures seem to exist in CTF-0, and they may have originated from some inter-crystal voids formed in the big aggregates.
On the other hand, CTF-1 with a larger pore size, which is condensed by p-dicyanobenzene was also explored. CTF-1 shows very similar characteristics to CTF-0 (Fig. S2†). Considering the (001) peak emerging at 26.3° in the diffraction pattern, the interlayer distance of CTF-1 is deduced to be 3.4 Å. Likewise, the layers of CTF-1 sheets prefer to assemble in an eclipsed manner. The resulting periodicity is supposed to reflect a hexagonal unit cell (a = b = 14.6 Å and c = 3.4 Å). Particularly, as confirmed by nitrogen adsorption experiments, micropores of ca. 1.5 nm, which are larger than those in CTF-0, dominate in CTF-1, facilitating research on pore-size dependent K-ion storage behavior.
Electrochemical characterization
A critical challenge in developing high-performance KIBs is overcoming the large volume expansion induced structural deterioration of the electrode material when K+ with a larger ionic radius than Li+ and Na+ is inserted/extracted into/from the electrode. This disqualifies the majority of outstanding LIB anode materials from being used in KIBs. The conjugated crystalline CTFs exhibit porosity and a periodic interlayer spacing of 0.34 nm, which raises hope that they will be capable of offering fast and smooth diffusion channels for K+ with minimum lattice distortion. Hence, we developed the CTF anode materials for KIBs to evaluate their energy storage performance.The electrochemical properties of CTF-0 and CTF-1 are displayed in Fig. 3. Fig. 3a shows the typical charge and discharge curves of CTF-0 and CTF-1 for the first several cycles at a current density of 50 mA g−1. Both of the CTFs exhibit a flat plateau at around 1.4 V during the first cycle, which is ascribed to the formation of a solid electrolyte interphase (SEI) film.42,43 And no obvious plateaus are observed in the following cycles for both CTFs. The irreversible cathodic peaks of the CTFs at about 1.4 V vs. K+/K during the first scan in cyclic voltammetry (CV) tests (Fig. S3†) are also consistent with the charge and discharge curves. Notably, CTF-0 displays almost twice the reversible capacity compared to CTF-1. Fig. 3b illustrates the cycling performance of the CTFs at a current density of 100 mA g−1. The specific capacity of CTF-0 attenuates from 154 mA h g−1 to 113 mA h g−1 after 200 cycles. In contrast, the specific capacity of CTF-1 is retained at around 60 mA h g−1 with slight variation. Additionally, CTF-0 not only possesses higher charge specific capacity but also retains higher coulombic efficiency (CE) in the initial several cycles. The higher CE of CTF-0 is also confirmed by the results of comparative CV tests in Fig. S3,† where it can be seen that CTF-0 exhibits a less obvious irreversible peak than CTF-1. Fig. 3c further compares the rate capabilities of the CTF materials. The specific capacities of CTF-0 are 152, 135, 114, 99, 84, 72 and 63 mA h g−1 at current densities of 0.05, 0.1, 0.2, 0.3, 0.5, 0.7 and 1 A g−1, respectively. However, the corresponding reversible capacities of CTF-1 are 73, 63, 52, 46, 39, 35, and 31 mA h g−1, respectively. It is supposed that CTF-0 delivers a higher reversible capacity than CTF-1.
