
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSnapshots of magnesium-centred diborane heterolysis by an outer sphere SN2 process†
Anne-Frédérique
Pécharman
,
Michael S.
Hill
 *,
Grace
McMullon
,
Claire L.
McMullin
* and
Mary F.
Mahon
*,
Grace
McMullon
,
Claire L.
McMullin
* and
Mary F.
Mahon
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bath.ac.uk
First published on 24th May 2019
Abstract
Reactions of the β-diketiminato magnesium diboranate derivative, [(BDI)Mg{pinB(n-Bu)Bpin}] (BDI = HC{(Me)CNDipp}2; Dipp = 2,6-i-Pr2C6H3), with N,N′-dialkyl and N,N′-diaryl carbodiimides provided the corresponding C-borylated magnesium borylamidinates. This reactivity occurs with the displacement of n-BuBpin and with the apparent addition of a nucleophilic {Bpin} anion to the electrophilic unsaturated carbodiimide carbon centres. In contrast, while analogous reactions of [(BDI)Mg{pinB(n-Bu)Bpin}] with N-alkyl or N-aryl aldimines and ketimines also resulted in facile displacement of n-BuBpin, they provided the organomagnesium products of {Bpin} addition to the imine nitrogen atom rather than the more electrophilic trigonal imine carbon. Computational assessment by density functional theory (DFT) indicated that, although the energetic differences are marginal, the organomagnesium products may be considered as the kinetic outcome of these reactions with respect to the generation of alternative amidomagnesium regioisomers. This latter deduction was borne out by the thermally-induced conversion of two such organomagnesium species to their C-borylated amidomagnesium isomers, both of which occur with negligible entropies of activation indicative of purely intramolecular processes. Detailed analysis by DFT of the reaction of [(BDI)Mg{pinB(n-Bu)Bpin}] with PhN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh indicated that B–N bond formation is initiated by attack of the imine nitrogen at the three-coordinate boron atom of the diboranate anion rather than the more crowded magnesium centre. Consistent with an effectively spontaneous reaction, the resultant cleavage of the B–B bond of the diboranate unit is accomplished via the traversal of two very modest barriers of only 8.3 and 6.7 kcal mol−1. This analysis was also supportive of a subsequent intramolecular B–N to B–C isomerisation process. Of greater general significance, however, the addition of the {Bpin}− anion to the reducible aldimine is best rationalised as a consequence of the electrophilic character of this three-coordinate boron centre rather than any intrinsic nucleophilicity associated with the B–B bond of the [pinBB(n-Bu)pin]− anion.
CHPh indicated that B–N bond formation is initiated by attack of the imine nitrogen at the three-coordinate boron atom of the diboranate anion rather than the more crowded magnesium centre. Consistent with an effectively spontaneous reaction, the resultant cleavage of the B–B bond of the diboranate unit is accomplished via the traversal of two very modest barriers of only 8.3 and 6.7 kcal mol−1. This analysis was also supportive of a subsequent intramolecular B–N to B–C isomerisation process. Of greater general significance, however, the addition of the {Bpin}− anion to the reducible aldimine is best rationalised as a consequence of the electrophilic character of this three-coordinate boron centre rather than any intrinsic nucleophilicity associated with the B–B bond of the [pinBB(n-Bu)pin]− anion.
Introduction
Although many boron-heteroatom (e.g. C, N, O) bonds may be generated by the reaction of an appropriate heteroelement nucleophile and a boron electrophile, the relative dearth of complementary anionic or nucleophilic boron species remains as something of a bottleneck in main group element synthesis. The viability of model lithioboranes was discussed by Schleyer and co-workers as long ago as 1995, their calculations suggesting that singlet boryl derivatives, X2B–Li, may be accessible through the introduction of stabilising electronegative X substituents (e.g. F, O, N).1 This supposition was ultimately borne out by Yamashita and Nozaki's pivotal isolation of the lithium diaminoboryl, compound 1,2–6 which has been shown to act as a competent nucleophile towards a wide range of electrophilic organic and inorganic reagents.7–15
While the synthesis of 1 instigated the development of a plethora of related anionic and neutral nucleophilic boron species, access to a vast majority of these derivatives, like 1 itself, requires the synthetically inconvenient reaction of a haloborane precursor with a potent alkali metal reductant.16–34 The realisation that the desymmetrisation of commercially available diboranes, e.g. the bis(pinacolato)diborane (B2pin2) adducts, 2 and 3, can provide surrogates for boryl anions is, thus, an important advance toward practicality that has prompted significant experimental and theoretical attention.35–52 Fernandez, Bo and co-workers, for example, performed density functional theory (DFT) calculations on the ‘transition metal-free’ diboration of non-activated alkenes and the hydroboration of enone substrates.39,44,53 Their proposed mechanism, leading to the hydroboration of methyl acrylate (Scheme 1), is predicated on an induced polarisation across the B–B bond such that one of the boron centres gains a strong nucleophilic character. A significant positive charge of +0.98 was calculated at the four-coordinate boron atom of the anionic Lewis base adduct between B2pin2 and MeO− (shown as the intermediate anion A in Scheme 1). Although a calculated charge of +0.56 also inferred that the remaining sp2 boron atom should be significantly electrophilic, the behaviour of A as a source of the [Bpin]− nucleophile was rationalised as a consequence of the resultant charge separation (Δq = 0.42) across the B(sp2)–B(sp3) bond. On this basis, nucleophilic addition of the {Bpin} moiety was mooted to take place by the assembly of a transition state via (TSAB, Scheme 1) involving overlap of the polarised B–B σ bond and the π* (LUMO) of the alkene. The neutral pinBOMe by-product is then released and formation of the β-borylation product (C) is achieved through protonation of the resultant anionic intermediate (B) by methanol, which is typically added in a five-fold excess to act as a source of methoxide anion. The explicit generation of any boryl anion, therefore, has neither been experimentally observed nor theoretically implicated during this nucleophilic borylation chemistry. Notably, the initial computational treatment of Bo and Fernandez and a more recent theoretical analysis of this ‘transition metal-free’ reactivity also disregard any explicit role for the alkali metal cation,53,54 despite its introduction, most commonly as Cs2CO3 or NaOt-Bu, as a necessary component of the catalytic mixture.
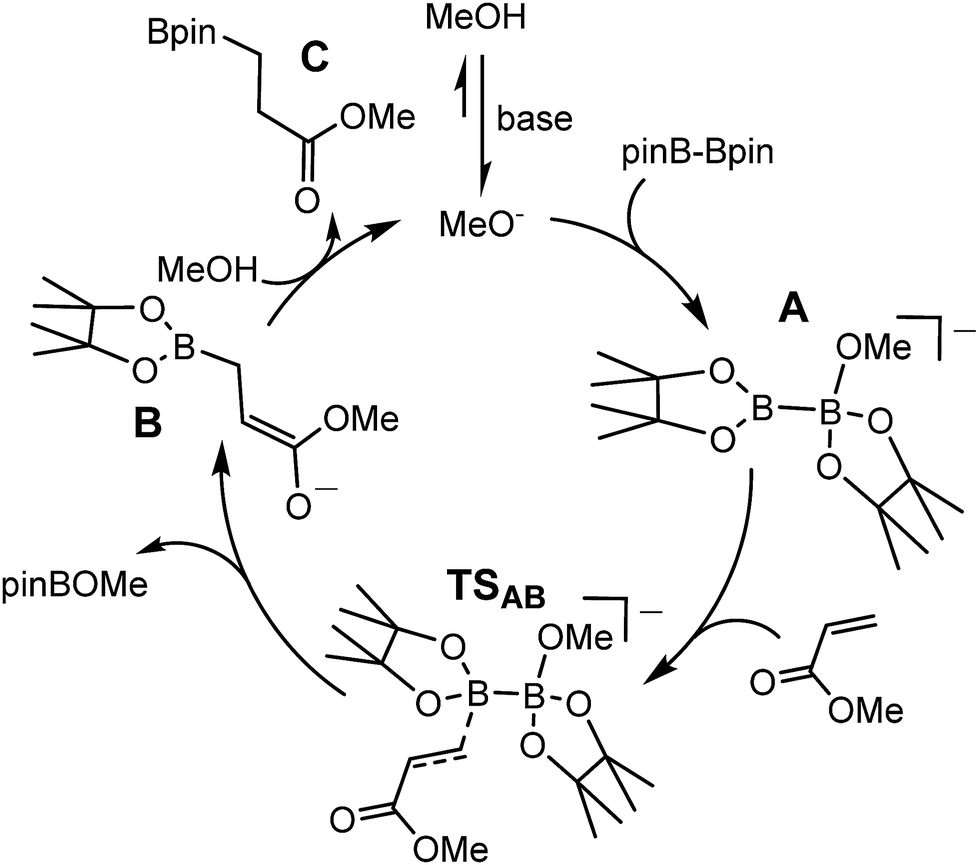 | ||
| Scheme 1 DFT calculated (BP86\\M06) reaction pathway proposed by Bo and Fernandez for the base initiated conjugate addition/hydroboration of methyl acrylate by B2pin2.53 | ||
We have recently reported that compounds such as 5, comprising B(sp2)–B(sp3) diboranate anions reminiscent of A, are easily synthesised by reactions of the β-diketiminato n-butylmagnesium derivative [(BDI)Mgn-Bu], 4, (BDI = HC{(Me)CNDipp}2; Dipp = 2,6-i-Pr2C6H3) with the relevant diborane (Scheme 2). Treatment of the resultant derivatives with 4-dimethylaminopyridine (DMAP) induces the heterolytic elimination of n-butyl boron pinacolate ester and the generation of terminal magnesium boryl derivatives such as compound 6.55,56 In our initial account of this chemistry we reported that compound 6 reacts as an unambiguous boryl nucleophile with a range of organic electrophiles including benzophenone and N,N′-di-iso-propylcarbodiimide to provide the B–C bonded borylalkoxide and borylamidinate derivatives, compounds 7 and 8. By analogy to the well-established reactivity of Mg–H or Mg–C bonds with similar heterocumulene electrophiles,57–60 these reactions are readily rationalised to result from insertion reactions of the CO and CN bonded substrates into the polarised Mg–B bond of 6. In subsequent research we have observed that the diboranate (5) itself is also an apparent source of the [Bpin] nucleophile.61–63 Of relevance to the current study, addition of 5 to organic isocyanates provided the anticipated C-boryl amidates (e.g.9) whereas reactions of diaryl ketones such as benzophenone resulted in aryl dearomatisation and the enolate reaction product (10) rather than thermodynamically preferred C-borylation of the electrophilic CO unit.62 In this contribution, we extend our study of the reactivity of 5 to representative carbodiimide and imine substrates. Although isoelectronic with the previously described isocyanate and carbonyl-containing small molecules, we speculated that the additional N-substitution of these substrates would enable greater kinetic discrimination and, in so doing, provide an opportunity for more direct experimental investigation of the nature of the B–B bond breaking process and the origin of the nucleophilic behaviour of B(sp2)–B(sp3) bonded species.
