
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnthracene-based mechanophores for compression-activated fluorescence in polymeric networks†
Christopher P.
Kabb
 a,
Christopher S.
O'Bryan
b,
Cameron D.
Morley
b,
Thomas E.
Angelini
bcd and
Brent S.
Sumerlin
*a
a,
Christopher S.
O'Bryan
b,
Cameron D.
Morley
b,
Thomas E.
Angelini
bcd and
Brent S.
Sumerlin
*a
aGeorge & Josephine Butler Polymer Research Laboratory, Center for Macromolecular Science & Engineering, Department of Chemistry, University of Florida, Gainesville, FL 32611, USA. E-mail: sumerlin@chem.ufl.edu
bDepartment of Mechanical and Aerospace Engineering, University of Florida, Gainesville, FL 32611, USA
cJ. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Gainesville, FL 32611, USA
dInstitute for Cell and Regenerative Medicine, University of Florida, Gainesville, FL 32611, USA
First published on 8th July 2019
Abstract
The recent attention given to functionalities that respond to mechanical force has led to a deeper understanding of force transduction and mechanical wear in polymeric materials. Furthermore, polymers have been carefully designed such that activation of “mechanophores” leads to productive outputs, such as material reinforcement or changes in optical properties. In this work, a crosslinker containing an anthracene–maleimide linkage was designed and used to prepare networks that display a fluorescence response when damaged. The pressure-dependent damage of poly(N,N-dimethylacrylamide) networks was monitored using solid-state fluorescence spectroscopy, with increasing compressive forces leading to higher degrees of mechanophore activation. When a stamp was used to compress the networks, only the areas in contact with the raised portion of the stamp underwent mechanophore activation, resulting in the generation of patterns that were only visible under UV light. Finally, an isomeric “flex” mechanophore was designed and used to prepare networks that were compressed and compared to the previously described networks.
Introduction
The burgeoning field of stimuli-responsive polymeric materials has typically focused on a relatively narrow range of commonly encountered stimuli,1 including heat,2–4 light,5,6 pH,7–10 and chemical triggers.11–15 Alternatively, the input of mechanical force to a polymer can also lead to changes in chemical properties. The principle of modifying materials via mechanochemical processes has long been studied,16–19 but its precise implementation into macromolecules had largely remained underutilized. A recent renaissance in the use of mechanical force as a means to modulate chemical reactivity has been motivated by the expanded practice of implementing principles of green chemistry into industrial processes.20 Concurrently, mechanochemically active linkages have been integrated into macromolecules to better understand fundamental aspects of force transduction in polymeric materials.21–23 Although studies related to mechanically facilitated degradation of polymers date back to the 1930s, until recently, those that harnessed mechanical force for specific modifications on the molecular level were sparse. The development of polymeric materials that feature functionalities capable of undergoing bond scission in response to an applied mechanical force (i.e., mechanophores) has led to exciting new findings, ranging from materials that toughen when damaged24,25 to the mechanically facilitated “unzipping” of a polymer backbone to yield a conducting polymer.26 The breadth of chemical functionalities that can be appended to mechanophores enables their incorporation into a variety of polymeric systems (e.g., vinyl networks,27,28 silicones,29 polyesters,30,31 and polyurethanes32), further enhancing their utility.To accurately assess the influence of mechanophores in polymeric materials, thermochemical and mechanochemical activation mechanisms must be effectively decoupled. A common strategy to achieve this is the use of thermally stable mechanophores, such as anthracene-based cycloadducts. For instance, anthracene–maleimide (Anth–Mal) cycloadducts display excellent thermal stability (up to 200 °C)2,33 and have been employed as mechanophores34 to examine the effects of macromolecular architecture35 or micellar aggregation36 on the extent of cycloreversion. In the former example, Boydston and coworkers examined the mechanochemical activation of centrally located Anth–Mal linkages in star and linear poly(methyl acrylate). Chain-scission rates were consistent with models that related the scission rate to the molecular weight of two chains emanating from the same point. Li et al. examined the effect of self-assembly of amphiphilic block copolymers on the mechanochemical activation of Anth–Mal groups that linked the two blocks.36 In a non-selective organic solution (i.e., where the polymer chains existed as unimers), the hydrophobic block length was insufficient to promote cycloreversion. However, when the polymers were dispersed in water, the assembly of amphiphilic chains into supramolecular assemblies increased the apparent chain length in the micellar core leading to efficient retro-Diels–Alder reactions with sonication. The findings outlined in both reports strongly support the notion that a critical chain length is necessary to generate sufficient force on a given point in a polymer chain to promote chain scission.37
Polymer mechanochemistry has also been extended to bulk materials, in which alternative modes of activation (e.g., shear, tensile, and compressive forces) are also pertinent. Often, mechanochromic materials (i.e., materials that change color in response to mechanical force) are utilized since optical characterization is generally straightforward, and a change in absorption, emission, or reflection can be used to visualize applied stresses.38,39 Responsive chromophores such as spiropyrans have been widely utilized due to their large bathochromic shift upon mechanically activated ring-opening to the corresponding merocyanine.29,40–42 Consequently, incorporation of color-changing mechanophores allows for detection of stresses that do not result in visible macroscopic damage. Anth–Mal cycloadducts lack absorption in the visible region, but upon cleavage via retro-Diels–Alder and liberation of anthracene, a yellow color and blue fluorescence emission is commonly observed. This “off–on” signal enables simple and sensitive detection of mechanical force using the emergence of color, rather than a more difficult-to-detect shift in the absorption spectrum. Furthermore, the fluorescence signal can only be observed when irradiated with a long-wave UV light source, rendering it useful as an “invisible ink” in polymeric materials.