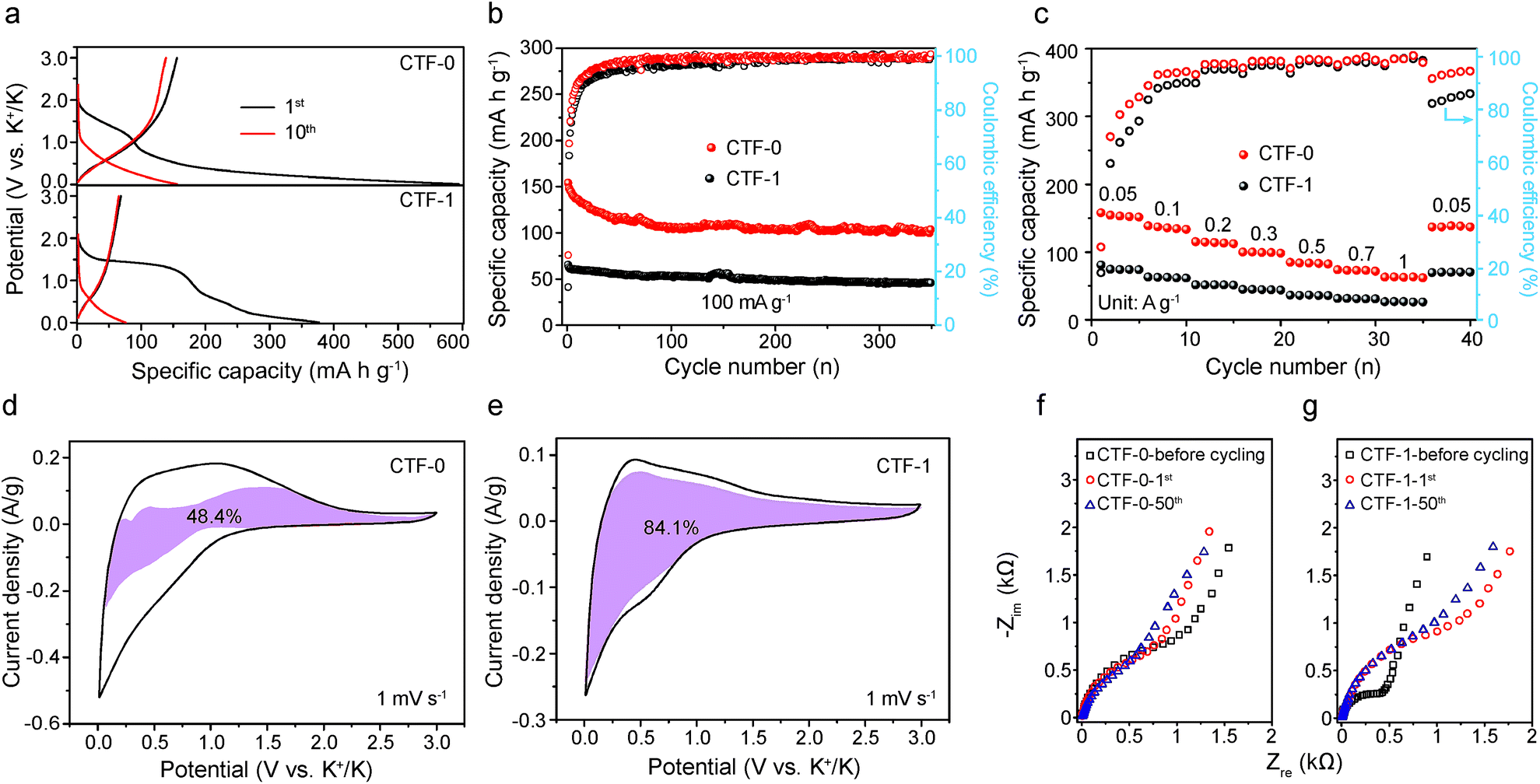 | ||
| Fig. 3 (a) Discharge/charge curves of the initial and the 10th curves at a current density of 50 mA g−1; (b) cycling performance of CTF-0 and CTF-1; and (c) rate capabilities of the CTFs. CV curves and the corresponding pseudocapacitive fractions illustrated by the purple region in (d) CTF-0 and (e) CTF-1. The EIS spectra of (f) CTF-0 and (g) CTF-1 before cycling and at the 1st and 50th cycles. | ||
In order to elucidate the reason why the K-ion storage performance differs along with the variation of pore size of CTFs, CV tests were conducted to study the kinetic process at scan rates of 0.1, 0.2, 0.3, 0.5 and 1 mV s−1 and the results are provided in Fig. S4a and b.† The results reveal that the peak current (i) of the CTFs is not proportional to the square root of the scan rate (v), suggesting that both faradaic and non-faradaic reactions occur in the K-ion storage process, and the following equations are used to qualitatively understand the reaction mechanism44–46
| i = avb | (1) |
log(i) = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logv + loga logv + loga | (2) |
Moreover, the calculation of the capacitive contribution at different scan rates is done on the basis of the formula given below:
| i = k1v + k2v0.5 | (3) |
The results of electrochemical impedance spectroscopy (EIS) tests of the CTFs in Fig. 3f and g demonstrate that the initial impedance of CTF-1 is apparently lower than that of CTF-0. This is also in accordance with the experimental results for the electronic conductivity of the CTFs, where CTF-1 possesses higher electronic conductivity than CTF-0 in the pressure range of 2 to 20 MPa (Fig. S5†). However, CTF-1 possesses higher impedance compared with CTF-0 after cycling. The phenomenon that the evident impedance increases after the first cycle of CTF-1 is in accordance with the CV test results in Fig. S3.† Results of further investigation of equivalent circuit fitting of the CTFs after 50 cycles are illustrated in Fig. S6a.† In terms of the specific fitting data after 50 cycles in Table S1,† the resistance of the SEI film (RSEI) of CTF-0 is apparently smaller than that of CTF-1, which is also consistent with the strong reduction peak of CTF-1 at the first cycle in Fig. S3.† Based on the inclined line in the Warburg region, the apparent potassium ion diffusion coefficient (Dapp) can be calculated using the equation given below:47,48
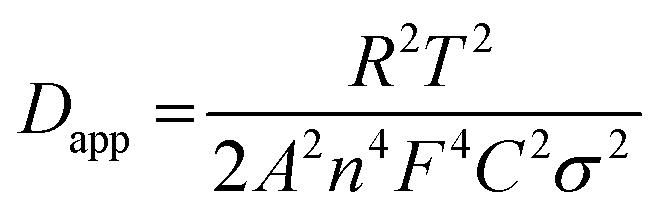 | (4) |
| Zre(or Zim) ∝ σω−1/2. | (5) |
The liner fitting and the corresponding apparent K ion diffusion coefficient of the CTFs are depicted in Fig. S6b and Table S2,† respectively. The Dapp value of CTF-0 is 2 orders of magnitude higher than that of CTF-1, which coincides with the prominently superior rate capability of CTF-0. Similar results can also be obtained from galvanostatic intermittent titration technique (GITT) tests (Fig. S7†), which further demonstrates that the CTF-0 has a higher Dapp value than CTF-1.
Molecular mechanistic simulations
