
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering protein polymers of ultrahigh molecular weight via supramolecular polymerization: towards mimicking the giant muscle protein titin†
Ruidi
Wang
ab,
Jiayu
Li
b,
Xiumei
Li
a,
Jin
Guo
b,
Junqiu
Liu
 a and
Hongbin
Li
*b
a and
Hongbin
Li
*b
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China
bDepartment of Chemistry, The University of British Columbia, Vancouver, BC V6T 1Z1, Canada. E-mail: hongbin@chem.ubc.ca
First published on 20th August 2019
Abstract
The giant muscle protein titin is the largest protein in cells and responsible for the passive elasticity of muscles. Titin, made of hundreds of individually folded globular domains, is a protein polymer with folded globular domains as its macromonomers. Due to titin's ultrahigh molecular weight, it has been challenging to engineer high molecular weight artificial protein polymers that mimic titin. Taking advantage of protein fragment reconstitution (PFR) of a small protein GB1, which can be reconstituted from its two split fragments GN and GC, here we report the development of an efficient, PFR-based supramolecular polymerization strategy to engineer protein polymers with ultrahigh molecular weight. We found that the engineered bifunctional protein macromonomers (GC-macromonomer-GN) can undergo supramolecular polymerization, in a way similar to condensation polymerization, via the reconstitution of GN and GC to produce protein polymers with ultrahigh molecular weight (with an average molecular weight of 0.5 MDa). Such high molecular weight linear protein polymers closely mimic titin and provide protein polymer building blocks for the construction of biomaterials with improved physical and mechanical properties.
1 Introduction
Naturally occurring elastomeric proteins are placed under mechanical stress during a wide range of biological processes, such as cell adhesion and muscle contraction, and serve as molecular springs to provide tissues with the desired mechanical properties, including elasticity, strength and toughness.1–8 A common feature of such elastomeric proteins is their tandem modular construction: they are composed of individually folded globular domains that are placed in tandem and their conformations can be viewed as pearls-on-a-string.1–7,9 From a polymer science perspective, such tandem modular proteins can be considered as protein polymers with individually folded globular protein domains as their macromonomers.The giant muscle protein titin is a representative of such tandem modular protein-based polymers. Titin has an ultrahigh molecular weight (MW): it is the largest single polypeptide chain in the cell and consists of more than 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 amino acid residues with a MW of more than 3.5 MDa.1,10,11 Titin is composed of more than two hundred individually folded immunoglobulin-like (Ig) and fibronectin-type III domains. These domains are each about 90 residues long and serve as the macromonomer for titin.11,12 The I-band part of titin is extensible and is largely responsible for the passive elastic properties of muscles.7,13 Truncation of titin, which leads to a much reduced MW, leads to the dysfunction of muscles, including cardiomyopathy.14
000 amino acid residues with a MW of more than 3.5 MDa.1,10,11 Titin is composed of more than two hundred individually folded immunoglobulin-like (Ig) and fibronectin-type III domains. These domains are each about 90 residues long and serve as the macromonomer for titin.11,12 The I-band part of titin is extensible and is largely responsible for the passive elastic properties of muscles.7,13 Truncation of titin, which leads to a much reduced MW, leads to the dysfunction of muscles, including cardiomyopathy.14
Due to its unique nanomechanical properties and its role in muscle mechanics, titin has attracted considerable interest as a potential building block for constructing protein-based biomaterials.15–17 However, it has been challenging to engineer titin-mimetic protein polymers with ultrahigh molecular weight. Even for the smallest titin isoform (the N2B cardiac titin), its I-band has a MW of ∼0.5 MDa with a contour length of ∼150 nm in its native state, due to the folded globular nature of the macromonomer.11 From the polymer length point of view, the I-band of human N2B cardiac titin is equivalent to a polystyrene of a MW of ∼110 kDa.
Various methods have been developed to engineer protein polymers, such as gene concatemerization and chemical conjugation (including disulfide bond formation and thiol–maleimide coupling).18–23 However, the resultant protein polymers have low degrees of polymerization. For example, the widely used polyprotein (I27)8 has a MW of 80 kDa and a contour length of ∼30 nm,15 which is equivalent to a polystyrene of a MW of only ∼21 kDa (a degree of polymerization of 200). Thus, such protein polymers in their folded states can only be considered as oligomers. The advantages/features associated with ultrahigh MW protein polymer titin thus cannot be fully realized in such oligomeric protein polymers. Here we report a protein fragment reconstitution-based supramolecular polymerization approach to synthesize artificial titin-mimetic protein polymers with ultrahigh molecular weight.
It is known that some proteins can be split into two fragments, which can then recognize each other via supramolecular interactions and reconstitute the folded conformation of the native protein spontaneously, a process called protein fragment reconstitution (PFR).24,25 The small protein GB1, the B1 IgG binding domain of protein G from Streptococcus (GB1), is one of such proteins.26 GB1 consists of 56 residues and assumes a β-grasp fold where an α-helix packs against a four stranded-β-sheet. When split into two fragments from the loop connecting the α-helix and β-strand 3, the two fragments A (1–40 aa) and B (41–56 aa) can reconstitute at a 1:1 ratio into the native GB1 fold, albeit with a nick in the loop.26–30 This non-covalent “GB1” fold has a dissociation constant Kd of ∼9 × 10−6 M.26 In our previous work, we engineered a loop elongation variant of GB1, termed GL5CC, where 5 residues were inserted into the unstructured loop and residues 42/44 were mutated to cysteines.31,32 When split into fragments GN (residue 1–42) and GC (residues 43–61), GN and GC can reconstitute into GB1's native fold (Fig. 1A). Under oxidizing conditions, the two cysteine residues can form a disulfide bond in the reconstituted GL5CC, converting the reconstituted noncovalent GL5CC to a covalently connected polypeptide chain.30 These unique properties make the PFR of GL5CC an attractive supramolecular driving force, which has been used to construct supramolecular hydrogels.30 Going a step further to fully explore the utility of PFR as a general supramolecular driving force, here we demonstrate the use of the PFR for engineering protein polymers with ultrahigh molecular weight via supramolecular polymerization. Our study not only enables the engineering of titin-mimetic protein polymers, but may also open up a new avenue toward addressing the long-standing challenge in engineering protein polymers with ultrahigh molecular weight for technological applications, such as spider silk production.