In this work, we study the compressive activation of thermally stable Anth–Mal linkages in bulk polymeric networks. Upon mechanically induced cleavage of the cycloadduct, liberated anthracene functionalities produce a detectable fluorescence signal under long-wave UV irradiation. The fluorescence emission was quantified using solid-state fluorescence spectroscopy, with the measured intensity correlating to the magnitude of the applied force. To demonstrate the application of Anth–Mal crosslinks as an “invisible ink,” we imprinted polymeric networks containing mechanochemically active crosslinking points. Upon irradiation with long-wave UV light, a persistent, localized fluorescence signal was observed in the imprinted area. After the visible deformation to the network had disappeared, the imprinted areas still displayed a fluorescence signal. Furthermore, we examined the differences between a classical mechanochemical activation pathway (i.e., bond stretching) with the recently reported “flex-activated” pathway (i.e., bond bending) by comparing isomeric crosslinkers designed with differing polymer attachment points. Although we observed mechanochemical response in networks crosslinked by classically oriented mechanophores, no response was detected for the flex-activated system, even at elevated temperatures. These results reinforce the need for a deeper understanding of the requirements for the design of a successful flex mechanophore.
Results and discussion
Incorporating a mechanophore into a crosslinking unit is a viable strategy for the preparation of polymeric materials that are mechanically responsive. As reported previously, anthracene–maleimide cycloadducts (Anth–Mal) can serve as mechanophores.34–36,43 In these examples, attachment of polymer chains to the nitrogen of the maleimide and the 9-position of the anthracene led to cycloreversion of the adduct under mechanical stress. Applied mechanical forces are localized along the axis of the anthracene–maleimide cycloadduct, leading to lengthening of the bridging bonds and subsequent cleavage. Therefore, the diacrylate crosslinker 1 was prepared with a centrally located Anth–Mal adduct and appropriate connectivity of the polymerizable functionalities (Fig. S1–S8†). The crosslinker was incorporated into poly(N,N-dimethylacrylamide) (PDMA) networks formed through photoinitiated radical polymerization. Upon compressing the networks, it was expected that the adduct would undergo a cycloreversion reaction. Cleavage and liberation of anthracene would then result in a fluorescence signal that could be observed under long-wave UV irradiation (∼365 nm, Fig. 1a). Control networks were also synthesized in which 1 was replaced with 1 mol% of di(ethylene glycol) diacrylate (DEGDA). This non-responsive crosslinker would still be expected to cleave when subjected to mechanical stress, but the scission events would not result in a fluorescence signal (Fig. 1b). Furthermore, to verify that the applied compressive force was responsible for cleaving the Anth–Mal adducts, the control monomer 2 was synthesized in which the alcohol of the maleimide terminus was converted