Given the above issues, detailed molecular mechanistic simulations were performed to explore the intrinsic mechanism for the pore size controlled K-ion storage performance. In order to understand the intercalation position of K+ in the crystalline CTF, several tentative stacking possibilities were proposed: (1) K+ resides in the center of the channel; (2) K+ is biased towards the nitrogen of the triazine in the channel; (3) K+ is located on the benzene in CTF-0; and (4) K+ is located on the triazine in CTF-0. The molecular structures and the corresponding calculated interaction energies are summarized in Fig. S8.† When the K+ resides in the center of the pore channel and in the middle of the interlayers, the composite architecture possesses the lowest energy, indicative of a thermodynamically optimal structure.Fig. 4 depicts the comparative molecular models as the K+ is intercalated into the CTFs. Due to the ultramicropores in CTF-0, only one K+ can be accommodated in the pore channels (Fig. 4a). And the K+ intercalation process induces an increase in structural energy (simulated enthalpy change ΔH = 6.05 kcal mol−1). In contrast, when the K+ is intercalated into the interlayers of CTF-1, the structural energy reduces correspondingly, as illustrated in Fig. 4b. Moreover, the larger pores in CTF-1 are capable of accommodating up to six K+ ions (simulated enthalpy change in potassiation ΔH = −7.32 kcal mol−1). From a comparison of the structural energies before and after K+ intercalation, it is concluded that the process of intercalation of K+ into the interlayers of CTF-0 is endothermic while the process of intercalation into CTF-1 is exothermic. Conversely, deintercalation of K+ from CTF-0 is exothermic, which implies an easier depotassiation process. However, deintercalation of K+ from CTF-1 proceeds with difficulty since the process of depotassiation from CTF-1 is endothermic. As a result, the KIBs with CTF-0 as the anode material deliver a higher reversible capacity than those with CTF-1.
 | ||
| Fig. 4 Simulated K+ storage mechanisms in CTF-0 (a) and CTF-1 (c) materials. ΔH represents the enthalpy change in the corresponding potassiation (Pot.) or depotassiation (Depot.) process. XPS spectra of C 1s of CTF-0 (b) and CTF-1(d), corresponding to the pristine anode after being immersed in electrolyte, in the discharged state of 0.01 V and in the charged state of 3.0 V, respectively. | ||
To further verify the intercalation/deintercalation mechanism, X-ray photoelectron spectroscopy (XPS) investigations were conducted. The peaks of the three carbon species located at 283.3 eV, 284.8 eV and 286.3 eV in the C 1s spectrum of the as-prepared CTF-0 anode after immersion in electrolyte are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C of the benzene ring and CO in the SEI, respectively (Fig. 4b). Notably, the CC groups disappear when the battery is discharged to 0.01 V, owing to the increased randomness of the CTF framework caused by the potassiation process.33 Then the intensity of the CC and C–C peaks recovers in the following charge process, implying a release of K+. Thus, the intercalation/deintercalation of K+ into/from CTF-0 is supposed to be reversible. In contrast, as for CTF-1, while the area ratio of CC and C–C species reduces in the discharged state, it remains nearly constant after the battery is charged to 3 V as depicted in Fig. 4d, which demonstrates that the deintercalation of K+ from CTF-1 is difficult, leading to a poor reversible capacity. At this point, the molecular mechanistic simulations along with the XPS analysis have shed light on the K-ion storage mechanism in CTF-0 and CTF-1.