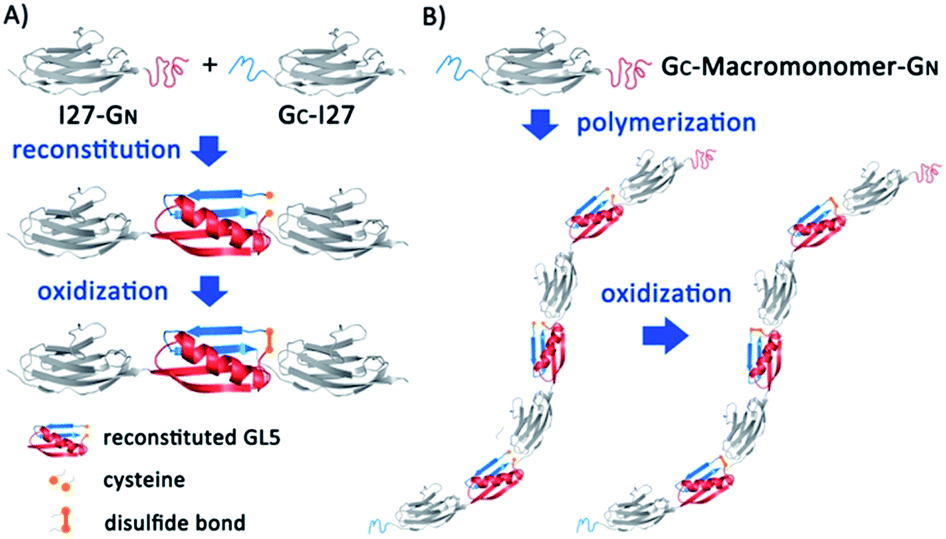 | ||
| Fig. 1 Schematic representation of protein-based polymer construction induced by supramolecular interactions. (A) Driven by protein fragment reconstitution, I27-GN and GC-I27 recognize and reconstitute to form native I27-(GN–GC)-I27 with a nick between two cysteines. After oxidation, the reconstituted GL5CC transforms into a covalently linked protein chain locked by the disulfide bond. (B) The bifunctional building block GC-macromonomer-GN assembles into a protein polymer spontaneously through the reaction between GN and GC, and disulfide bonds forming under oxidizing conditions can further stabilize the whole structure. | ||
2 Experimental
2.1 Protein engineering
GN and GC are two fragments from GL5CC we constructed previously.30,32 GL5CC is split into two fragments at residue 42: GN is the N-terminal fragment (residues 1–42) and GC is the C-terminal fragment (residues 43–61). Both GN and GC were amplified with the template of GL5CC–27w34f through polymerase chain reaction (PCR). In our design, GN was flanked with 5′ BamHI and 3′ KpnI restriction sites, respectively, while GC was flanked with 5′ BamHI and 3′ BglII-KpnI restriction sites, respectively. I27w34f carrying 5′ BamHI and 3′ BglII-KpnI restriction sites was constructed as described previously.33 For simplicity, I27 was referred to as I27w34f in this study. Digestion of GN with restriction endonuclease BamHI and KpnI resulted in overhanging “sticky ends” whose sequence corresponded to that of the pQE80L-I27 vector digested with BglII and KpnI, and the sticky-ended GN insert can be ligated into the digested pQE80L-I27 vector to form pQE80L-I27-GN. In a similar way, we also constructed pQE80L-GC-I27 and pQE80L-GC-I27-GN plasmids.After transforming plasmids containing the desired gene into Escherichia coli strain DH5α, protein overexpression was carried out. The overnight starter was inoculated into the LB liquid medium with 100 μg mL−1 ampicillin and incubated until OD600 is about 0.6–0.8 at 37 °C. Then, 1 mM isopropyl-1-β-D-thiogalactoside was added to the culture to induce protein overexpression. Protein overexpression continued for about 4 hours. Then the cells were harvested by centrifugation at 5000 rpm, 4 °C for 10 min. The cells were lysed with 100 mg mL−1 lysozyme. Proteins were purified as soluble monomers from the supernatant using Co2+ affinity chromatography. Excess salts in the eluted proteins samples were removed by dialysis against water for 24 hours and the dialyzed protein samples were then lyophilized. Amino acid sequences of all the constructs used in this study are detailed in Table S1 in the ESI.† It is important to note that a temperature of 37 °C and the reducing cytoplasm of E. coli are essential in preventing the polymerization of GC-I27-GN in E. coli and ensure that GC-I27-GN is expressed as a soluble monomer, as the melting temperature of the reconstituted, reduced GL5CC is 23 °C.
2.2 Supramolecular polymerization
To polymerize GC-I27-GN into high molecular weight protein polymers, lyophilized GC-I27-GN was first dissolved in 1 × PBS (pH = 7.4) to a concentration of 30 μM, and the supramolecular polymerization via PFR was carried out at 4 °C overnight. To obtain covalently linked protein polymers, the solution was allowed to be oxidized by air oxygen. In order to analyze the time course of polymerization, the dissolved protein was first reduced by 3 mM DTT for 2 h at room temperature. Then DTT was removed by passing the protein solution through a desalting column. The protein solution was then incubated at 4 °C for various time periods (10 min, 30 min, 70 min, 3 h, 6 h, 1 d and 2 d). During this process, the protein was oxidized by air oxygen.2.3 Stopped-flow spectrofluorimetry measurements
Stopped-flow experiments were carried out on a Biologic SFM-4 stopped-flow instrument. Through monitoring the tryptophan fluorescence at 350 nm, association of I27-GN and GC-I27 and dissociation of the I27-(GN–GC)-I27 complex were monitored. Since I27 (the I27w34f) does not carry tryptophan, the fluorescence of tryptophan reflects the association/dissociation of GN and GC. In the association measurements, I27-GN and GC-I27 solution with the same initial protein concentration containing 3 mM DTT were mixed in a 1:1 ratio. For unfolding measurements, equal amounts of I27-GN and GC-I27 were mixed and reduced by 3 mM DTT first, then 4 M GdmCl solution was added to the I27-(GN–GC)-I27 complex in order to denature the reconstitution domain. The same processes were carried out for individual I27-GN and GC-I27 as negative controls. Protein concentrations in all tests were adjusted to 20 μM. The association kinetics curve was fitted to the second-order rate law. The dissociation kinetics curves were fitted to the first-order rate law.