to an acetyl group rather than a polymerizable acryloyl group (Fig. S9–S16†). This monomer would therefore result in Anth–Mal linkages that are pendent to the polymer backbone, rather than being present at crosslinking junctions. As such, the applied force would not be localized on the Anth–Mal linkage and would not be expected to result in cycloreversion (Fig. 1c). To ensure the stability of Anth–Mal linkages under the polymerization conditions and investigate the extent of incorporation into the polymer networks, the linear polymer P(DMA0.99-co-20.01) was prepared. The bulk copolymerization yielded a polymer that showed no indication of changes to the cycloadduct during polymerization, as evidenced by 1H NMR spectroscopy (Fig. S17†) and GPC analysis (Fig. S18†). Furthermore, the composition of the purified copolymer matched with the feed ratio of monomers, indicating successful copolymerization of the two monomers.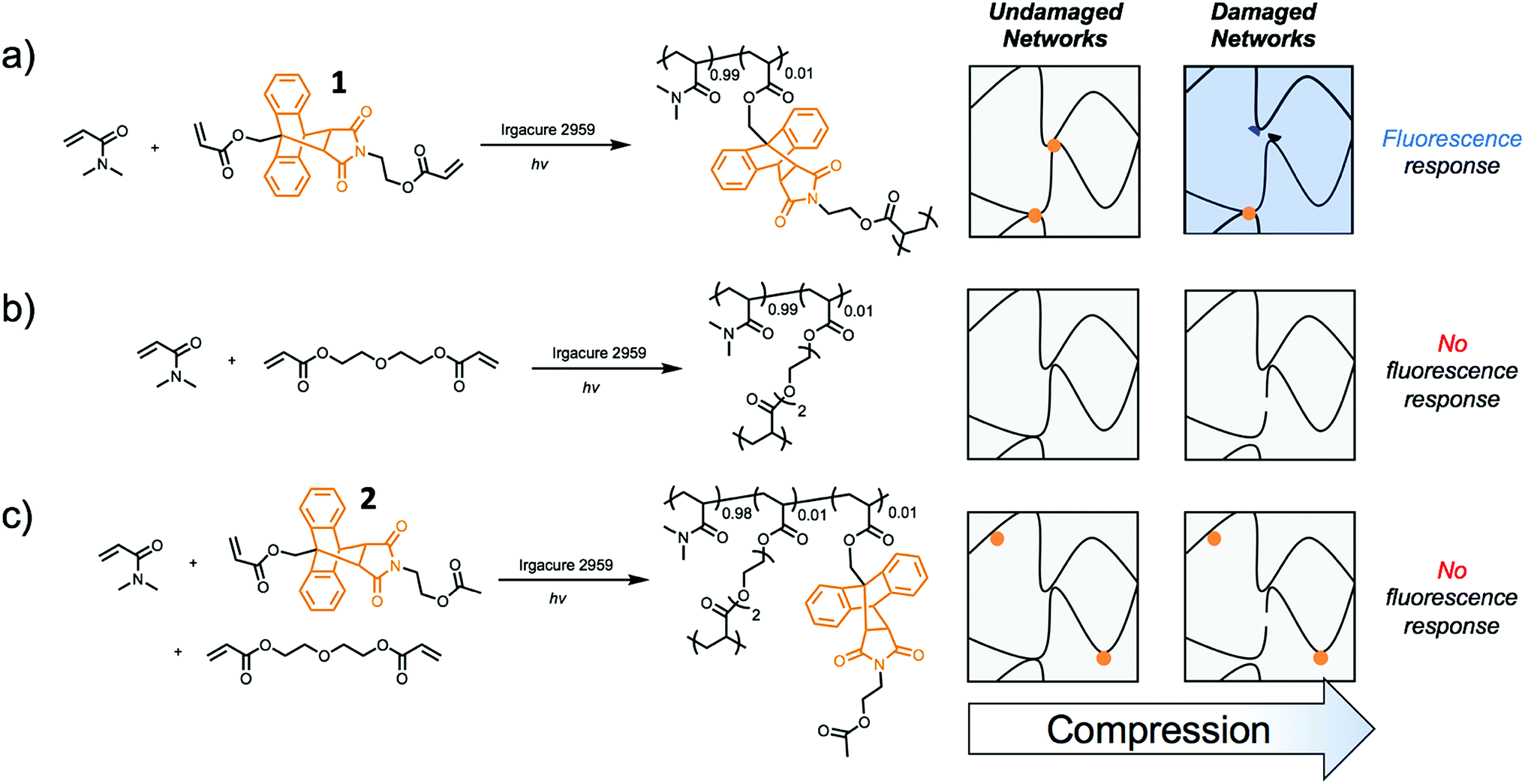 | ||
Fig. 1 Expected response to compressive stress for networks composed of (a) 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMA:1, (b) 99:1 DMA:DEGDA, and (c) 98:1:1 DMA:DEGDA:2. DMA:N,N-dimethylacrylamide; DEGDA: di(ethylene glycol) diacrylate; 1: mechanophore-containing crosslinker; 2: mechanophore-containing monomer. :1 DMA:1, (b) 99:1 DMA:DEGDA, and (c) 98:1:1 DMA:DEGDA:2. DMA:N,N-dimethylacrylamide; DEGDA: di(ethylene glycol) diacrylate; 1: mechanophore-containing crosslinker; 2: mechanophore-containing monomer. | ||
As the fluorescence intensity of our samples exposed to compressive forces is expected to be proportional to the number of Anth–Mal adducts that break, solid-state fluorescence spectroscopy was used to quantify the extent of damage to the network. Thin, cylindrical networks were prepared (d ∼ 0.7 mm, t ∼ 1.5 mm) and compressed with pressures ranging from 50 MPa to 1065 MPa (Fig. 2a). Using a petite integrating sphere attachment, we measured the fluorescence intensity of the compressed samples. Each sample was measured in three discrete locations to account for spatial heterogeneity (Fig. S19–S28†), and the averaged spectra for a network crosslinked with 1 mol% 1 (99:1 DMA:1) are shown in Fig. 2b. The intensity at 415 nm was plotted as a function of applied pressure (Fig. 2c), with higher pressures leading to greater degrees of network damage and cycloreversion. The same experiments were performed with DEGDA-crosslinked networks (99:1 DMA:DEGDA), and as expected, the incorporation of this non-responsive crosslinker led to a lack of fluorescence signal in the damaged materials. Furthermore, networks containing pendent Anth–Mal groups (98:1:1 DMA:DEGDA:2) were absent of fluorescence following compression. These results suggest that for cycloreversion to occur, the polymer chains must be attached to both ends of the mechanophore. The applied stress is then localized on the crosslinking points as the polymer backbone is extended, leading to elongation of the Anth–Mal bonds. If only one end of the mechanophore is covalently connected to the polymeric network, force is not transduced to the cycloadduct, and it remains intact.