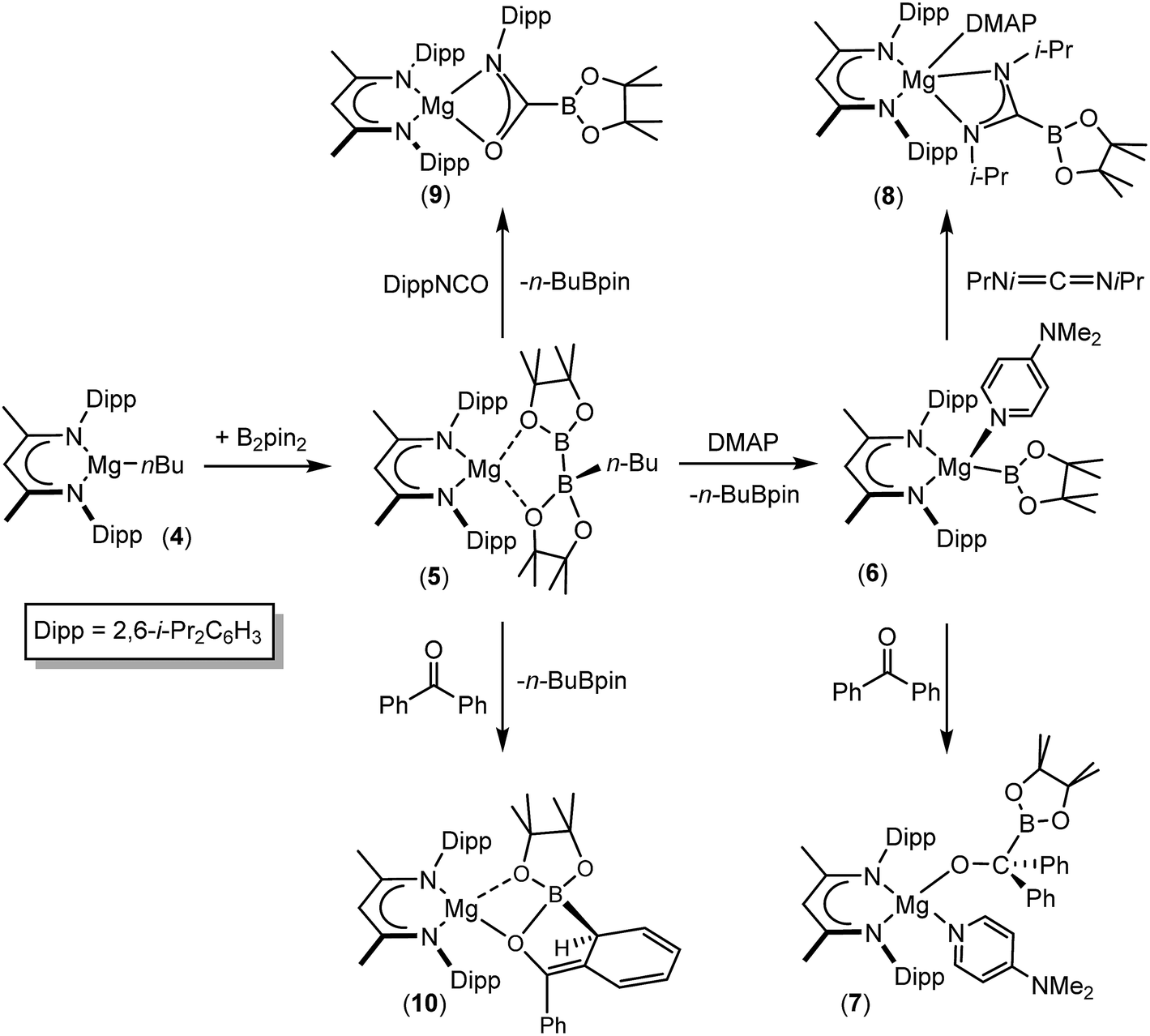 | ||
| Scheme 2 Synthesis and previously reported reactivity of compounds 5 and 6. | ||
Results and discussion
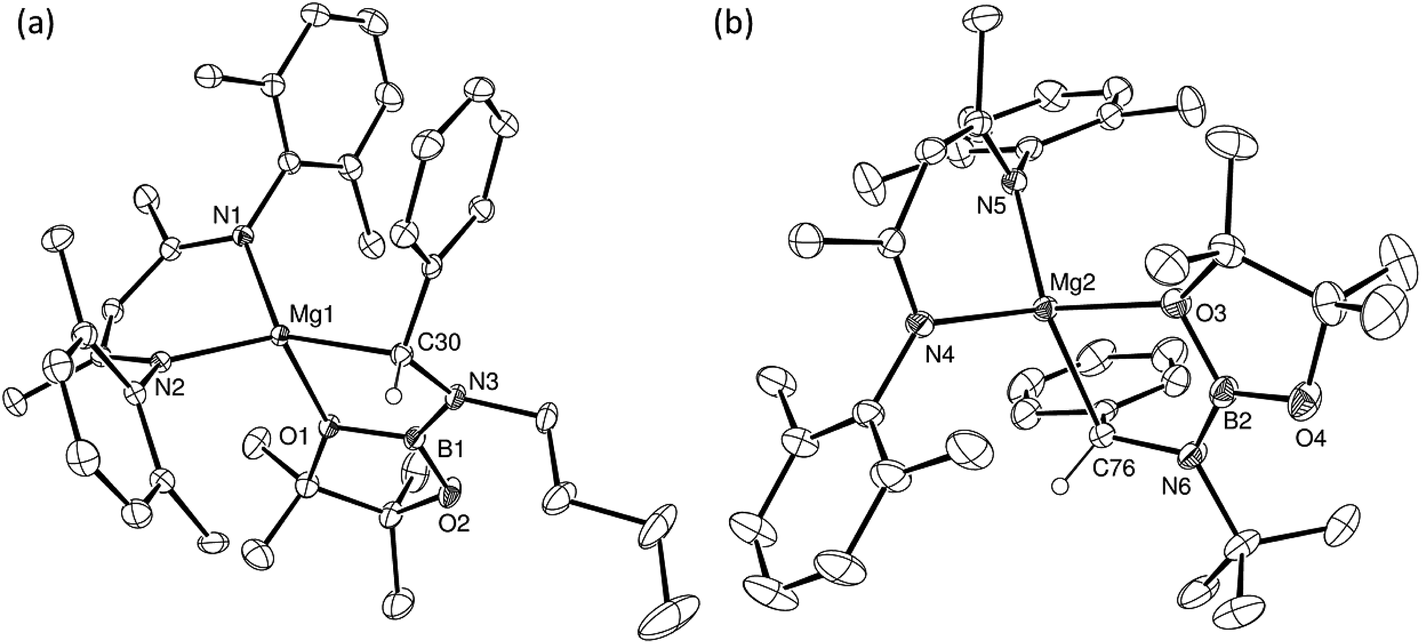
An initial reaction was performed in toluene between equimolar quantities of compound 5 and N,N′-di-tert-butylcarbodiimide. Heating to 80 °C induced complete consumption of the starting materials through the elimination of n-BuBpin [δ(11B) = 34.3 ppm] and the formation of a single new symmetrical β-diketiminate species (11). This latter compound was most clearly characterised by the appearance of new singlet resonances at δ 1.10 and 1.24 ppm in the resultant 1H NMR spectrum. These signals integrated with respective intensities of 12H and 18H relative to the 1H BDI methine proton observed at 4.89 ppm allowing the identification of compound 11 as a magnesium C-borylamidinate resulting from formal boryl anion addition to the sp-hybridised carbodiimide carbon atom. Although the end result of this process is analogous to the synthesis of compound 8 (Scheme 2), B–C bond formation is enabled without the observable generation of a magnesium boryl and it is apparent that addition of the carbodiimide initiates the necessary heterolysis of the B(sp2)–B(sp3) bond of compound 5. The identity of compound 11 was confirmed by removal of the reaction solvent and crystallisation of the resultant solid from hexane at −35 °C. This procedure yielded single crystals suitable for X-ray diffraction analysis, the results of which are displayed in Fig. 1a.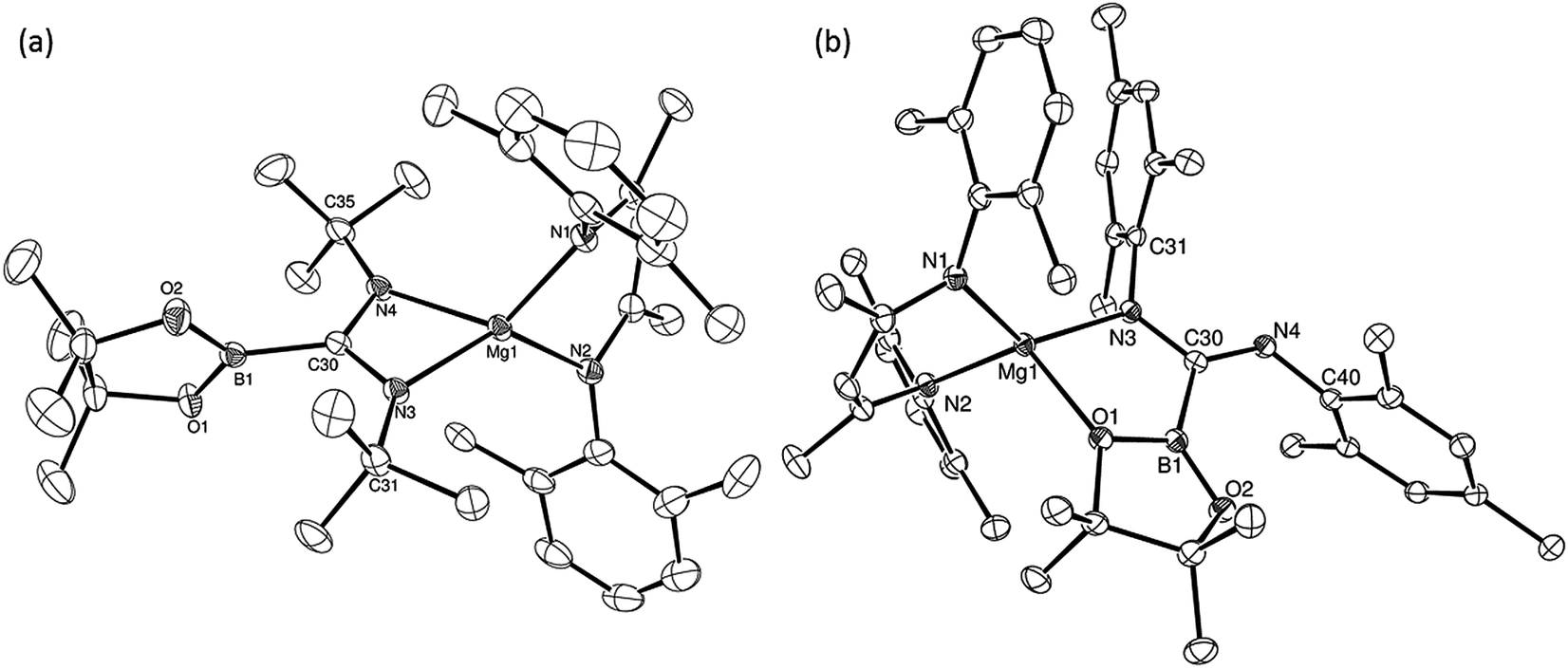 | ||
| Fig. 1 ORTEP representations (25% probability ellipsoids) of (a) compound 11 and (b) compound 14. Hydrogen atoms and iso-propyl methyl carbon atoms are removed for clarity. Selected bond lengths (Å) and angles (°): (11) Mg1–N1 2.0394(18), Mg1–N2 2.0384(19), Mg1–N3 2.0193(17), Mg1–N4 2.0638(18), N3–C30 1.344(3), N4–C30 1.338(3), C30–B1 1.593(3), N2–Mg1–N1 94.72(8), N2–Mg1–N4 122.70(8), N1–Mg1–N4 119.70(8), N3–Mg1–N1 123.37(8), N3–Mg1–N2 