2.4 Fast protein liquid chromatography (FPLC)
FPLC experiments were carried on an Akta FPLC system and samples were separated by using a HiLoad Superdex200 pg preparative size exclusion chromatography column (GE Healthcare). The samples were analyzed and eluted with a 20 mM, pH = 7.5 phosphate buffer containing 100 mM NaCl at a constant flow rate of 1 mL min−1. For samples needed to be kept under reduced conditions, 1 mM DTT was also included in mobile phase buffer. In each run, 5 mL of 0.2 mM protein solution was injected. A UV detector was set to monitor 280 nm.The SEC calibration was achieved by using the following protein standards: (1) amylase: 200 kDa; (2) alcohol dehydrogenase: 150 kDa; (3) bovine serum albumin: 66 kDa; (4) carbonic anhydrase: 29 kDa and (5) cytochrome c: 12.4 kDa.
2.5 Dynamic light scattering (DLS) experiments
DLS experiments were carried out on a NanoBrook Omni particle size and zeta potential analyser (Brookhaven Instruments). Lyophilized proteins were dissolved in deionized water at a concentration of 1 mg mL−1. Before experiments, protein solutions were filtered using 0.22 μm syringe filters. All measurements were performed in triplicate at 25 °C and the time duration of each measurement was 120 s. The diffusion-averaged Mw was calculated from the Mark–Houwink–Sakurada (MHS) equation D = K × Mwα, where D is the diffusion coefficient, and macromolecule dependent constants K and α were determined using proteins of known molecular weight.2.6 Single-molecule force spectroscopy experiments
Single-molecule atomic force microscopy (AFM) measurements were performed on a custom-built AFM as described previously.33 1 μL protein polymer solution (0.2 mM) was deposited onto a clean glass coverslip covered with about 50 μL of PBS and was allowed to adsorb onto the glass coverslip for ∼5 min before the force-extension measurements. Experiments were performed at room temperature in PBS buffer (pH 7.4). The spring constant of each individual cantilever (Si3N4 cantilevers from Brucker, with a typical spring constant of 40 pN nm−1) was calibrated in PBS buffer using the equipartition theorem before each experiment. The pulling experiments were carried out in the constant speed mode. The pulling speed used in the AFM experiments was 400 nm s−1. The technical details of the AFM pulling experiments can be found elsewhere.18,33 Briefly, in a typical experiment, the cantilever was brought into contact with the protein sample at a contact force of ∼1 nN, and the cantilever was then allowed to retract. Due to nonspecific physisorption, some protein polymers could be picked up and stretched by the AFM cantilever. The resulting force-extension curve was recorded and analysed using custom-written codes in Igor Pro.2.7 Hydrogel preparation
Protein hydrogels were prepared using the well-developed Ru(II)(bpy)32+-mediated photochemical crosslinking method. The lyophilized protein samples were dissolved in PBS at the desired concentration and were allowed to be oxidized by air oxygen. Then ammonium persulfate (APS) and Ru(II)(bpy)32+ were added to the protein solution to a final concentration of 50 mM and 0.2 mM, respectively. A 200 W fibre optic white light source was used to irradiate the sample for 10 min at a height of 10 cm to crosslink the protein aqueous solution into hydrogels. To redissolve the hydrogel, the hydrogel was incubated in PBS buffer containing 30 mM dithiothreitol (DTT) at room temperature overnight and then at 50 °C for 1 hour.3 Results and discussion
3.1 Design principle
To use PFR as a supramolecular driving force to engineer titin-mimetic protein polymers, we used the 27th immunoglobulin-like domain12 (I27, also called I91 according to ref. 11) from human cardiac titin as the macromonomer and the GN and GC fragments as two reactive functional groups to build bifunctional macromonomers. We reason that the PFR between GN and GC should allow the bifunctional macromonomers to undergo supramolecular polymerization in a way similar to condensation polymerization (Fig. 1).The PFR of GN and GC is directional, thus placing them in proper orientation relative to the macromonomer is essential for the construction of protein polymers with a high degree of polymerization. Fusing GN to the N-terminus of the protein macromonomer and GC to the C-terminus, the PFR of GL5CC will place the protein macromonomers right next to each other, resulting in a large steric hindrance that will likely prevent further reactions (Fig. S1†). In contrast, fusing GC to the N-terminus and GN to the C-terminus of the protein macromonomer will result in no steric hindrance, thus facilitating further reactions (Fig. 1).
To test if such an arrangement can lead to efficient PFR, we constructed mono-functional fusion proteins I27-GN and GC-I27. Since PFR relies on noncovalent supramolecular interactions, we used native polyacrylamide gel electrophoresis (PAGE), size-exclusion fast protein liquid chromatography (SE-FPLC) and stopped-flow fluorimetry to confirm the PFR of the I27-GN and GC-I27 under native and reducing conditions, where the disulfide bond formation is prevented. As shown in Fig. 2A, upon mixing I27-GN with GC-I27 at a 1:1 molar ratio, the original bands for I27-GN (lane 1) and GC-I27 (lane 2) disappeared. Instead, a new band emerged (lane 3), indicating that GN-I27 and I27-GC can recognize each other under reducing conditions and undergo effective protein fragment reconstitution. It is well known that protein mobility in native PAGE is determined by a combination of factors, including protein conformation, surface charge and MW; native PAGE is less suitable to determine the MW.34 Thus, the relative position of the three bands cannot be used to directly compare their MW.