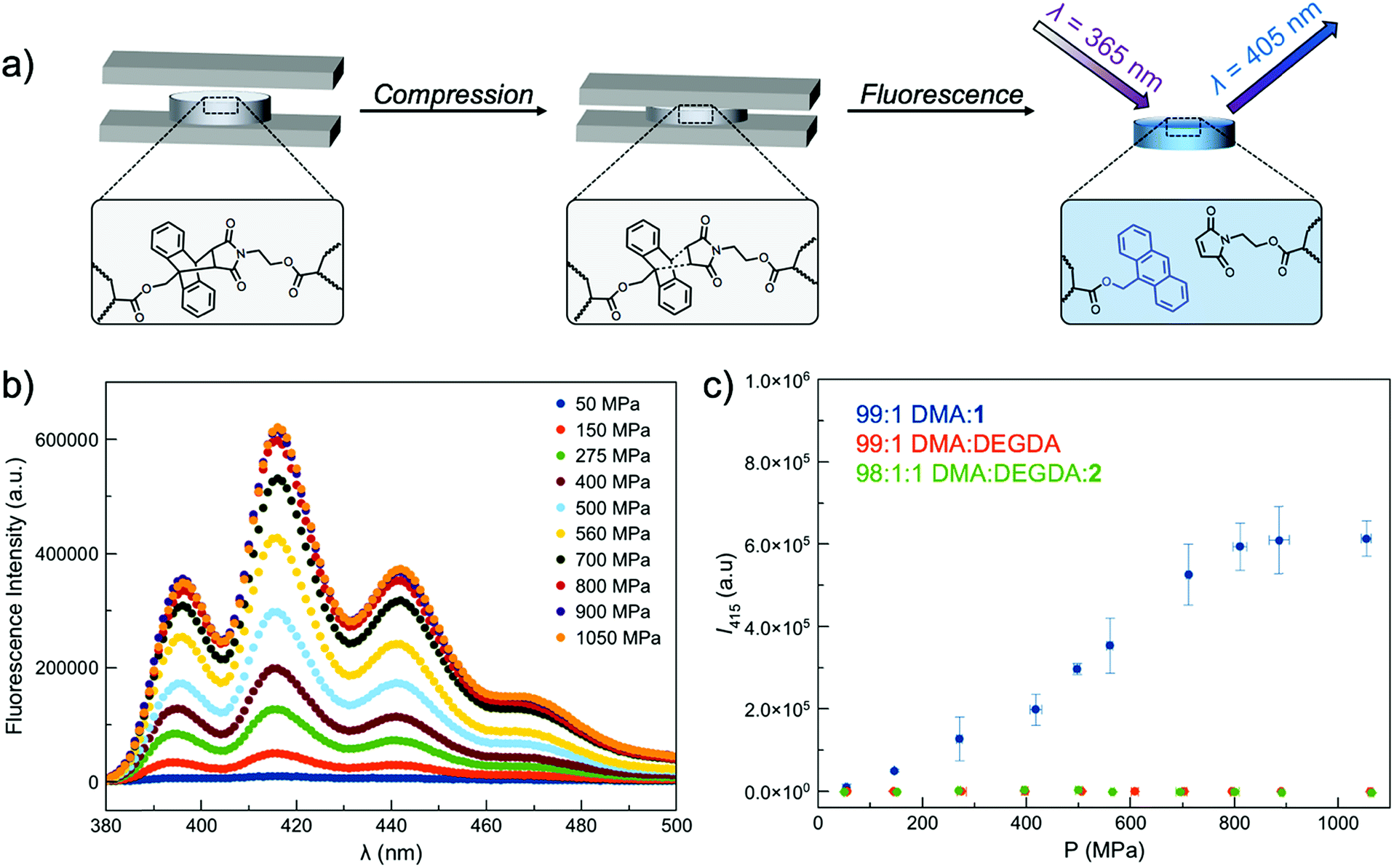 | ||
| Fig. 2 (a) Schematic of Anth–Mal cycloreversion under compression, which leads to liberation of anthracene and a detectable fluorescence signal. (b) Solid-state fluorescence spectra of compressed PDMA networks. (c) Plot of maximum fluorescence intensity (λ = 415 nm) for compressed PDMA networks. DMA:N,N-dimethylacrylamide; DEGDA: di(ethylene glycol) diacrylate; 1: mechanophore-containing crosslinker; 2: mechanophore-containing monomer. | ||
We expected the ability of the network to dissipate an applied stress would be diminished with increasing crosslink density, as stresses would be more readily localized at crosslinking junctions, leading to more rapid failure of Anth–Mal bonds.44 When a force is applied, elongation of the polymer chains leads to chain rupture once a critical rupture force is reached. This process becomes more likely as the length of the stretched chain approaches the contour length of the polymer, and is therefore more pronounced with shorter chains.44 Thus, as the crosslink density of the network is increased and the length of the parent chains (i.e., linear segments connected by crosslinking junctions) consequently decreases, greater degrees of activation are expected.45 To test this hypothesis, we prepared networks with high crosslink densities (i.e., 5 and 10 mol%) by adding the desired amount of the non-responsive crosslinker DEGDA (i.e., 4 and 9 mol%) along with the responsive 1 (1 mol% for all samples, Fig. S29–S48†). The Young's moduli of the networks were measured by indenting 1 mm thick samples with a hemispherical glass indentation tip and fitting the Hertz contact model to the resulting force vs. displacement curves (Fig. S49†). As expected, increasing the crosslink density resulted in an increased modulus, ranging from E* = 1.33 MPa for 1 mol% crosslinked networks to E* = 9.00 MPa for 10 mol% crosslinked networks (Fig. S50†). After compression with a hydraulic press, the fluorescence intensity for each was compared against a calibration curve of 9-anthracenemethanol embedded in PDMA networks (Fig. S51†) to normalize for the initial concentration