C, C–C of the benzene ring and CO in the SEI, respectively (Fig. 4b). Notably, the CC groups disappear when the battery is discharged to 0.01 V, owing to the increased randomness of the CTF framework caused by the potassiation process.33 Then the intensity of the CC and C–C peaks recovers in the following charge process, implying a release of K+. Thus, the intercalation/deintercalation of K+ into/from CTF-0 is supposed to be reversible. In contrast, as for CTF-1, while the area ratio of CC and C–C species reduces in the discharged state, it remains nearly constant after the battery is charged to 3 V as depicted in Fig. 4d, which demonstrates that the deintercalation of K+ from CTF-1 is difficult, leading to a poor reversible capacity. At this point, the molecular mechanistic simulations along with the XPS analysis have shed light on the K-ion storage mechanism in CTF-0 and CTF-1.
In addition, the theoretical capacity of a material is determined by the number of charge carriers and the molar weight of the specific material.49 It can be calculated using the following formula (with mA h g−1 units):
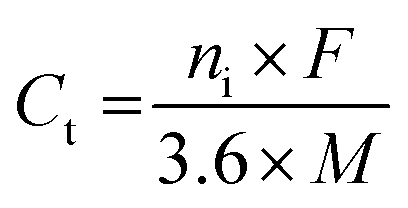 | (6) |
Conclusions
In essence, two homologous CTFs with different pore sizes were developed as anode materials for high-performance KIBs. Benefiting from the porosity and van der Waals-bonded interlayers of CTFs, the intercalation/deintercalation of K ions does not cause serious volume change or deformation of the framework, contributing to the high rate capability and long cycle life. Specifically, the electrochemical performance dramatically relies on the pore size of the CTFs. As the deintercalation of K ions from CTF-0 with smaller pores is exothermic, the depotassiation process could proceed more easily. The ultramicropores in CTF-0 are conducive to the reversible intercalation/deintercalation of K ions and account for the enhanced electrochemical performance. Our results provide an important insight into the mechanism of K-ion storage and rational design of COF anode materials for KIBs with superb energy storage performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21802017, 21531003 and 91622106),the “111” project (B18012), the China Postdoctoral Science Foundation (Grant No. 2018M631848), and the Education Department of Jilin Province “13th Five-Year” Science and Technology Research Project (JJKH20190273KJ).Notes and references
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS.
- G. Wang, F. Wang, P. Zhang, J. Zhang, T. Zhang, K. Müllen and X. Feng, Adv. Mater., 2018, 30, 1802949 CrossRef.
- Y. You, S. Xin, H. Y. Asl, W. Li, P.-F. Wang, Y.-G. Guo and A. Manthiram, Chem, 2018, 4, 2124–2139 CAS.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2014, 7, 19 CrossRef.
- I. Sultana, T. Ramireddy, M. M. Rahman, Y. Chen and A. M. Glushenkov, Chem. Commun., 2016, 52, 9279–9282 RSC.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- Z. Jian, Z. Xing, C. Bommier, Z. Li and X. Ji, Adv. Energy Mater., 2016, 6, 1501874 CrossRef.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552–557 RSC.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- X. Wang, X. Xu, C. Niu, J. Meng, M. Huang, X. Liu, Z. Liu and L. Mai, Nano Lett., 2017, 17, 544–550 CrossRef CAS.
- D.-S. Bin, Z.-X. Chi, Y. Li, K. Zhang, X. Yang, Y.-G. Sun, J.-Y. Piao, A.-M. Cao and L.-J. Wan, J. Am. Chem. Soc., 2017, 139, 13492–13498 CrossRef CAS.
- T. Deng, X. Fan, J. Chen, L. Chen, C. Luo, X. Zhou, J. Yang, S. Zheng and C. Wang, Adv. Funct. Mater., 2018, 28, 1800219 CrossRef.