131.68(8), N3–C30–Mg1 55.02(10), N3–C30–B1 123.89(17), N4–C30–Mg1 56.91(10), N4–C30–N3 111.93(17), N4–C30–B1 124.17(18), B1–C30–Mg1 178.91(14); (14) N1–Mg1 2.043(2), N2–Mg1 2.053(2), N3–Mg1 2.005(2), O1–Mg1 2.1043(17), C30–B1 1.605(3), C30–N3 1.354(3), C30–N4 1.299(3), N1–Mg1–N2 96.84(8), N1–Mg1–O1-111.57(8), N2–Mg1–O1 116.31(8), N3–Mg1–N1 127.47(9), N3–Mg1–N2 122.14(9), N3–Mg1–O1 83.10(7), N3–C30–B1 110.48(19), N4–C30–B1 126.0(2), N4–C30–N3 123.4(2). | ||
The structure of compound 11 comprises a C-borylamidinate ligand formed by the delivery of a [pinB]− anion to the electrophilic carbon centre of t-BuNCNt-Bu. The ligand adopts an N,N-chelated κ2-coordination mode to magnesium, which is analogous to the previously reported derivative, compound 8. In the absence of the DMAP co-ligand, however, the magnesium centre of 11 is four-coordinate with the coordination environment of Mg1 completed solely by the bidentate BDI anion. Most likely a result of the lower coordination number of the magnesium centre, the Mg–N distances to the boryl amidinate ligand of 11 [Mg1–N3 2.0193(17), Mg1–N4 2.0638(18) Å] are appreciably shorter than the corresponding bond lengths observed in 8 [2.1100(10), 2.2124(10) Å]. In both compounds, however, the C–B bond lengths are effectively identical [8: 1.5972(17); 11: 1.593(3) Å], while the relevant C–N bond lengths [8: 1.3383(14), 1.3228(15); 11: 1.344(3), 1.338(3) Å] are indicative of a similar level of delocalisation across the amidinato chelate.
Although the basic constitution of the C-borylamidinate anions of both 8 and 11 is broadly reminiscent of alkyl or aryl amidinates resulting from the reaction of carbodiimides with the M–C bonds of polar σ-organometallics,64,65 the only other precedent for directly analogous C-borylamidinate formation is provided by a scandium derivative, which was itself generated by use of compound 1.66 We, thus, assayed the generality of the reactivity of compound 5 with a further range of readily available N,N′-dialkyl and diaryl carbodiimides (Scheme 3).