of 1 in each responsive network. Over the range of crosslinking densities considered, there is not a significant difference in activation profiles (Fig. 3). Furthermore, it was observed that each of the samples plateau around a similar value, ranging from 4–5% mechanophore activation. While these results do not clarify the definitive role of crosslinking density on mechanophore activation, cycloreversion is observed in each case to an extent such that the emitted light is intense enough to be observed by eye when irradiated with long-wave UV light.
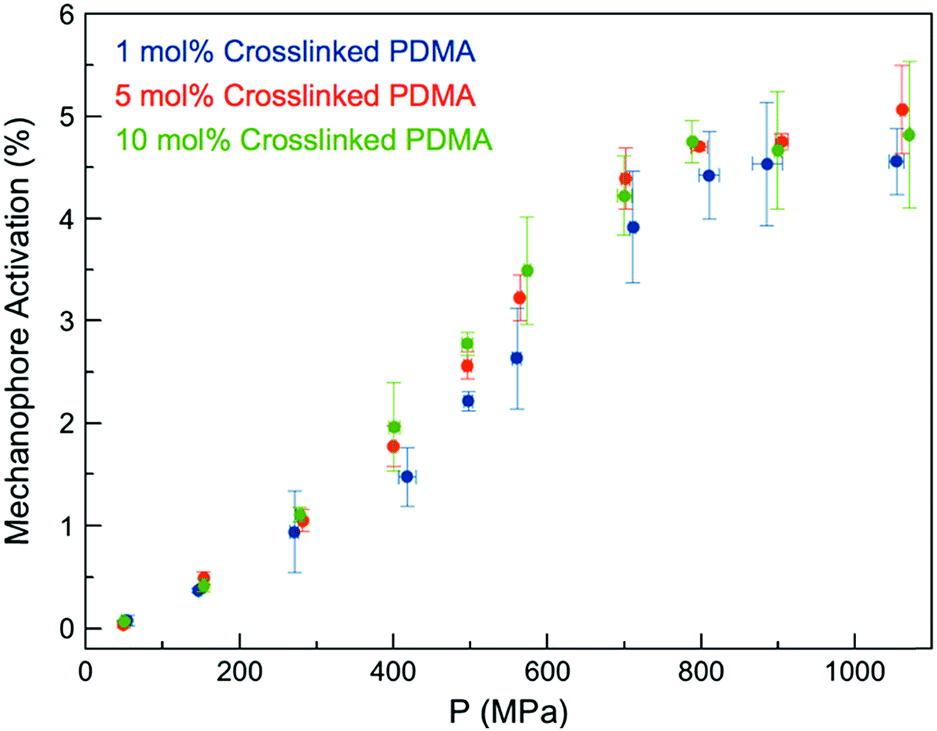 | ||
| Fig. 3 Anth–Mal mechanophore activation in AnthMalDA-crosslinked PDMA networks with varying crosslink densities. | ||
The mechanochemical activation of Anth–Mal linkages provides a unique opportunity to encode information into polymeric materials using an “invisible ink.” Patterns stamped into the networks under compression may be invisible or nearly invisible unless illuminated with UV light (Fig. 4a). To examine this process, we prepared a stamp with a raised anthracene pattern (see ESI for details†). A rectangular network consisting of 99:1 DMA:1 was prepared. Before compression, no large defects were observed (Fig. 4b), and no fluorescence was detected when irradiated with a UV lamp (Fig. 4c). After stamping under a pressure of approximately 150 MPa, the pattern was visible under white light (Fig. 4d) and fluoresced blue when irradiated with 365 nm light (Fig. 4e and f). Interestingly, the pattern was no longer visible under white light two days after compression, but the fluorescence could still be observed under UV irradiation (Fig. S52†). This indicates that even after relaxation of the polymer network, the anthracene–maleimide bonds do not reform to a significant extent, so the fluorescence signal of anthracene is still detected. The control network, consisting of P(DMA0.99-co-DEGDA0.01), also showed no defects (Fig. 4g) or response under UV irradiation (Fig. 4h). After stamping the material, the anthracene imprint is clearly observed (Fig. 4i), but no fluorescence signal is detected under UV irradiation (Fig. 4j and k).