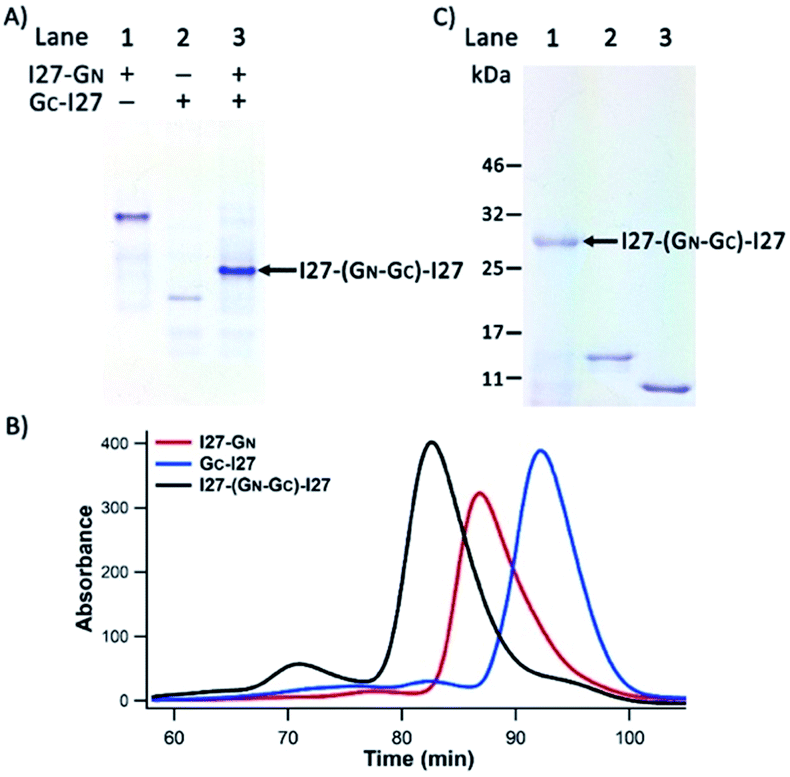 | ||
| Fig. 2 I27-GN and GC-I27 associated via PFR. (A) A photograph of 10% native-PAGE showed the PFR of I27-GN and GC-I27 into I27-(GN–GC)-I27 at 1:1 stoichiometry. Reduced I27-GN and GC-I27 were mixed at a 1:1 molar ratio (each at 20 μM), then analysed via native-PAGE at 4 °C. (B) SE-FPLC profiles of reduced I27-GN (red), GC-I27 (blue) and I27-(GN–GC)-I27 (black). The elution time is 89 min, 93 min and 82 min for I27-GN, GC-I27 and I27-(GN–GC)-I27, respectively. (C) Non-reducing 12% SDS-PAGE analysis of reconstituted I27-(GN–GC)-I27 under oxidizing conditions. Lane 1: the reconstituted mixture sample oxidized by air oxygen overnight; lane 2: reduced I27-GN and lane 3: reduced GC-I27. | ||
To further confirm the native PAGE results, we carried out SE-FPLC experiments (Fig. 2B). The 1:1 mixture of I27-GN and GC-I27 eluted at the shortest time, followed by I27-GN and GC-I27. This result clearly indicated the successful reconstitution of I27-GN and GC-I27 at a 1:1 ratio, and is consistent with the expected MW of I27-GN, GC-I27 and their 1:1 reconstituted supramolecular complex. The stopped-flow spectrofluorimetry results also confirmed the successful and rapid reconstitution of I27-GN and GC-I27 under reducing conditions (Fig. S2†).
To further stabilize the reconstituted noncovalent GN–GC complex, we used air oxygen to oxidize the two engineered cysteines in GN and GC to a disulfide bond in the reconstituted GN–GC complex30,31 (Fig. 1A), thus converting a non-covalently associated GN–GC complex into a covalently linked folded protein domain. Fig. 2C shows the non-reducing sodium dodecyl sulfate (SDS)-PAGE results. After boiling and SDS denaturation, the oxidized reconstituted complex I27-(GN–GC)-I27 appeared as a single band with a MW of ∼28 kDa, the sum of the two interacting protein fragments (I27-GN: 15 kDa and GC-I27: 13 kDa), corroborating that GN–GC indeed formed a covalently linked reconstituted protein domain after oxidation.
3.2 PFR of the bi-functional GC-macromonomer-GN leads to the engineering of protein polymers of ultrahigh molecular weight
To engineer titin-mimetic protein polymers, we engineered the bi-functional protein macromonomer: GC-I27-GN, where GC and GN were fused to the macromonomer at its N- and C-termini to minimize steric hindrance, respectively. Through PFR, the bi-functional protein macromonomers should react with each other in a way similar to the condensation polymerization reaction for polymers, to allow for the construction of supramolecular protein polymers. To convert the supramolecular protein polymer into a covalently linked protein polymer, we performed oxidation to form the disulfide bond within the reconstituted GN–GC complex.Indeed, we found that the macromonomers spontaneously started to polymerize in aqueous solution right after the protein purification step. Fig. S3† shows the native PAGE image of the resultant polymeric assemblies under reducing conditions, which correspond to the non-covalent supramolecular protein polymers formed by the PFR. Gradual oxidation of the reconstituted GL5CC complex by air oxygen resulted in the formation of high molecular weight, covalently linked protein polymers. It is evident from SDS-PAGE that most macromonomer GC-I27-GN reacted with each other and was converted into protein polymers with a high degree of polymerization (Fig. 3A).
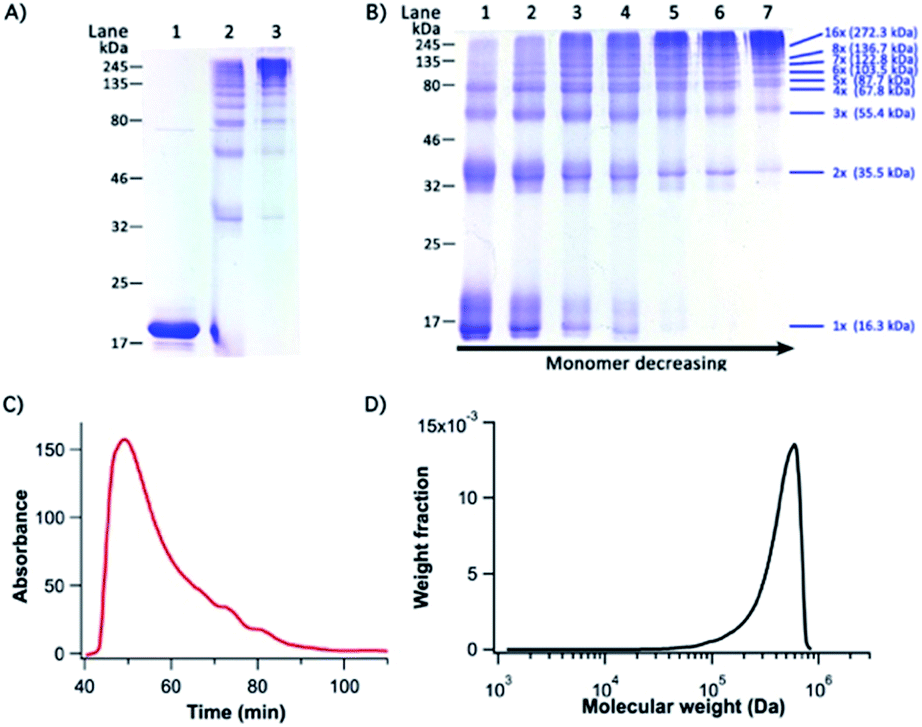 | ||
| Fig. 3 GC-I27-GN polymer characterization by SDS-PAGE and SE-FPLC (A) a photograph of a 12% SDS-PAGE characterizing GC-I27-GN-based supramolecular protein polymers. Lanes 1–3 are reduced GC-I27-GN, freshly prepared GC-I27-GN and GC-I27-GN-based protein polymers oxidized by air oxygen overnight, respectively. Lanes 2 and 3 are non-reduced SDS-PAGE. (B) Analysis for time course of polymerization of GC-I27-GN-based supramolecular protein polymers by non-reducing 12% SDS-PAGE. Lanes 1–7 are samples after incubation for 10 min, 30 min, 70 min, 3 h, 6 h, 1 d and 2 d. In both (A) and (B), 30 μM GC-I27-GN was used in the polymerization. (C) SE-FPLC elution time plot and (D) molecular weight distribution curve of polymerized GC-I27-GN-based protein polymers. In SE-FPLC experiments, 200 μM GC-I27-GN was used in the polymerization. | ||
Fig. 3B shows the time course of the polymerization by PFR. It is evident that as the reaction proceeded, the amount of protein polymers with higher MW increased while the fraction of the macromonomer and lower MW species (dimer and trimer) decreased with time and even got completely consumed. This is characteristic of the classical condensation polymerization and confirms that the supramolecular polymerization via PFR indeed follows a mechanism similar to that of condensation polymerization. It is clear that after one day of reaction, a high degree of polymerization was achieved, and only a trace amount of dimers was still present. A ladder of protein polymers with different degrees of polymerization was clearly visible. Protein polymers with a degree of polymerization higher than 10 were not well-resolved in the SDS-PAGE. To characterize the MW of the protein polymers, we carried out SE-FPLC (Fig. 3C and D). Fig. 3D shows the molecular weight distribution of the resultant protein polymers. The number average molecular weight (Mn) and weight average molecular weight (MW) of the protein polymers are ∼505 kDa and 542 kDa, respectively, giving rise to a MW polydispersity index (PDI) of 1.09. Dynamic light scattering measurements revealed a diffusion averaged MW of 458 kDa, further corroborating the high molecular weight of the engineered protein polymers (Fig. S4, ESI†). Such a high molecular weight of the protein polymers constructed from the PFR is much larger than that of any other protein polymer reported so far constructed by either gene concatenation18,33 or chemical conjugation,19,20 representing an efficient and successful attempt to engineer a protein polymer with high molecular weight that is close to that of the I-band cardiac N2B titin.
To further prove that the polymerization is through the head-to-tail supramolecular condensation reaction, we included monofunctional species in the reaction mixture. The introduction of monofunctional reactants into a condensation polymerization reaction will terminate the growing polymer chain and significantly reduce the MW of the resultant polymers. Indeed, in the PRF-based supramolecular condensation reaction, the inclusion of 17% GC-I27 in the reaction mixture (the ratio of GC-I27-GN:GC-I27 is 5:1) significantly reduced the amount of the high molecular weight products (Fig. S5†). Further increasing the concentration of GC-I27 eliminated the high molecular weight products completely. These results confirmed that the titin-mimetic protein polymers are indeed obtained via the PRF-based supramolecular condensation polymerization reaction.
Since the protein polymers are obtained via the PRF-based supramolecular condensation polymerization reaction, protein monomer concentration may play important roles in determining MW and MW polydispersity. Our preliminary results showed that a lower concentration of the GC-I27-GN monomer led to a lower degree of polymerization. For example, the degree of polymerization of GC-I27-GN at 5 μM is quite low (Fig. S6A†). When the monomer concentration is high enough, further increasing monomer concentration appeared to increase the polydispersity of the molecular weight (Fig. S6B and C, Table S2†). However, to gain a better understanding of this supramolecular polymerization reaction, a detailed study of the effect of protein concentration on polymer length and distribution is needed.
3.3 Single molecule spectroscopy experiments confirmed the nature of the protein polymer
We carried out single-molecule force spectroscopy experiments to further confirm that the titin-mimetic protein polymers constructed from PRF-based supramolecular polymerization are covalently linked, continuous polymer chains. As shown in Fig. 4, stretching the protein polymer (GC-I27-GN)n resulted in characteristic sawtooth-like force-extension curves, where the sawtooth peak corresponds to the mechanical unfolding of individual I27 domains and the reconstituted GL5CC domains in the protein polymer chain.18,33 Fitting the sawtooth peaks to the worm-like chain (WLC) model of polymer elasticity clearly revealed two populations of unfolding events: blue events with a contour length increment (ΔLc) of ∼18 nm, and events colored in red with a ΔLc of ∼28 nm, corresponding to the unfolding of the oxidized reconstituted GL5CC and I27 domains, respectively.32,33,35 The observed number of unfolding force peaks of GL5CC and I27 domains in a force-extension curve is often more than 13, indicating that the stretched protein polymer is at least a septamer of (GC-I27-GN)n. These observations indicated that the stretched protein polymers are indeed continuous protein polymers made of tandem repeats of (GC-I27-GN) linked by covalent bonds. It is well known that in single-molecule force spectroscopy experiments, a protein polymer is picked up randomly along the contour of the protein polymer chain; thus the observed number of unfolding force peaks can only provide a rough estimate of the length of the protein polymer chain.18 For example, stretching native titin, which contains hundreds of Ig and FnIII domains, often resulted in force-extension curves of ∼20–30 unfolding force peaks.36 Thus, our experimental results provide a lower estimate of the size of the constructed protein polymers.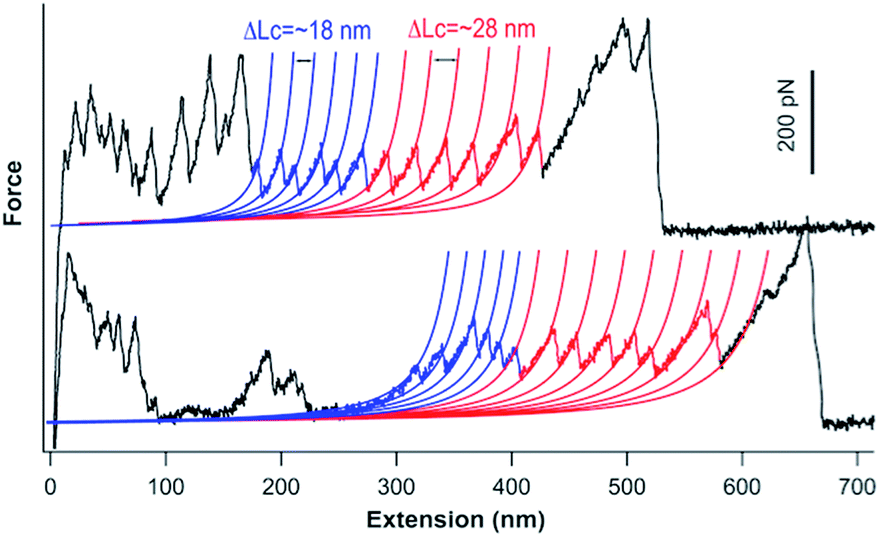 | ||
| Fig. 4 Representative force-extension curves of the GC-I27-GN-based protein polymers (200 μM GC-I27-GN was used in the polymerization). Blue and red lines correspond to the WLC fits to the experimental data with a ΔLc of ∼18 nm and 28 nm, respectively. Unfolding events colored in blue correspond to the unfolding of the reconstituted GL5CC, and events colored in red correspond to the unfolding of I27 domains. | ||
3.4 Ultrahigh molecular weight protein polymers allow for engineering protein hydrogels at lower concentrations with improved mechanical properties
In our previous work, we used protein polymers, such as (GB1)8, to construct protein-based hydrogels using a well-developed Ru(II)(bpy)32+-mediated photocrosslinking strategy, which crosslinked two tyrosine residues in proximity into dityrosine adducts under white light illumination.37,38 The lowest gelation point for these hydrogels typically was around 5% (50 mg mL−1), and such 5% hydrogels showed a storage modulus of ∼4 kPa.39 Compared with the high molecular weight protein polymers we engineered here, the protein polymers such as (GB1)8 we used can only be considered as oligomers, thus lacking advantages that could be entailed by high molecular weight protein polymers.The ultrahigh molecular weight titin-mimetic protein polymers we engineered here should allow for the engineering of protein hydrogels at much reduced protein concentration and with improved mechanical properties. To demonstrate this, we engineered protein-based hydrogels using the Ru(II)(bpy)32+-mediated photocrosslinking strategy. We found that GC-I27-GN-based polymer solution can be readily crosslinked into a solid hydrogel at a protein concentration as low as 1% (Fig. 5), and 1% protein hydrogels showed a storage modulus of 0.6 kPa. Higher protein concentrations lead to a higher storage modulus of the protein hydrogels (Fig. 5C). Compared with (GB1)8-based hydrogels (with the lowest gelation point of 5% and storage modulus of 4 kPa), the reduction in gelation concentration and improved mechanical properties of GC-I27-GN-based protein polymer hydrogels can be readily rationalized by the increased molecular weight of the protein polymers.
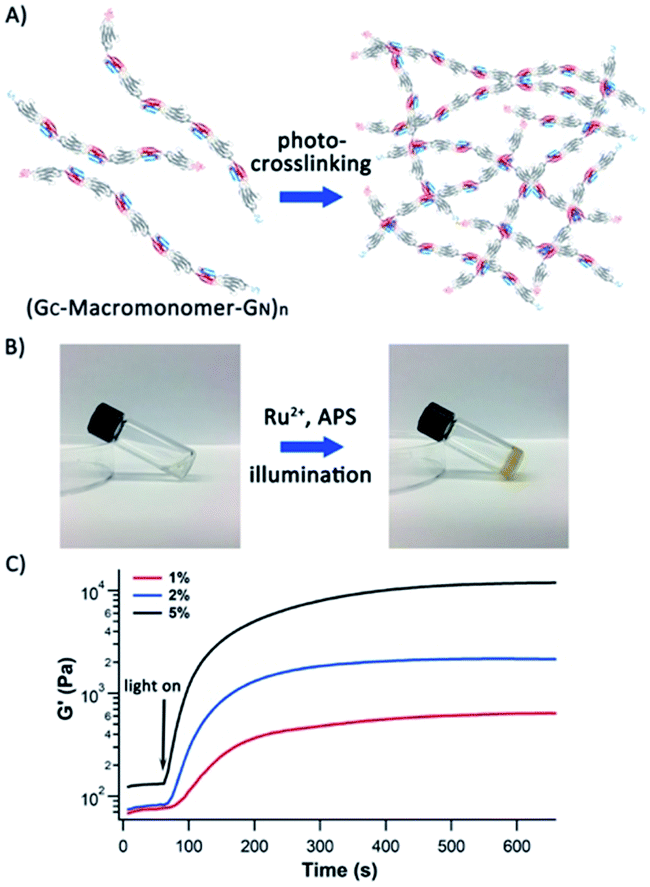 | ||
| Fig. 5 Protein hydrogels based on high molecular weight titin-mimetic protein polymers can be prepared at lower concentrations with improved mechanical properties. (A) A schematic of protein hydrogels constructed via the Ru2+-mediated photocrosslinking method. The protein building blocks are titin-mimetic (GC-macromonomer-GN)n-based protein polymers engineered via PFR. (B) 1% oxidized (GC-I27-GN)n solution can be readily photochemically crosslinked into solid protein hydrogels. (C) Photo-rheology of (GC-I27-GN)n based protein hydrogels. It is evident that the protein polymer solution can be crosslinked into protein hydrogels rapidly (typically less than two minutes). The storage moduli are ∼0.6 kPa, 2 kPa and 11 kPa for 1%, 2% and 5% hydrogels, respectively. | ||
Another unique feature of this protein hydrogel lies in its responsiveness to the redox potential. Under oxidizing conditions, the GC-macromonomer-GN-based protein polymer is a covalently linked single polypeptide chain. However, in the reduced state, it is a supramolecular protein polymer assembled from the non-covalently associated GC–GN complex via PFR when the temperature is lower than the melting temperature Tm of the reconstituted reduced GL5CC (which is ∼23 °C).30 When the temperature is higher than Tm, the reconstituted GC–GN complex will dissociate into GN and GC, leading to the depolymerization of the protein polymer (GC-I27-GN)n. This property should entail the protein polymer and protein hydrogels with temperature and redox potential responsiveness. As shown in native PAGE (Fig. S7†), upon incubation in PBS buffer containing 5 mM DTT at 37 °C for 30 minutes, the protein polymers depolymerized to the monomer state. The protein polymer hydrogel showed similar behaviors. As shown in Fig. S8,† when incubated in PBS buffer containing 30 mM DTT overnight at room temperature (∼20 °C), the (GC-I27-GN)n hydrogel remained intact. However, when incubated in the same buffer at 50 °C for one hour, the protein hydrogel was completely dissolved, resulting from the disassembly of the non-covalent supramolecular polymer and the crosslinked network structure.
4 Discussion
Ultrahigh molecular weight is a hallmark for many protein polymers, including titin, silkworm protein fibroin and spider silk protein, and essential for the physical and mechanical properties of such protein-based materials/tissues.8 Engineering ultrahigh molecular weight bio-mimetic protein polymers of these natural protein polymers has attracted considerable efforts, and some significant progress has been made. For example, high molecular weight spider dragline silk protein has been produced using a metabolically engineered E. coli expression host and fibres made of such engineered proteins showed mechanical properties close to those of native spider dragline silk.40 Using an elegantly designed protein shackle, which is based on an isopeptide tag system derived from pili of Streptococcus pyogenes, as the building block, ultrathin protein polymers of ultrahigh molecular weight were successfully engineered.23 Despite this progress, it remains challenging to engineer protein polymers of ultrahigh molecular weight using either biotechnology or chemical methods.Here we have developed an efficient, protein fragment reconstitution-based supramolecular polymerization strategy to accomplish this goal. The bifunctional monomer state can be readily controlled via redox potential. Through supramolecular polymerization under ambient conditions, protein polymers of ultrahigh molecular weight (with a MW of 0.5 MDa), which is close to that of the I-band part of human cardiac titin, have been constructed. This study represents a successful attempt to engineer titin-mimetic protein polymers of ultrahigh molecular weight, which allows for the engineering of protein hydrogels with low protein polymer concentration and improved mechanical properties. Moreover, the engineered disulfide bond provides a redox-based method to depolymerize the engineered protein polymers to fully recover the monomers.
This PRF-based supramolecular polymerization is a step growth polymerization process in nature. Thus, the temperature and monomer concentration can, in principle, be used to tune the polymerization process to better control the molecular weight and molecular weight distribution. In particular, temperature will likely have an interesting effect on this polymerization reaction, as temperature will affect not only the GC–GN reconstitution (due to the relatively low melting temperature of the reconstituted GC–GN complex,∼23 °C), but also the oxidation kinetics to form a covalent GC–GN complex. For this, a detailed understanding of the rate law of this supramolecular polymerization is essential.
Moreover, this supramolecular polymerization approach is completely genetically encoded and modular. Different protein macromonomers, including protein chimera, can be readily fused with GC and GN to produce protein polymers with ultrahigh molecular weight. Thus, this method is general and should be applicable to a wide range of protein macromonomers. Compared with the protein shackle approach,23 which allows for the incorporation of other proteins as “side chains” into the protein shackle, the PRF-based method enables the incorporation of other proteins into the protein polymer backbone, thus enriching the toolbox for engineering high molecular weight protein polymers. Therefore, our approach may open up a new avenue, which can serve as an alternative to metabolic engineering, towards engineering proteins of ultrahigh molecular weight for technological applications, such as the engineering of artificial extracellular matrix protein fibronectin and spider dragline silk proteins. Thus, we anticipate that this PFR-based supramolecular polymerization method will find a broad range of applications in constructing protein-based biomaterials and functional assemblies.
5 Conclusions
Using protein fragment reconstitution (PFR) of GB1 as a supramolecular driving force, we used the I27 domain of titin as the bifunctional macromonomer to engineer protein polymers of ultrahigh molecular weight via the supramolecular condensation polymerization reaction. The resultant protein polymers showed a MW of 0.5 MDa, close to the molecular weight of the I-band part of human cardiac titin. The engineered protein polymers can be depolymerized via redox potential. Such high molecular weight linear protein polymers can serve as building blocks to engineer protein hydrogels with improved mechanical properties and reduced minimum gelation point. Due to its genetically encoded and modular nature, this PFR-based method will open new avenues towards engineering protein polymers with ultrahigh molecular weight, which can entail protein-based biomaterials and functional assemblies with improved physical and mechanical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. C. Yip, K. Ryan, and M. MacLachlan for their generous help in TEM, FPLC and DLS measurements. This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the National Natural Sciences Foundation of China (21420102007) and the Changjiang Scholar Program. R. Wang acknowledges the fellowship support of the China Scholarship Council. J. Li acknowledges the fellowship support of the NanoMat CREATE Training Program.Notes and references
- S. Labeit and B. Kolmerer, Science, 1995, 270, 293 CrossRef CAS PubMed.
- A. F. Oberhauser, P. E. Marszalek, H. P. Erickson and J. M. Fernandez, Nature, 1998, 393, 181 CrossRef CAS PubMed.
- B. L. Smith, T. E. Schäffer, M. Viani, J. B. Thompson, N. A. Frederick, J. Kindt, A. Belcher, G. D. Stucky, D. E. Morse and P. K. Hansma, Nature, 1999, 399, 761 CrossRef CAS.
- A. S. Tatham and P. R. Shewry, Trends Biochem. Sci., 2000, 25, 567 CrossRef CAS.
- G. Lee, K. Abdi, Y. Jiang, P. Michaely, V. Bennett and P. E. Marszalek, Nature, 2006, 440, 246 CrossRef CAS.
- L. Tskhovrebova and J. Trinick, Nat. Rev. Mol. Cell Biol., 2003, 4, 679 CrossRef CAS PubMed.
- W. A. Linke, D. E. Rudy, T. Centner, M. Gautel, C. Witt, S. Labeit and C. C. Gregorio, J. Cell Biol., 1999, 146, 631 CrossRef CAS PubMed.
- J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp and K. Savage, Philos. Trans. R. Soc., B, 2002, 357, 121 CrossRef CAS PubMed.
- M. Sotomayor, D. P. Corey and K. Schulten, Structure, 2005, 13, 669 CrossRef CAS PubMed.
- K. Wang, Adv. Biophys., 1996, 33, 123 CrossRef CAS PubMed.
- M. L. Bang, T. Centner, F. Fornoff, A. J. Geach, M. Gotthardt, M. McNabb, C. C. Witt, D. Labeit, C. C. Gregorio, H. Granzier and S. Labeit, Circ. Res., 2001, 89, 1065 CrossRef CAS PubMed.
- S. Improta, A. S. Politou and A. Pastore, Structure, 1996, 4, 323 CrossRef CAS PubMed.
- H. Granzier and S. Labeit, J. Physiol., 2002, 541, 335 CrossRef CAS PubMed.
- J. T. Hinson, A. Chopra, N. Nafissi, W. J. Polacheck, C. C. Benson, S. Swist, J. Gorham, L. Yang, S. Schafer, C. C. Sheng, A. Haghighi, J. Homsy, N. Hubner, G. Church, S. A. Cook, W. A. Linke, C. S. Chen, J. G. Seidman and C. E. Seidman, Science, 2015, 349, 982 CrossRef CAS PubMed.
- H. Li, W. A. Linke, A. F. Oberhauser, M. Carrion-Vazquez, J. G. Kerkvliet, H. Lu, P. E. Marszalek and J. M. Fernandez, Nature, 2002, 418, 998 CrossRef CAS PubMed.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69 CrossRef CAS PubMed.
- F. Saqlain, I. Popa, J. M. Fernandez and J. Alegre-Cebollada, Macromol. Mater. Eng., 2015, 300, 369 CrossRef CAS PubMed.
- M. Carrion-Vazquez, A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3694 CrossRef CAS PubMed.
- H. Dietz, M. Bertz, M. Schlierf, F. Berkemeier, T. Bornschlogl, J. P. Junker and M. Rief, Nat. Protoc., 2006, 1, 80 CrossRef CAS PubMed.
- P. Zheng, Y. Cao and H. Li, Langmuir, 2011, 27, 5713 CrossRef CAS PubMed.
- D. E. Meyer and A. Chilkoti, Biomacromolecules, 2002, 3, 357 CrossRef CAS PubMed.
- X. Li, Y. Bai, Z. Huang, C. Si, Z. Dong, Q. Luo and J. Liu, Nanoscale, 2017, 9, 7991 RSC.
- R. Matsunaga, S. Yanaka, S. Nagatoishi and K. Tsumoto, Nat. Commun., 2013, 4, 2211 CrossRef PubMed.
- A. Galarneau, M. Primeau, L.-E. Trudeau and S. W. Michnick, Nat. Biotechnol., 2002, 20, 619 CrossRef CAS PubMed.
- T. K. Kerppola, Chem. Soc. Rev., 2009, 38, 2876 RSC.
- N. Kobayashi, S. Honda, H. Yoshii, H. Uedaira and E. Munekata, FEBS Lett., 1995, 366, 99 CrossRef CAS PubMed.
- N. Kobayashi, S. Honda and E. Munekata, Biochemistry, 1999, 38, 3228 CrossRef CAS PubMed.
- M. C. Bauer, W.-F. Xue and S. Linse, Int. J. Mol. Sci., 2009, 10, 1552 CrossRef CAS PubMed.
- S. Honda, N. Kobayashi, E. Munekata and H. Uedaira, Biochemistry, 1999, 38, 1203 CrossRef CAS PubMed.
- N. Kong and H. Li, Adv. Funct. Mater., 2015, 25, 5593 CrossRef CAS.
- Q. Peng, N. Kong, H.-C. E. Wang and H. Li, Protein Sci., 2012, 21, 1222 CrossRef CAS PubMed.
- Y. Wang, X. Hu, T. Bu, C. Hu, X. Hu and H. Li, Langmuir, 2014, 30, 2761 CrossRef CAS PubMed.
- Y. Cao and H. Li, Nat. Mater., 2007, 6, 109 CrossRef CAS PubMed.
- C. Arndt, S. Koristka, H. Bartsch and M. Bachmann, Methods Mol. Biol., 2012, 869, 49 CrossRef CAS PubMed.
- T. Bu, H.-C. E. Wang and H. Li, Langmuir, 2012, 28, 12319 CrossRef CAS PubMed.
- M. Rief, M. Gautel, F. Oesterhelt, J. M. Fernandez and H. E. Gaub, Science, 1997, 276, 1109 CrossRef CAS PubMed.
- C. M. Elvin, A. G. Carr, M. G. Huson, J. M. Maxwell, R. D. Pearson, T. Vuocolo, N. E. Liyou, D. C. Wong, D. J. Merritt and N. E. Dixon, Nature, 2005, 437, 999 CrossRef CAS PubMed.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69 CrossRef CAS PubMed.
- J. Fang and H. Li, Langmuir, 2012, 28, 8260 CrossRef CAS PubMed.
- X. X. Xia, Z. G. Qian, C. S. Ki, Y. H. Park, D. L. Kaplan and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14059 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02128k |
| This journal is © The Royal Society of Chemistry 2019 |