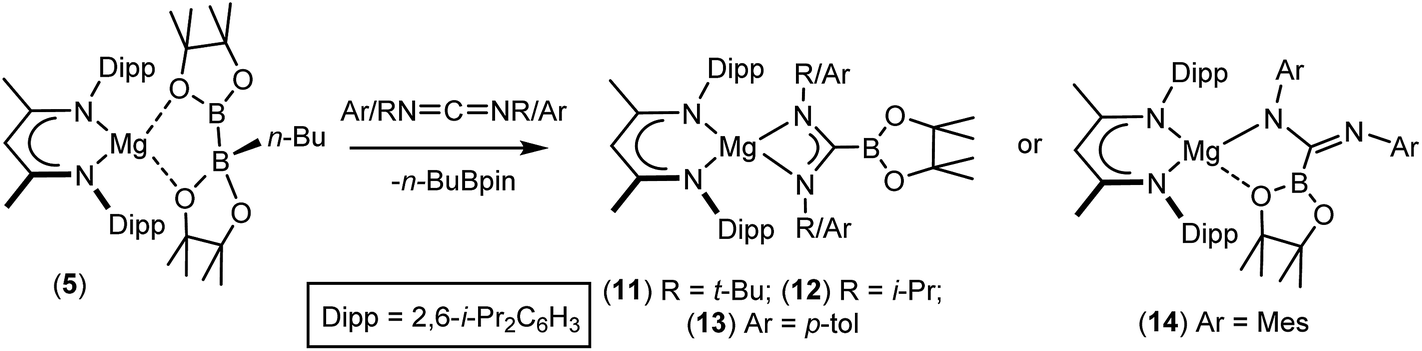 | ||
| Scheme 3 Synthesis of compounds 11–14. | ||
As was the case for the synthesis of compound 11, reaction with i-PrNCN-i-Pr required heating to 80 °C to ensure complete conversion to the borylamidinate (12). In contrast, both reactions with the N-aryl carbodiimides proceeded smoothly at room temperature, irrespective of the steric demands of the N-aryl substituent. Although the respective 1H NMR spectra of the N-i-Pr and N-p-tolyl borylamidinate derivatives, compounds 12 and 13, were analogous to that observed for compound 11 and, thus, consistent with the adoption of similar pseudo-C2 symmetric structures in solution, the high solubility of these compounds precluded the crystallisation of pure bulk samples. In contrast, addition of MesNCNMes to a solution of 5 resulted in the immediate deposition of needle-like single crystals of compound 14. Although the subsequent insolubility of these crystals prohibited the acquisition of meaningful NMR data, they were found to be suitable for single crystal X-ray diffraction analysis. The results of this analysis (Fig. 1b) revealed that the N,N′-mesityl-C-borylamidinate adopts an unusual bidentate κ2-N,O coordination mode in which the ligand binds to magnesium through one of the mesityl-bonded nitrogen atoms and a single oxygen of the pinacolate unit. As a consequence of this unsymmetrical binding mode, and in contrast to the delocalised structures adopted by compounds 8, 9 and 11, the C30–N3 [1.354(3) Å] and C30–N4 [1.299(3) Å] distances are consistent with localised single and double bonds, respectively. Although we have previously observed similar Mg–O interactions between the Bpin units of a number of magnesium formamidinatoborate [2.007(3) Å],58 formamidatoborate [1.9631(12) Å]60 and aldimidoborate [1.9714(11) Å]59 derivatives, all of these compounds comprised a 4-coordinate hydridoborate as the formally charged component of the coordinated ligand. The Mg1–O1 bond length [2.1043(17) Å] of 14, in which the appended Bpin boron centre remains three-coordinate, is consequently elongated, albeit less so than in a further closely related magnesium borylamide [(BDI)MgNDipp(Bpin)] [2.3737(18) Å] in which the κ2-N,O binding is provided by a more strained 3-membered N–B–O chelate.67
Having confirmed its potential for B–C bond formation at the electrophilic carbon centres of carbodiimides, we turned our attention to the reactivity of compound 5 with the potentially broader substrate scope provided by N-alkyl and N-aryl imines. Initial reactions were performed between compound 5 and the N-alkyl aldimines, N-n-butyl- and N-t-butyl-1-phenylmethanimine. Assessment of both reactions after ten minutes at room temperature by 1H and 11B{1H} NMR spectroscopy indicated that complete consumption of 5 had again occurred with the liberation of n-BuBpin and the respective formation of two new β-diketiminate compounds, 15BN and 16BN (Scheme 4). In contrast to the data provided by compounds 11–13, however, the 1H NMR spectra of compounds 15BN and 16BN presented separate signals for all four BDI iso-propyl substituents and both BDI ‘backbone’ methyl environments. The latter of these resonances appeared as pairs of unique singlets at ca. δ 1.5–1.6 ppm, each with intensities corresponding to three protons by relative integration. These observations indicate that, like the starting complex 5, these species possess neither a plane of symmetry orthogonal to nor a C2 axis in the plane of the β-diketiminate magnesium chelate. Guided by these observations and the precedent provided by the synthesis of compounds 11–14, it was anticipated that both compounds 15BN and 16BN would be the amide products resulting from formal C-borylation of the electrophilic aldimine carbon centre. This expectation was refuted, however, by the definitive solid state identification provided by single crystal X-ray diffraction analysis performed on both compounds after crystallisation from hexane at −35 °C. The results of these analyses (Fig. 2) established that both reactions had yielded an alternative regiochemistry, and the formation of aminomethylmagnesium products resulting from attachment of the boryl unit to the more electron rich nitrogen atoms of the aldimine double bonds. The compounds are, thus, denoted as 15BN and 16BN indicative of the B–N bonded isomers (Scheme 4).
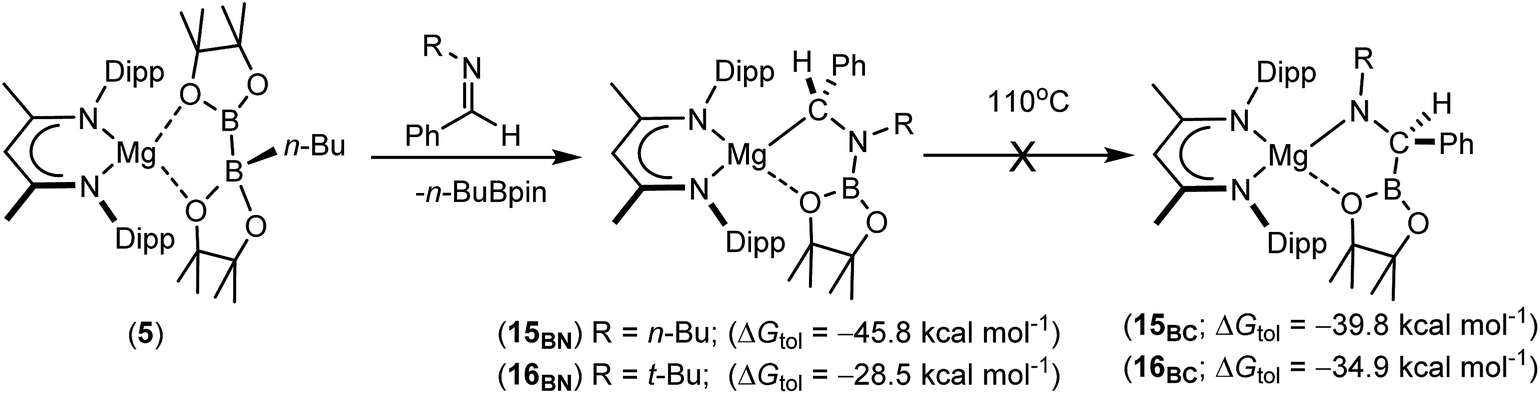 | ||
| Scheme 4 Synthesis of compounds 15BN and 16BN with their DFT-computed energies relative to complex 5 and those of the experimentally unobserved magnesium amide isomers, 15BC and 16BC. | ||
 | ||
| Fig. 2 ORTEP representations (25% probability ellipsoids) of (a) compound 15BN and (b) the Mg2-containing molecule of compound 16BN. Hydrogen atoms except for those attached to C30 (15BN) and C76 (16BN), iso-propyl methyl groups and the Mg1-containing molecule of compound 16BN have been removed for clarity. Selected bond lengths (Å) and angles (°): (15BN) Mg1–N1 2.0460(11), Mg1–N2 2.0301(11), Mg1–C30 2.1549(13), Mg1–O1 2.1250(9), N3–B1 1.3904(18), N3–C30 1.4754(16), N2–Mg1–N1 96.36(4), N2–Mg1–C30 123.40(5), N1–Mg1–O1 107.37(4), N1–Mg1–C30 129.17(5), N2–Mg1–O1 116.00(4), N2–Mg1–N1 96.36(4), N3–B1–O1 121.82(12); (16BN) Mg2–O3 2.1061(13), Mg2–N4 2.0660(15), Mg2–N5 2.0556(15), Mg2–C76 2.1668(18), O3–B2 1.426(3), N6–C76 1.474(2), N6–B2 1.388(3), O3–Mg2–C76 82.44(6), N4–Mg2–O3 118.29(6), N4–Mg2–C76 114.77(7), N6–C76–Mg2 108.67(11), N6–B2–O3 122.30(17). | ||
Despite the differing steric demands of the N-alkyl substituents, the structures of compounds 15BN and 16BN are very similar. Both compounds comprise four-coordinate magnesium centres in which the alkaline earth coordination spheres are satisfied by a bidentate BDI ligand and the relevant N-boryl(phenyl)aminomethyl anions. The latter ligands are bidentate due to coordinative interactions from one of the pincacolato oxygen atoms of the N-appended [Bpin] units. In both 15BN and 16BN, the Mg–C bond lengths [ca. 2.16 Å] are entirely typical of σ-bonded magnesium organometallics, while the Mg–O bond distances [ca. 2.15 Å] are commensurate with that observed for compound 14 and, thus, may be judged to display similar basic characteristics to those discussed above.
The unexpected regioselectivity of this N-borylation process was investigated by density functional theory (DFT). Initial calculations performed at the BP86 level of theory (see ESI† for full methodology) provided optimised geometries for 15BN and 16BN, which corresponded closely with the experimental solid state structures and revealed the reactions of compound 5 with both N-alkyl aldimine substrates to be significantly exergonic (Scheme 4). Furthermore, the structure of 15BN was computed to lie 6.0 kcal mol−1 lower in energy than its C-borylated isomer, the putative magnesium amide, 15BC, and these results, therefore, identify 15BN as the thermodynamic outcome of the reaction of compound 5 with the N-n-butyl aldimine substrate. In contrast, analogous calculations performed on the more sterically encumbered N-t-butyl species indicated isomerisation of the experimentally identified magnesium alkyl species (16BN) to the amide derivative (16BC) to be more exergonic by −6.4 kcal mol−1. Despite these contrasting thermodynamic preferences, monitoring of d8-toluene solutions of neither 15BN nor 16BN by 1H NMR spectroscopy evidenced any observable tendency toward isomerisation to the alternative C-borylated species, even after heating at 110 °C for five days (Scheme 4).
Further reactions of compound 5 with the N-phenyl aldimine, PhNCHPh, and the N-phenyl ketimine, PhNCPh2, were monitored at room temperature by 1H and 11B{1H} NMR spectroscopy. In both cases, two new compounds (17BN and 18BN) were formed in good yields (Scheme 5). Like the other aldimine-derived derivatives, 15BN and 16BN, compound 17BN displayed 1H and 13C{1H} NMR spectra consistent with the absence of a plane of symmetry orthogonal to the BDI chelate. In contrast, the data provided by the ketamine derivative, compound 18BN, were characteristic of a symmetrically disposed diphenylmethylmagnesium species. These deductions were confirmed by single crystal X-ray diffraction analysis, which established that both compounds 17BN and 18BN were again the N-borylated organomagnesium products formed through formal addition of a boryl anion to the previously unsaturated imine nitrogen centre (Fig. 3). Although there are some minor variations across the individual Mg–C, Mg–O and B–N bond lengths, these measurements are within the accepted norms for single bonds of these types and necessitate no further comment.
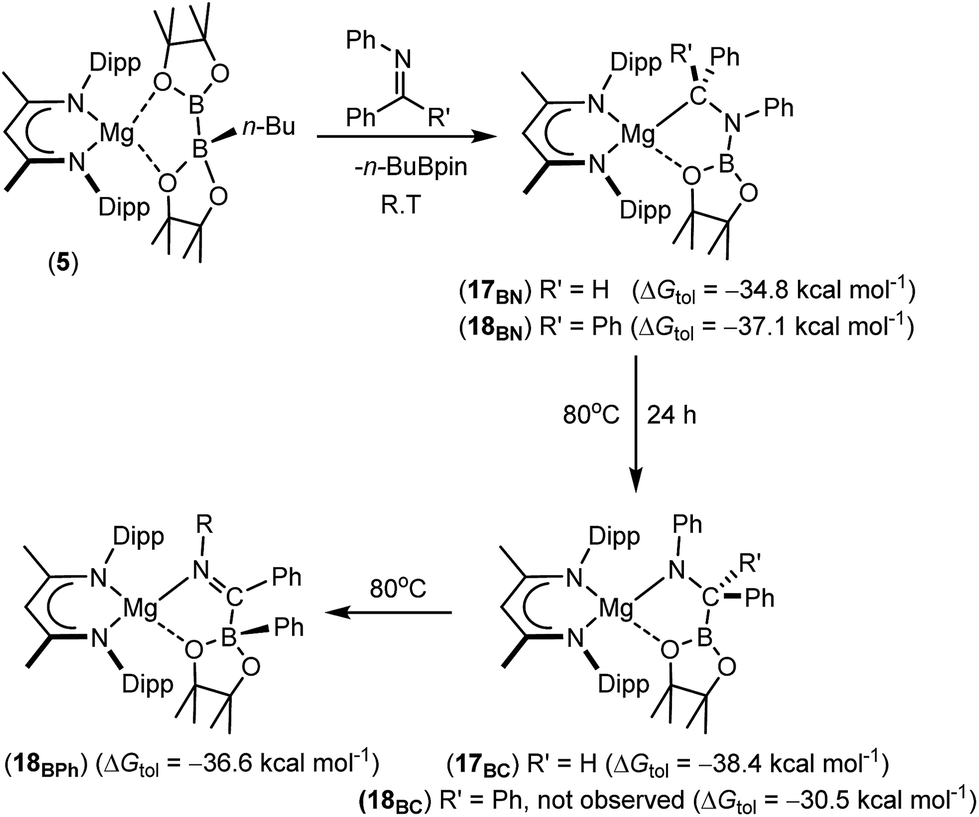 | ||
| Scheme 5 Synthesis of compounds 17BN and 18BN and their isomerisation to compounds 17BC and 18BPh, respectively. DFT-computed energies are given in parenthesis relative to complex 5. | ||
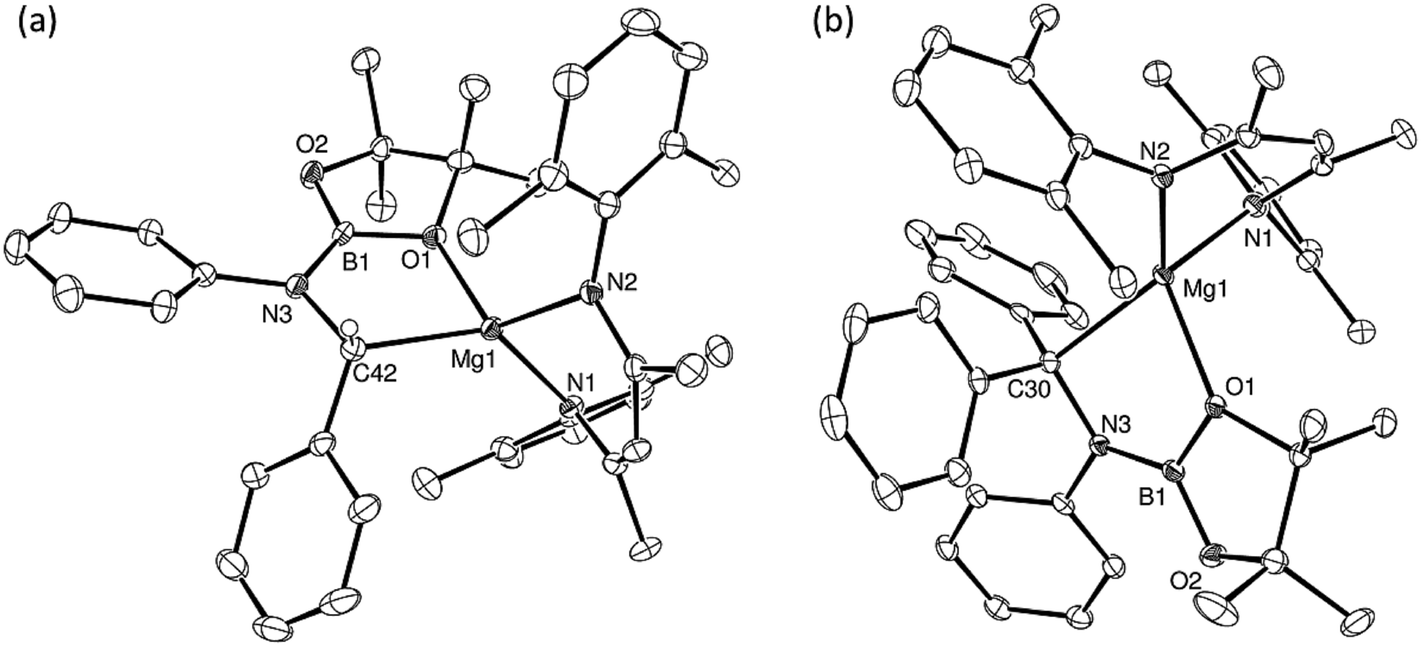 | ||
| Fig. 3 ORTEP representations (25% probability ellipsoids) of (a) compound 17BN and (b) compound 18BN. Hydrogen atoms except H42 in 17BN, iso-propyl methyl groups and occluded solvent in 18BN are removed for clarity. Selected bond lengths (Å) and angles (°): (17BN) Mg1–O1 2.018(3), Mg1–N1 2.039(3), Mg1–N2 2.051(3), Mg1–C42 2.197(3), N3–C42 1.491(4), N3–B1 1.397(5), O1–Mg1–N1 113.03(12), O1–Mg1–N2 119.93(12), O1–Mg1–C42 82.00(11), N1–Mg1–N2 94.59(12), N1–Mg1 C42 131.04(13), N2–Mg1–C42 118.58(13), B1–O1–Mg1 113.5(2); (18BN) Mg1–O1 2.1188(15), Mg1–N1 2.0940(18), Mg1–N2 2.0600(17), Mg1–C30 2.2343(18), N3–C30 1.493(2), N3–B1 1.417(3), O1–Mg1–C30 80.92(6), N1–Mg1–O1 111.90(7), N1–Mg1–C30 126.79(7), N2–Mg1–O1 112.77(6), N2–Mg1–N1 95.62(7), N2–Mg1–C30 127.91(8). | ||
The reactions providing both 17BN and 18BN from 5 were calculated to be significantly exergonic (17BN, ΔGtol = −34.8 kcal mol−1; 18BN, ΔGtol = −37.1 kcal mol−1). As was the case for the N-n-butyl product, 15BN, this computational assessment indicated that 18BN described the more stable regioisomer (18BC, ΔGtol = −30.5 kcal mol−1) by 6.6 kcal mol−1. In contrast, the aldimine reaction product, 17BN, was, like 16BN, identified as the kinetic reaction product and calculated to lie 3.6 kcal mol−1 higher in free energy than the corresponding C-borylated amide species, 17BC (Scheme 5). Counter to the expectation provided by these differing outcomes, heating of both organomagnesium compounds, 17BN and 18BN, overnight in d8-toluene at 80 °C resulted in their clean conversion to two further new β-diketiminato magnesium derivatives, designated as compounds 17BC and 18BPh, respectively. Although compound 17BC continued to display 1H and 13C{1H} NMR signatures reminiscent of its precursor, compound 17BN, the apparent C2 symmetry provided to compound 18BN by its diphenylmethyl anion was lost upon conversion to compound 18BPh. The origin of these observations was again resolved by isolation of samples of compounds 17BC and 18BPh suitable for X-ray diffraction analysis, the results of which are illustrated in Fig. 4.
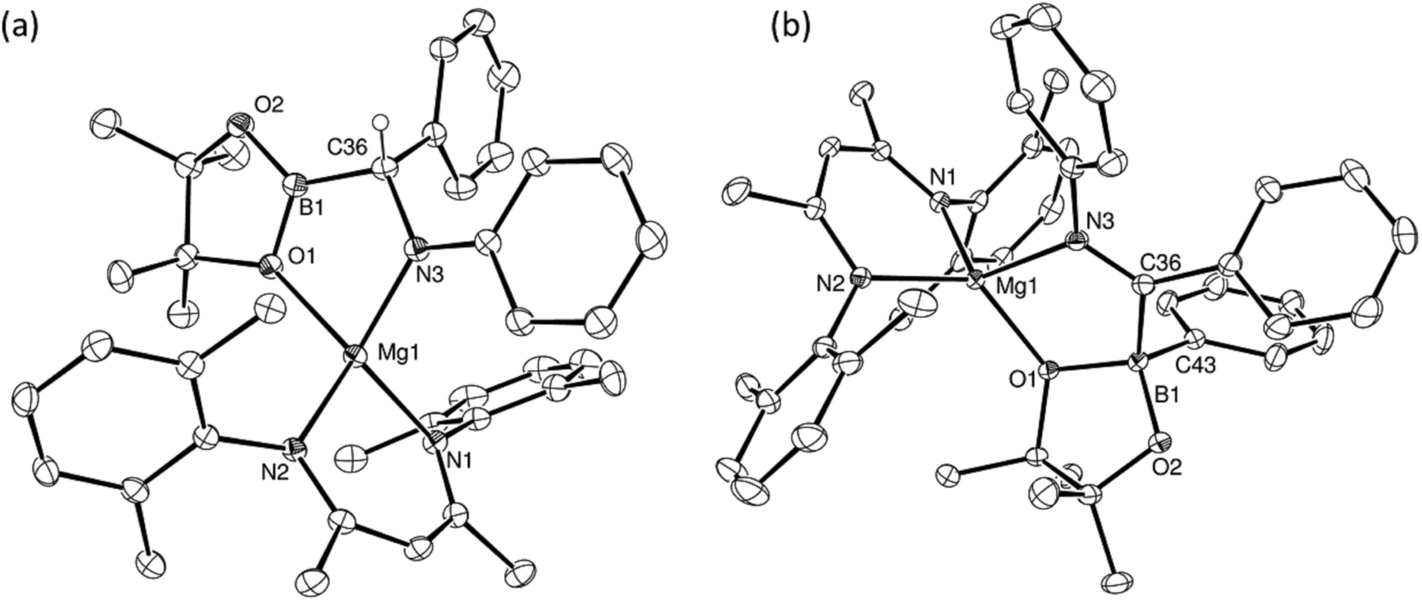 | ||
| Fig. 4 ORTEP representations (25% probability ellipsoids) of (a) compound 17BC and (b) compound 18BPh. Hydrogen atoms except H36 in 17BC and iso-propyl methyl groups are removed for clarity. Selected bond lengths (Å) and angles (°): (17BC) Mg1–O1 2.1679(18), Mg1–N1 2.072(2), Mg1–N2 2.0615(19), Mg1–N3 2.004(2), O1–B1 1.397(3), N3–C36 1.465(3), C36–B1 1.562(3), N1–Mg1–O1 129.95(8), N2–Mg1–O1 114.67(8), N2–Mg1–N1 93.78(8), N3–Mg1–O1 83.59(8), N3–Mg1–N1 113.24(9), N3–Mg1–N2 125.52(8), N3–C36–B1 108.73(19); (18BPh) Mg1–O1 1.9854(10), Mg1–N1 2.0612(12), Mg1–N2 2.0649(12), Mg1–N3 2.1218(12), O1–B1 1.5343(17), N3–C36 1.2967(19), C36–B1 1.6505(19), C43–B1 1.621(2), O1–Mg1–N1 120.14(5), O1–Mg1–N2 131.08(5), O1–Mg1–N3 83.93(4), N1–Mg1–N2 93.18(5), N1–Mg1–N3 116.63(5), N2–Mg1–N3 113.76(5), C36–N3–Mg1 113.37(9), N3–C36–B1 119.08(12), C43–B1–C36 108.28(11), O1–B1–C36 106.72(11), B1–O1–Mg1 115.32(8). | ||
The solid-state structure of compound 17BC confirmed its constitution as a C-borylated magnesium amide and the product of nitrogen-to-carbon transfer of the {Bpin} unit from compound 17BN. The resultant structure displayed a close correspondence to the optimised geometry of 17BC with B1–C36 [1.562(3) Å], C36–N3 [1.465(3) Å] and Mg1–N3 [2.004(2) Å] distances that are consistent with their assignment as single bonds within the normal ranges. Notably, the angle subtended by B1–C36–N3 [108.73(19) Å] is also close to the tetrahedral value expected for this completely saturated unit. In contrast, compound 18BPh is a further amidomagnesium isomerisation product resulting from intramolecular carbon-to-boron phenyl transfer between the previously magnesium-bound diphenylmethanide carbon centre and the boron of the {Bpin} unit (Scheme 5). The asymmetry apparent in the solution NMR analysis of compound 18BPh is, therefore, readily ascribed to the generation of the stereogenic phenyl(pinacolato)borate unit and the resultant loss of the plane of symmetry associated with the chelated anion. The short N3–C36 [1.2967(19) Å] bond distance and the almost trigonal geometry at C36 [N3–C36–B1 119.08(12)°; Σ(angles subtended at C36) = 359.68°] also indicate that the magnesium-bound N–C–B–O chelate is best described as arising from coordination of an N-phenyl-C-iminoborate. Although no magnesium compounds containing this precise type of anion appear to have been reported previously, the C–N, C–B and Mg–B bond lengths show reasonable correspondence with the relevant comparable distances within several organomagnesium derivatives of a tris(4,4-dimethyl-2-oxazolinyl)phenylborate spectator ligand that have been applied by Sadow and co-workers in a variety of catalytic processes.68–73
The kinetics of the transformations of compounds 17BN to 17BC and of 18BN to 18BPh were monitored by 1H NMR spectroscopy over a range of temperatures (60, 65, 70, 75 °C). In both systems, only the starting materials and the final products were observed throughout the reactions and no further intermediates or side products could be identified. Plots of ln[17] and ln[18] were found to be linear for >3 half-lives at all four temperatures of study and the resultant first order rate constants enabled the construction of Arrhenius and Eyring plots and determination of the macro- and microscopic activation parameters presented in Table 1. Both the activation energies and enthalpies of activation are consistent with the mild thermal conditions necessary to induce both transformations. Although entropies of activation are notoriously open to interpretation,74 the negligible ΔS≠values associated with both processes suggest that the gross constitutions of the starting materials are maintained and that the isomerisation reactions are intramolecular in origin.
| E a (kcal mol−1) | ΔH≠ (kcal mol−1) | ΔS≠ (cal K−1 mol−1) | |
|---|---|---|---|
| 17BN → 17BC | 22.3 (0.2) | 21.6 (0.2) | −0.01 (0.6) |
| 18BN → 18BPh | 25.3 (0.3) | 24.7 (0.3) | −0.001 (1.0) |
Further insight into the formation and transformation of compounds 17BN and 18BN was provided by DFT (BP86) calculations, which were performed to interrogate the nature and energetics of any plausible intermediates and transition states. Multiple attempts were made to optimise a geometry in which the less sterically encumbered imine, PhNCHPh, coordinates through its C or N atom at the Mg centre of 5. Irrespective of the starting orientation, however, the alkaline earth atom was found to be too crowded to allow any type of coordination due to the steric requirements of the N- and C-bonded phenyl groups, which hinder the direct approach of the imine to magnesium.
The NPA charges of the three- and four-coordinate boron centres of the [pinBBpin(nBu)]− ligand of 5 provided results reminiscent of those described by Bo, Fernandez and co-workers for the [pinBBpin(OMe)]− anion.39 The three-coordinate boron of the {Bpin} unit was found to manifest a substantial positive charge (+0.848), which, although less positive than the charge borne by the sterically encumbered magnesium centre (+1.578), is significantly more electron rich (+0.582) than the four-coordinate boron atom of the {pinB(n-Bu)} component of the diboranate complex anion. These observations militate against the reactivity of compound 5 as a source of nucleophilic [Bpin]− and not only infer that the coordinatively unsaturated {Bpin} unit displays considerable electrophilic potential but also that the B–B bond is significantly polarised toward the higher coordinate boron centre. Prompted by these observations, an alternative ‘outer sphere’ diboranate activation process invoking nucleophilic imine addition at the three-coordinate boron atom was assessed.
Fig. 5 illustrates the resultant free energy profile for the reaction of 5 with PhNCHPh and the subsequent isomerisation of 17BN to 17BC. The formation of the B–N bond of 17BN is initiated by attack of the imine nitrogen at the three-coordinate boron centre of the diboranate anion of 5. The formation of compound 17BN ensues through assembly of an adduct species, [INT(5-17BN)], which is 3.7 kcal mol−1 higher in energy than the adduct, 5·PhNCHPh. Consistent with the effectively spontaneous generation of 17BN, however, accomplishment of this significantly exergonic transformation (ΔGtol = −34.8 kcal mol−1) necessitates the traversal of two very modest barriers, [TS(5-17BN)1 and TS(5-17BN)2], of only 8.3 and 6.7 kcal mol−1, respectively. This process occurs within the magnesium coordination sphere to effect the cleavage of the B–B bond of the diboranate unit, which is observed to elongate from 1.83 Å [INT(5-17BN)] to 2.44 Å at TS(5-17BN)2 (Fig. 6a) en route to its complete rupture and the formation of 17BN. Indicative of the necessary redistribution of electron density, the calculated charges of the now four-coordinate nitrogen- and carbon-bound boron centres (+1.078 and +1.024, respectively) effectively equalise during the traversal of TS(5-17BN)2. This transformation, therefore, may be viewed as an obverse SN2 process resulting in the displacement of the nBuBpin component of the diboronate anion by the similarly neutral aldimine substrate. From this perspective, the addition of the [Bpin]− anion to the reducible aldimine is best rationalised as a consequence of the electrophilic character of this three-coordinate boron centre rather than any intrinsic nucleophilicity associated with the B–B bond of the [pinBB(n-Bu)pin]− anion. Although a further transition state [TS(5-17BCN)] invoking effective attack of the aldimine sp2 carbon on the electrophilic boron centre of 5 was also located, this was very much higher in energy (30.3 kcal mol−1) and ultimately led to the C-borylated magnesium amide, 17BC (via17BCN, vide infra), rather than the experimentally observed organomagnesium derivative (17BN) (Fig. 5).
 | ||
| Fig. 5 DFT calculated free energy (kcal mol−1) profile for the reaction of PhNCHPh with compound 5 (in toluene), relative to 5 and the free substrates. | ||
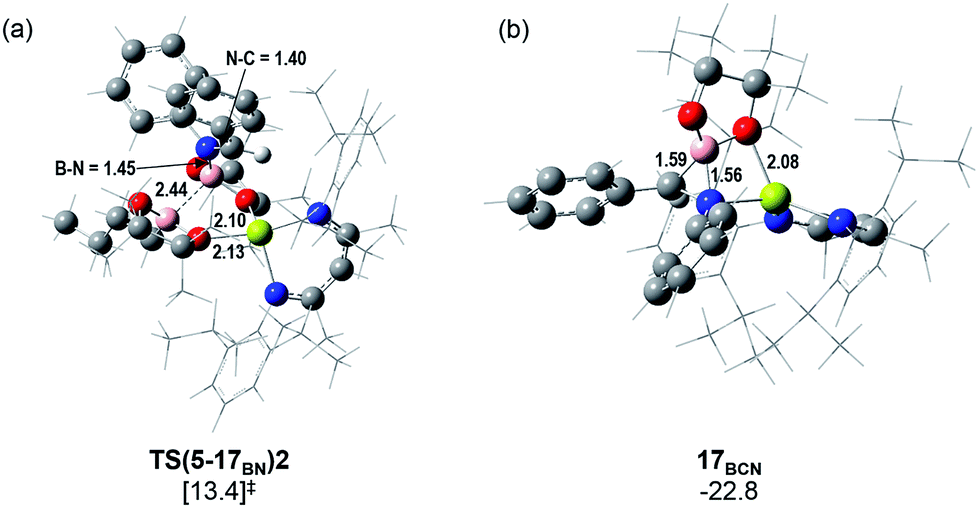 | ||
| Fig. 6 The DFT calculated (a) transition state associated with B–B bond cleavage during the transformation of compound 5 to compound 17BN; (b) intermediate structure of 17BCN (grey = C; red = O; pink = B; blue = N; yellow = Mg). | ||
In correspondence with the thermal conditions necessary for the observation of the reaction, an activation barrier of some 26.2 kcal mol−1 [TS(17BN-17BCN)] must be overcome to induce the isomerisation of 17BN to the amidomagnesium derivative, 17BC. Consistent with the negligible entropy of activation deduced for this transformation (Table 1), the transferral of the {Bpin} unit between nitrogen and carbon is purely intramolecular in origin. This process invokes the intermediacy of the azaboranuidacyclopropane species, 17BCN (Fig. 6b), which is generated by the breaking of the aminobenzyl C–Mg bond of 17BN. Although 17BCN is stabilised by a significant interaction between the magnesium centre and one of the pinacolato oxygen atoms (2.08 Å), it is unobservable under the experimental conditions and, most likely as a result of its inherent ring strain, is transformed immediately to 17BCvia a negligible barrier [TS(17BCN-17BC)] of 4.1 kcal mol−1. Although a related nitrogen-to-carbon rearrangement has been implicated in the decomposition of PhCH2CH(Bpin)(NH2) to PhCH2CH2NHBpin,75 to the best of our knowledge, the transformation of 17BN to 17BC provides the first documented example of a retro aza-bora-Brook rearrangement.76,77
The isomerisation of the diphenylmethanide derivative, compound 18BN, to the borate derivative, 18Ph, (Scheme 5) was also examined computationally (Fig. S38†). Consistent with the experimental analysis summarised in Table 1, a rate determining kinetic barrier [TS(18BN-18BCN)] of 21.6 kcal mol−1 must be surmounted during an initial intramolecular isomerisation step to an azaboranuidacyclopropane species, 18BCN. This species is analogous to 17BCN (Fig. 6b) and is similarly labile with respect to its onward transformation to a putative amide derivative, 18BC. In contrast to the greater thermodynamic viability deduced of compound 17BC over 17BN, compound 18BC was found to lie marginally higher (6.6 kcal mol−1) in energy than its isolable organomagnesium isomer, 18BN. While we suggest that this is a likely consequence of the enhanced benzhydrylic stabilization of the Mg–C interaction, any generation of 18BC will be fleeting with respect to the formation of the ultimate, and experimentally confirmed, reaction product, 18Ph, which effects carbon-to-boron phenyl migration viaTS(18BC-18BPh) with a barrier of 19.4 kcal mol−1.
Conclusion
The results described herein confirm that a magnesium diboranate complex (5) derived from commercially available B2pin2 can, like previously reported desymmetrised diborane esters, act as an apparent source of an anionic boryl nucleophile. Consistent with the transference of a [Bpin]− anion displaying significant nucleophilic character, reactions with carbodiimides result in the formation of C-borylamidinate species as the only magnesium-containing reaction products. Treatment of compound 5 with a variety of N-alkyl or aryl aldimine or ketimine substrates, however, provided consistent access to N-borylated organomagnesium compounds. In these cases, the B–B bond breaking event is initiated by displacement of charge neutral n-BuBpin through an approach of the imine nitrogen atom to the three-coordinate {Bpin} component of the diboranate anion. This process enables production of a boron–nitrogen bonded isomer not only when the magnesium organometallic is thermodynamically more stable but also when the alternate C-bonded magnesium amide isomer is lower in free energy. In such instances, the N–boron bonded kinetic reaction products can be isolated but may be converted to their thermodynamically more stable isomers via an accessible intramolecular pathway. While more direct nucleophilic attack at magnesium cannot be discounted for less sterically encumbered bases, and we do not suggest that this mode of heterolytic activation is operant in all instances when diborane desymmetrisation results in generation of boryl anion equivalents, our results suggest that this outer sphere mechanism is a viable route for consideration. We are continuing to study the implications of these observations, particularly with regard to the rational design of further reactive alkaline earth and group 13-centred systems.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Engineering and Physical Sciences Research Council (UK) for their generous support of this research (Grants: EP/N014456/1 and EP/R020752/1). This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath.References
- M. Wagner, N. Hommes, H. Noth and P. V. Schleyer, Inorg. Chem., 1995, 34, 607–614 CrossRef CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed.
- T. B. Marder, Science, 2006, 314, 69–70 CrossRef CAS PubMed.
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef CAS PubMed.
- M. Yamashita and K. Nozaki, in Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, 2015, vol. 49, pp. 1–37 Search PubMed.
- H. Braunschweig, Angew. Chem., Int. Ed., 2007, 46, 1946–1948 CrossRef CAS PubMed.
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570–9571 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS PubMed.
- T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162–14163 CrossRef CAS PubMed.
- T. Arnold, H. Braunschweig, W. C. Ewing, T. Kramer, J. Mies and J. K. Schuster, Chem. Commun., 2015, 51, 737–740 RSC.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M. J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315–319 CrossRef CAS PubMed.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 14159–14163 CrossRef CAS PubMed.
- R. Frank, J. Howell, J. Campos, R. Tirfoin, N. Phillips, S. Zahn, D. M. P. Mingos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 9586–9590 CrossRef CAS PubMed.
- Y. Okuno, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 920–923 CrossRef CAS PubMed.
- B. L. Wang, Y. X. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef CAS PubMed.
- W. Lu, H. T. Hu, Y. X. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef CAS PubMed.
- H. Braunschweig, C. W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 CrossRef CAS PubMed.
- H. Braunschweig, M. Burzler, R. D. Dewhurst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5650–5653 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2016, 55, 436–440 CrossRef CAS PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 RSC.
- L. B. Kong, Y. X. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 CrossRef CAS PubMed.
- L. B. Kong, R. Ganguly, Y. X. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893–2902 RSC.
- D. Wu, L. B. Kong, Y. X. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS PubMed.
- T. Imamoto and T. Hikosaka, J. Org. Chem., 1994, 59, 6753–6759 CrossRef CAS.
- L. B. Kong, W. Lu, Y. X. Li, R. Ganguly and R. Kinjo, Inorg. Chem., 2017, 56, 5586–5593 CrossRef CAS PubMed.
- J. Monot, A. Solovyev, H. Bonin-Dubarle, E. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacote, Angew. Chem., Int. Ed., 2010, 49, 9166–9169 CrossRef CAS PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 CrossRef CAS PubMed.
- E. Bernhardt, V. Bernhardt-Pitchougina, H. Willner and N. Ignatiev, Angew. Chem., Int. Ed., 2011, 50, 12085–12088 CrossRef CAS PubMed.
- J. Landmann, J. A. P. Sprenger, R. Bertermann, N. Ignat'ev, V. Bernhardt-Pitchougina, E. Bernhardt, H. Willner and M. Finze, Chem. Commun., 2015, 51, 4989–4992 RSC.
- J. Landmann, P. T. Hennig, N. V. Ignat'ev and M. Finze, Chem. Sci., 2017, 8, 5962–5968 RSC.
- J. Landmann, F. Keppner, D. B. Hofmann, J. A. P. Sprenger, M. Haring, S. H. Zottnick, K. Muller-Buschbaum, N. V. Ignat'ev and M. Finze, Angew. Chem., Int. Ed., 2017, 56, 2795–2799 CrossRef CAS PubMed.
- H. Ito, H. Yamanaka, J. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821–6825 CrossRef CAS.
- C. Kleeberg, L. Dang, Z. Y. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS PubMed.
- M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478–3481 CrossRef CAS PubMed.
- K. S. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253–7254 CrossRef CAS PubMed.
- A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyas and E. Fernandez, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed.
- M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. L. Santos, J. Org. Chem., 2011, 76, 3997–4007 CrossRef CAS PubMed.
- S. B. Thorpe, X. Guo and W. L. Santos, Chem. Commun., 2011, 47, 424–426 RSC.
- H. Gulyas, A. Bonet, C. Pubill-Ulldemolins, C. Sole, J. Cid and E. Fernandez, Pure Appl. Chem., 2012, 84, 2219–2231 CAS.
- J. Cid, H. Gulyas, J. J. Carbo and E. Fernandez, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC.
- N. Miralles, J. Cid, A. B. Cuenca, J. J. Carbo and E. Fernandez, Chem. Commun., 2015, 51, 1693–1696 RSC.
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Y. Mo, D. Qiu, M. S. Cheung, L. Dang, J. B. Wang, U. Radius, Z. Y. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed.
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- J. H. Zheng, Z. H. Li and H. D. Wang, Chem. Sci., 2018, 9, 1433–1438 RSC.
- J. Cid, A. Hermann, J. E. Radcliffe, L. D. Curless, H. Braunschweig and M. J. Ingleson, Organometallics, 2018, 37, 1992–1998 CrossRef CAS.
- A. B. Cuenca, R. Shishido, H. Ito and E. Fernandez, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- L. Garcia, J. Sendra, N. Miralles, E. Reyes, J. J. Carbo, J. L. Vicario and E. Fernandez, Chem.–Eur. J., 2018, 24, 14059–14063 CrossRef CAS PubMed.
- N. Miralles, R. Alam, K. J. Szabo and E. Fernandez, Angew. Chem., Int. Ed., 2016, 55, 4303–4307 CrossRef CAS PubMed.
- C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyas and E. Fernandez, Chem.–Eur. J., 2012, 18, 1121–1126 CrossRef CAS PubMed.
- F. Haeffner, Comput. Theor. Chem., 2018, 1131, 90–98 CrossRef CAS.
- A. F. Pecharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef CAS PubMed.
- A.-F. Pécharman, M. S. Hill and M. F. Mahon, Dalton Trans., 2018, 47, 6 RSC.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem.–Eur. J., 2016, 22, 7158–7162 CrossRef CAS PubMed.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC.
- Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, C. Weetman and M. S. Hill, Chem. Sci., 2017, 8, 3529–3537 RSC.
- A. F. Pecharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef CAS PubMed.
- A.-F. Pécharman, M. S. Hill, C. L. McMullin and M. F. Mahon, Organometallics, 2018, 37, 4457–4464 CrossRef.
- A. F. Pecharman, M. S. Hill and M. F. Mahon, Angew. Chem., Int. Ed., 2018, 57, 10688–10691 CrossRef CAS PubMed.
- F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- F. T. Edelmann, in Adv. Organometal. Chem., ed. A. F. Hill and M. J. Fink, 2013, vol. 61, pp. 55–374 Search PubMed.
- S. H. Li, J. H. Cheng, Y. H. Chen, M. Nishiura and Z. M. Hou, Angew. Chem., Int. Ed., 2011, 50, 6360–6363 CrossRef CAS PubMed.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362–13365 CrossRef CAS PubMed.
- J. F. Dunne, D. B. Fulton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 17680–17683 CrossRef CAS PubMed.
- S. R. Neal, A. Ellern and A. D. Sadow, J. Organomet. Chem., 2011, 696, 228–234 CrossRef CAS.
- J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782–16785 CrossRef CAS PubMed.
- D. Mukherjee, N. L. Lampland, K. K. Yan, J. F. Dunne, A. Ellern and A. D. Sadow, Chem. Commun., 2013, 49, 4334–4336 RSC.
- D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959–964 RSC.
- N. L. Lampland, A. Pindwal, S. R. Neal, S. Schlauderaff, A. Ellern and A. D. Sadow, Chem. Sci., 2015, 6, 6901–6907 RSC.
- R. Sanjeev, D. A. Padmavathi and V. Jagannadham, World J. Chem. Educ., 2018, 6, 78–81 CrossRef CAS.
- D. S. Matteson and K. M. Sadhu, Organometallics, 1984, 3, 614–618 CrossRef CAS.
- For an authenticated example of a bora-Brook rearrangement involving carbon-to-oxygen migration of a boryl group, see; H. Kisu, H. Sakaino, F. Ito, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2016, 138, 3548–3552 CrossRef CAS PubMed.
- For a very recent observation of 1,2-boryl carbon-to-nitrogen migration, see; Z. Li, L. Zhang, M. Nishiura and Z. Hou, ACS Catal., 2019, 9, 4388–4393 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation data, X-ray crystallographic information, computational details, and tables of Cartesian coordinates (PDF). Crystallographic (CIF) data for compounds 11, 14, 15BN, 16BN, 17BN, 18BN, 17BC and 18Ph are available from the Cambridge Structural Database as CCDC 1910885–1910892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02087j |
| This journal is © The Royal Society of Chemistry 2019 |