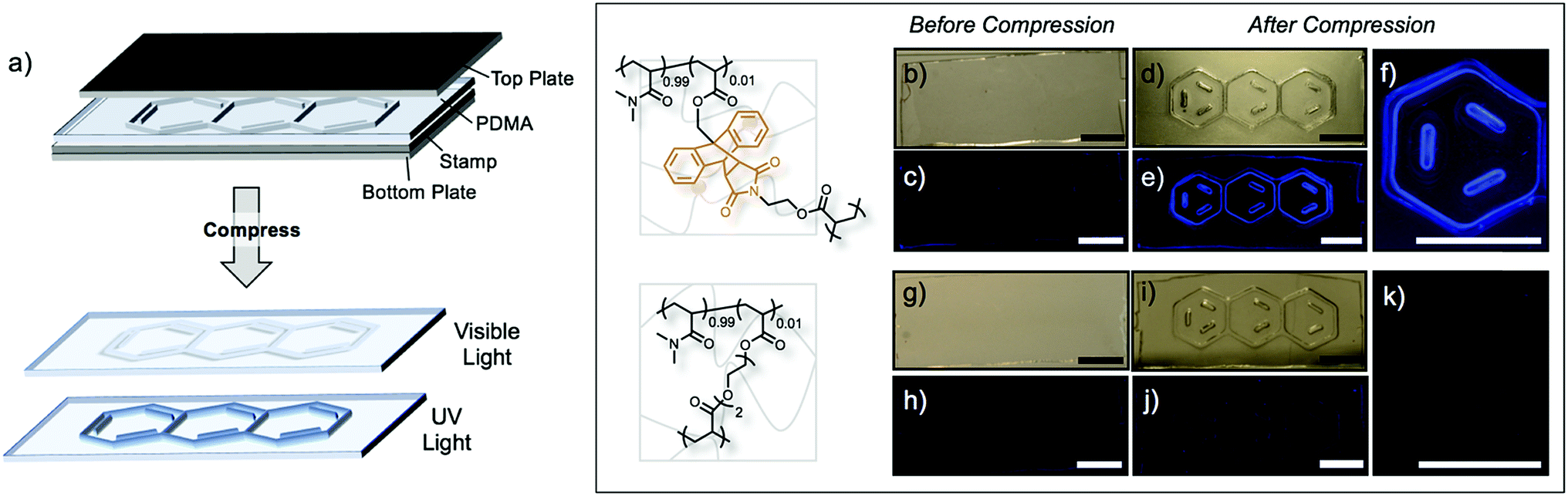 | ||
| Fig. 4 (a) Stamping “invisible ink” into PDMA networks crosslinked by AnthMalDA leads to a fluorescence signal in the shape of the pattern. 99:1 DMA:AnthMalDA networks before compression under (b) white light and (c) UV light show no defects. After stamping, the anthracene pattern is apparent under (d) white light and (e and f) UV light. 99:1 DMA:DEGDA networks before compression under (g) white light and (h) UV light show no defects. After stamping, the anthracene pattern is apparent under (i) white light, but no signal is detectable under UV light (j and k). | ||
Traditionally, mechanophores are designed in such a way that the elongated polymer chains are in alignment with the labile bond. As such, applied forces lead to bond elongation and ultimately chain scission. Alternatively, Boydston and coworkers have introduced the nascent concept of “flex” activation, in which the labile bonds are orthogonal to the elongated polymer backbone.27,28,32 In this mechanism, the resultant bond bending and/or bond angle distortion leads to cleavage of the mechanophore and release of a small molecule (Scheme 1).
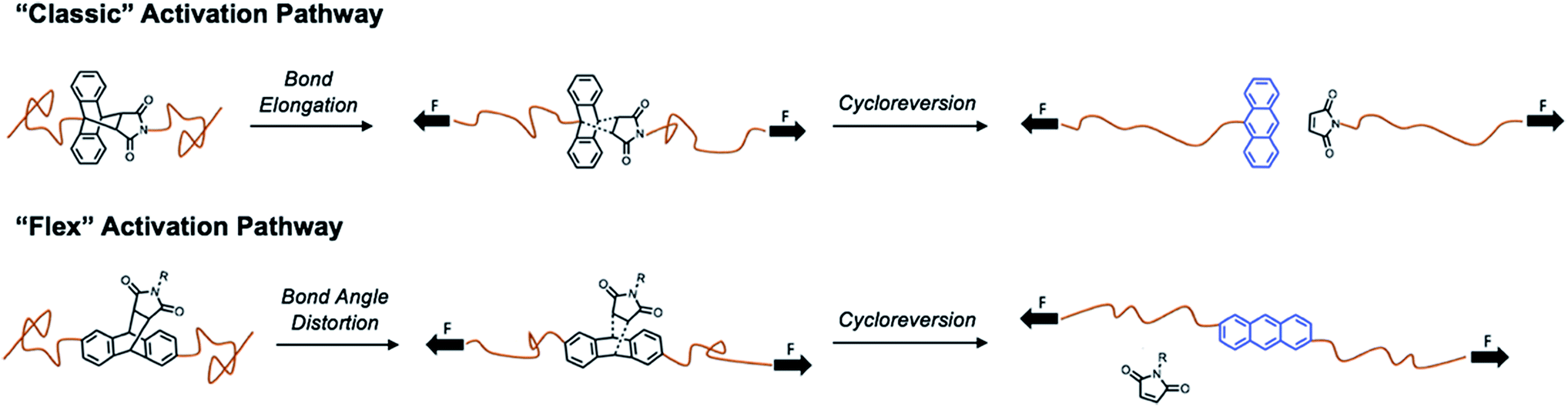 | ||
| Scheme 1 Comparison between “classic” mechanophore activation pathway, in which bond stretching promotes cycloreversion, and “flex” activation, in which the force is directed perpendicular to the axis of cycloreversion, such that bond bending leads to cycloreversion. | ||
By design, this pathway does not lead to destruction of network integrity upon activation. All examples to date have relied on the use of [4 + 2] cycloadducts, which have been shown to undergo cycloreversion due to deformations induced by mechanical force. Therefore, we designed an isomeric “flex” crosslinker in which the polymer chains would be connected via the 2- and 6-positions of the anthracene ring (Fig. S53–S68†). Networks crosslinked with the flex mechanophore were prepared and subjected to compression as described above. However, under the conditions studied, no cycloreversion was detectable by solid-state fluorescence spectroscopy (Fig. S69 and S70†). Furthermore, the samples were heated to 75 and 100 °C and subjected to 1050 MPa compressive loads. No fluorescence signal was observed (Fig. S71†), indicating a lack of cycloreversion, even under these harsh conditions. When heated to 200 °C, a small percentage of free anthracene is noted, but compression at this temperature yields a negligible increase in the degree of cycloreversion. This is not unprecedented, as work by Craig and coworkers on Anth–1,2,4-triazoline-3,5-dione adducts necessitated the use of high temperatures under tension to achieve small degrees of activation.29 Alternatively, in Boydston's pioneering work, adducts of furans and alkynes have much lower energies of activation, and therefore are more applicable as flex mechanophores.27 From our comparison, it is apparent that the necessary energy for flex activation is larger than classic activation, and further examination is needed to elucidate the pertinent design criteria for a successful flex mechanophore.
Conclusions
In summary, we have prepared networks crosslinked by mechanoresponsive Anth–Mal adducts. When the networks are subjected to compressive forces, cycloreversion of the adduct occurs, liberating fluorescent anthracene groups. As expected, the extent of cycloreversion is strongly dependent on the applied pressure; higher fluorescence intensities are measured with larger pressures. When compressed with a patterned stamp, the raised areas of the stamp imprinted the network. In the imprinted areas, cycloreversion to anthracene yielded a fluorescent signal. We demonstrated this technique by preparing an anthracene-shaped stamp. After the polymeric network had relaxed, the imprinted pattern was invisible under white light. However, under UV irradiation, the damaged crosslinking points fluoresced in the shape of the pattern.Mechanophores that result in a visual output when damaged have proven valuable in many applications. For example, damage-reporting objects28,46 have been developed, in which stresses on the material can be easily visualized. The use of mechanophores has been extended into prototyping using additive manufacturing, in which optimization of object design can be achieved by visualizing potential failure points. This visualization allows for the creation of more robust objects that are reinforced at areas that have a high likelihood of failure. Furthermore, it may be possible to use the fluorescent response to compression to aid in recycling, where plastics can be stamped and sorted based on the output from the imprinted area, or anticounterfitting applications. The type and/or the optical response may be tuned simply by altering the mechanophores. In the case of anthracene derivatives, substitutions along the aromatic rings can greatly influence the electronics, providing access to a wide range of colors. Finally, further understanding of the principles that guide mechanophore design (namely, the criteria for a successful flex mechanophore) will provide access to mechanophores that imbue numerous beneficial properties to polymeric networks.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted with Government support under and awarded by DoD through the ARO (W911NF-17-1-0326). The authors would like to thank Dr James D. Bullock for his assistance with the solid-state fluorescence spectrometer setup.References
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14 CrossRef CAS PubMed.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Nat. Chem., 2017, 9, 817 CrossRef CAS PubMed.
- G. B. Lyon, A. Baranek and C. N. Bowman, Adv. Funct. Mater., 2016, 26, 1477 CrossRef CAS.
- G. J. Berg, T. Gong, C. R. Fenoli and C. N. Bowman, Macromolecules, 2014, 47, 3473 CrossRef CAS.
- C. P. Kabb, C. S. O'Bryan, C. C. Deng, T. E. Angelini and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2018, 10, 16793 CrossRef CAS PubMed.
- M. B. Gordon, J. M. French, N. J. Wagner and C. J. Kloxin, Adv. Mater., 2015, 27, 8007 CrossRef CAS PubMed.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678 CrossRef CAS PubMed.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277 CrossRef CAS.
- S. Mukherjee, M. R. Hill and B. S. Sumerlin, Soft Matter, 2015, 11, 6152 RSC.
- C. C. Deng, W. L. A. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Soft Matter, 2009, 5, 2347 RSC.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142 CrossRef CAS.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098 CrossRef CAS.
- J. J. Cash, T. Kubo, D. J. Dobbins and B. S. Sumerlin, Polym. Chem., 2018, 9, 2011 RSC.
- H. Staudinger, Ber. Dtsch. Chem. Ges. B, 1930, 63, 921 CrossRef.
- H. Staudinger and H. F. Bondy, Ber. Dtsch. Chem. Ges. B, 1930, 63, 734 CrossRef.
- H. Staudinger and E. O. Leupold, Ber. Dtsch. Chem. Ges. B, 1930, 63, 730 CrossRef.
- W. Kauzmann and H. Eyring, J. Am. Chem. Soc., 1940, 62, 3113 CrossRef CAS.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13 CrossRef CAS PubMed.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423 CrossRef CAS PubMed.
- C. E. Diesendruck, G. I. Peterson, H. J. Kulik, J. A. Kaitz, B. D. Mar, P. A. May, S. R. White, T. J. Martínez, A. J. Boydston and J. S. Moore, Nat. Chem., 2014, 6, 623 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181 CrossRef CAS PubMed.
- J. Wang, I. Piskun and S. L. Craig, ACS Macro Lett., 2015, 4, 834 CrossRef CAS.
- M. B. Gordon, S. Wang, G. A. Knappe, N. J. Wagner, T. H. Epps and C. J. Kloxin, Polym. Chem., 2017, 8, 6485 RSC.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189 CrossRef CAS PubMed.
- B. Cao, N. Boechler and A. J. Boydston, Polymer, 2018, 152, 4 CrossRef CAS.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216 CrossRef CAS.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577 CrossRef CAS PubMed.
- G. I. Peterson, M. Turtoglu, M. B. Larsen, S. L. Craig, M. A. Ganter, D. W. Storti and A. J. Boydston, Rapid Prototyp. J., 2015, 21, 520 CrossRef.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2014, 136, 1276 CrossRef CAS PubMed.
- J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Price and D. M. Haddleton, Polym. Chem., 2010, 1, 102 RSC.
- J. Li, T. Shiraki, B. Hu, R. A. E. Wright, B. Zhao and J. S. Moore, J. Am. Chem. Soc., 2014, 136, 15925 CrossRef CAS PubMed.
- D. C. Church, G. I. Peterson and A. J. Boydston, ACS Macro Lett., 2014, 3, 648 CrossRef CAS.
- H. Li, R. Göstl, M. Delgove, J. Sweeck, Q. Zhang, R. P. Sijbesma and J. P. A. Heuts, ACS Macro Lett., 2016, 5, 995 CrossRef CAS.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177 CrossRef CAS.
- C. Calvino and C. Weder, Small, 2018, 14, 1802489 CrossRef PubMed.
- B. R. Crenshaw, M. Burnworth, D. Khariwala, A. Hiltner, P. T. Mather, R. Simha and C. Weder, Macromolecules, 2007, 40, 2400 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed.
- C. K. Lee, B. A. Beiermann, M. N. Silberstein, J. Wang, J. S. Moore, N. R. Sottos and P. V. Braun, Macromolecules, 2013, 46, 3746 CrossRef CAS.
- C. M. Degen, P. A. May, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2013, 46, 8917 CrossRef CAS.
- A. R. Sulkanen, J. Sung, M. J. Robb, J. S. Moore, N. R. Sottos and G.-y. Liu, J. Am. Chem. Soc., 2019, 141, 4080 CrossRef CAS PubMed.
- F. J. Vernerey, R. Brighenti, R. Long and T. Shen, Macromolecules, 2018, 51, 6609 CrossRef CAS.
- G. J. Lake and A. G. Thomas, Proc. R. Soc. London, Ser. A, 1967, 300, 108 CrossRef CAS.
- G. R. Gossweiler, C. L. Brown, G. B. Hewage, E. Sapiro-Gheiler, W. J. Trautman, G. W. Welshofer and S. L. Craig, ACS Appl. Mater. Interfaces, 2015, 7, 22431 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02487e |
| This journal is © The Royal Society of Chemistry 2019 |