- W. Wang, J. Zhou, Z. Wang, L. Zhao, P. Li, Y. Yang, C. Yang, H. Huang and S. Guo, Adv. Energy Mater., 2018, 8, 1701648 CrossRef.
- K. Beltrop, S. Beuker, A. Heckmann, M. Winter and T. Placke, Energy Environ. Sci., 2017, 10, 2090–2094 RSC.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. K. Jung, H.-D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef.
- D.-H. Yang, Z.-Q. Yao, D. Wu, Y.-H. Zhang, Z. Zhou and X.-H. Bu, J. Mater. Chem. A, 2016, 4, 18621–18627 RSC.
- Y. Liang, C. Luo, F. Wang, S. Hou, S.-C. Liou, T. Qing, Q. Li, J. Zheng, C. Cui and C. Wang, Adv. Energy Mater., 2019, 9, 1802986 CrossRef.
- L. Fan, Q. Liu, Z. Xu and B. Lu, ACS Energy Lett., 2017, 2, 1614–1620 CrossRef CAS.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef CAS.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS.
- X. Zhan, Z. Chen and Q. Zhang, J. Mater. Chem. A, 2017, 5, 14463–14479 RSC.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS.
- H. Liao, H. Ding, B. Li, X. Ai and C. Wang, J. Mater. Chem. A, 2014, 2, 8854–8858 RSC.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, P. C. Lan and S. Ma, Adv. Mater., 2019, 31, 1900008 CrossRef.
- X. Zhang, G. Zhu, M. Wang, J. Li, T. Lu and L. Pan, Carbon, 2017, 116, 686–694 CrossRef CAS.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS.
- Z. A. Ghazi, L. Zhu, H. Wang, A. Naeem, A. M. Khattak, B. Liang, N. A. Khan, Z. Wei, L. Li and Z. Tang, Adv. Energy Mater., 2016, 6, 1601250 CrossRef.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L.-P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef.
- X. Chen, H. Zhang, C. Ci, W. Sun and Y. Wang, ACS Nano, 2019, 13, 3600–3607 CrossRef CAS PubMed.
- H. W. Chen, H. Y. Tu, C. J. Hu, Y. Liu, D. R. Dong, Y. F. Sun, Y. F. Dai, S. L. Wang, H. Qian, Z. Y. Lin and L. W. Chen, J. Am. Chem. Soc., 2018, 140, 896–899 CrossRef CAS.
- K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef.
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef.
- T. S. Naidu, H. T. Hoon, J. S. Hyun, B. Onur, C. J. Wook and C. Ali, Angew. Chem., Int. Ed., 2016, 128, 3158–3163 CrossRef.
- K. A. See, S. Hug, K. Schwinghammer, M. A. Lumley, Y. Zheng, J. M. Nolt, G. D. Stucky, F. Wudl, B. V. Lotsch and R. Seshadri, Chem. Mater., 2015, 27, 3821–3829 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- X. Zou and G. Zhu, Adv. Mater., 2018, 30, 1700750 CrossRef.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef.
- Y. Liu, Z. Tai, Q. Zhang, H. Wang, W. K. Pang, H. K. Liu, K. Konstantinov and Z. Guo, Nano Energy, 2017, 35, 36–43 CrossRef CAS.
- D. Chao, P. Liang, Z. Chen, L. Bai, H. Shen, X. Liu, X. Xia, Y. Zhao, S. V. Savilov, J. Lin and Z. X. Shen, ACS Nano, 2016, 10, 10211–10219 CrossRef CAS.
- Y.-Y. Wang, B.-H. Hou, J.-Z. Guo, Q.-L. Ning, W.-L. Pang, J.-W. Wang, C.-L. Lü and X.-L. Wu, Adv. Energy Mater., 2018, 8, 1703252 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS.
- X.-L. Wu, Y.-G. Guo, J. Su, J.-W. Xiong, Y.-L. Zhang and L.-J. Wan, Adv. Energy Mater., 2013, 3, 1155–1160 CrossRef CAS.
- X.-H. Zhang, W.-L. Pang, F. Wan, J.-Z. Guo, H.-Y. Lü, J.-Y. Li, Y.-M. Xing, J.-P. Zhang and X.-L. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 20650–20659 CrossRef CAS.
- K. Ghatak, S. Basu, T. Das, V. Sharma, H. Kumar and D. Datta, Phys. Chem. Chem. Phys., 2018, 20, 22805–22817 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and supplementary figures. See DOI: 10.1039/c9sc02340b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |