
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide-induced super-assembly of biocatalytic metal–organic frameworks for programmed enzyme cascades†
Jieying
Liang
 ab,
Federico
Mazur
a,
Chuyang
Tang
a,
Xunan
Ning
*b,
Rona
Chandrawati
*a and
Kang
Liang
*ac
ab,
Federico
Mazur
a,
Chuyang
Tang
a,
Xunan
Ning
*b,
Rona
Chandrawati
*a and
Kang
Liang
*ac
aSchool of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: rona.chandrawati@unsw.edu.au; kang.liang@unsw.edu.au
bSchool of Environmental Science and Engineering, Guangdong University of Technology, Guangzhou 510006, China. E-mail: ningxunan666@126.com
cGraduate School of Biomedical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 22nd July 2019
Abstract
Despite the promise of metal–organic frameworks (MOFs) as functional matrices for enzyme stabilization, the development of a stimulus-responsive approach to induce a multi-enzyme cascade reaction in MOFs remains a critical challenge. Here, a novel method using peptide-induced super-assembly of MOFs is developed for programmed enzyme cascade reactions on demand. The super-assembled MOF particles containing different enzymes show remarkable 7.3-fold and 4.4-fold catalytic activity enhancements for the two-enzyme and three-enzyme cascade reactions, respectively, as compared with the unassembled MOF nanoparticles. Further digestion of the coiled-coil forming peptides on the MOF surfaces leads to the MOF superstructure disassembly and the programmed enzyme cascade reaction being “switched-off”. Research on these stimuli-responsive materials with controllable and predictable biocatalytic functions/properties provide a concept to facilitate the fabrication of next-generation smart materials based on precision chemistry.
Introduction
Multi-enzyme catalytic cascades play key roles in biological signal transduction and metabolic pathways,1,2 and have also attracted significant interest in the field of enzymatic fuel cells,3 biosensors,4 disease diagnostics5 and the synthesis of pharmaceuticals.6 Despite the unprecedented selectivity and reactivity of enzymes as compared to synthetic catalysts, challenges still remain in the full utilization of enzymes for commercial scale applications because of their poor reusability, nominal thermal stability and diminished performance in organic media.7 To overcome this problem, a rational approach is to encapsulate the enzymes in suitable matrices, which significantly improves their stability and reusability.8,9 Very recently, metal–organic frameworks (MOFs) have emerged as one of the most promising candidate matrices for enzyme incorporation to yield structures that can protect them from inhospitable external environments (e.g. elevated temperatures or proteolytic media), while facilitating size-selective transport of substrates to the active site via their ordered and tailorable pore network.10–14 Constructed from Zn and 2-methylimidazole with a sodalite (SOD) topology, zeolitic imidazolate framework-8 (ZIF-8) demonstrates superior stability under aqueous physiological conditions15 and its surface can easily be functionalized to tailor it for different applications.16 Moreover, high enzyme encapsulation efficiency could be achieved via a modular, single-step synthetic process.17–20In the multi-enzyme catalytic cascade reactions, the intermediate needs to be efficiently transported from one enzyme to another. In other words, positioning different enzymes in close proximity could minimize the diffusion limitation and maximize the enzymatic cascade activity.21 For enzymes encapsulated in MOFs, the physical barrier of MOF frameworks and their disorder states significantly hinder substrate diffusion and lead to a decrease in the reaction rate.22 Accordingly, recent studies on encapsulating multiple enzymes in a single MOF particle have received significant attention.18,19,22–24 This method, by introducing an enzyme cocktail into one MOF particle, shows enhanced multi-enzyme stability and adequate biocatalytic activity. However, challenges remain in designing a smart and stimulus-responsive approach to program a multi-enzyme cascade reaction in MOFs on demand. Success in this endeavour is expected to unlock new potential for biocatalytic MOF systems toward smart biosensor and diagnostic device applications, where a stimulus-responsive and on-demand enzymatic cascade reaction is imperative.
Self-assembly is a promising and practical route for the creation of stimuli-responsive nanomaterials with novel properties suitable for applications in biosensing,25–31 drug delivery32 and therapeutics.33 Peptides have the inherent ability to self-assemble,34 which is driven by noncovalent intermolecular interactions (van der Waals, hydrophobic, hydrogen bonding, electrostatic, and aromatic π-stacking) through molecular recognition.35,36 Peptide-based biosensors have been developed for the detection of various analytes, including ions,25,32 proteins,33,37,38 and cells.39 Among them, helical hetero-dimers have wide applications in the design of three-dimensional (3D) nanostructures. For example, complementary dimeric coiled-coil peptides can induce the self-assembly of artificial viral capsids.40,41 Protein–protein helical hetero-dimers were used in colloidal assembly of polystyrene microparticles42 and protein super-structures.43 In addition, super-assembled gold nanoparticles induced by dimeric coiled-coil peptides exhibited stimuli-responsive characteristics in response to pH and temperature changes.44 In these studies, surface functionalized complementary peptide sequences drove particle super-assembly. The peptides were engineered with a cysteine residue to facilitate their covalent attachment onto nanoparticle surface via gold-thiol or other chemistry (e.g. thiol-maleimide).44–46 These studies highlight the immense utility of helical hetero-dimers in the design and fabrication of self-assembled and smart functional materials.
Here, we demonstrate the on-demand multi-enzyme cascade reaction in MOFs via a novel peptide-induced MOF super-assembly process. Single-enzyme-containing MOF particles are surface-functionalized with complementary peptides, where the self-assembled peptide coiled-coil structure induces MOF particle super-assembly, which brings different enzyme-containing MOF particles to proximity to trigger two- or three-enzyme cascade reactions (Scheme 1). Further disruption of the peptide binding leads to disassembly of the MOF superstructures and hence termination of the enzyme cascade reaction. This super-assembly approach for a programmed “on/off” multi-enzyme cascade is expected to facilitate further development of smart stimuli-responsive biocatalytic MOF systems for advanced energy and biotechnology applications.
 | ||
| Scheme 1 Schematic illustration of (a) two-enzyme (GOx/ZIF-8A and HRP/ZIF-8A) and (b) three-enzyme (β-gal/ZIF-8A, GOx/ZIF-8A and HRP/ZIF-8A) cascades “switch on” driven by complementary coiled-coil forming peptide (JR2EK and JR2KC) induced MOF superstructures and then “switch off” by chemically disrupting the coiled-coil superstructure. | ||
Results and discussion
Enzyme-containing ZIF-8 nanocrystals (enzyme/ZIF-8) were synthesized by mixing aqueous solutions of zinc nitrate (0.0135 g, 1 mL), enzyme (0.4 mL, 5 mg mL−1) i.e. glucose oxidase (GOx), horseradish peroxidase (HRP) or β-galactosidase (β-gal), and 2-methylimidazole (HmIm, 0.2830 g, 4 mL), and then stirring for 30 min at room temperature, followed by centrifugation and washing cycles. A pH ranging from 7.0–9.6 upon enzyme/ZIF-8 synthesis has a negligible effect on GOx and HRP, while the activity of β-gal decreases with increasing pH (Table S1 and Fig. S1†). Nevertheless, all the enzymes retained their full activity once the pH was adjusted back to 7.0. A post-synthetic ligand exchange method (PSM)47 was then carried out to functionalize ZIF-8 with primary amines by introducing 3-amino-1,2,4-triazole (Atz) into the framework (ZIF-8A) to enable further peptide functionalization. As revealed by scanning electron microscopy (SEM) images (Fig. 1a), ZIF-8A and enzyme-containing ZIF-8A (enzyme/ZIF-8A) present a rhombic dodecahedral morphology with average diameters of 700–800 nm and display a similar morphology to those of the pure ZIF-8 and enzyme/ZIF-8 crystals (Fig. S2a†). The similar XRD patterns of ZIF-8, ZIF-8A and enzyme/ZIF-8A indicated that ZIF-8A prepared by the PSM retained the crystal structure of the parent ZIF-8 and the incorporation of enzyme did not affect the long-range ordered structure of ZIF-8 (Fig. 1b).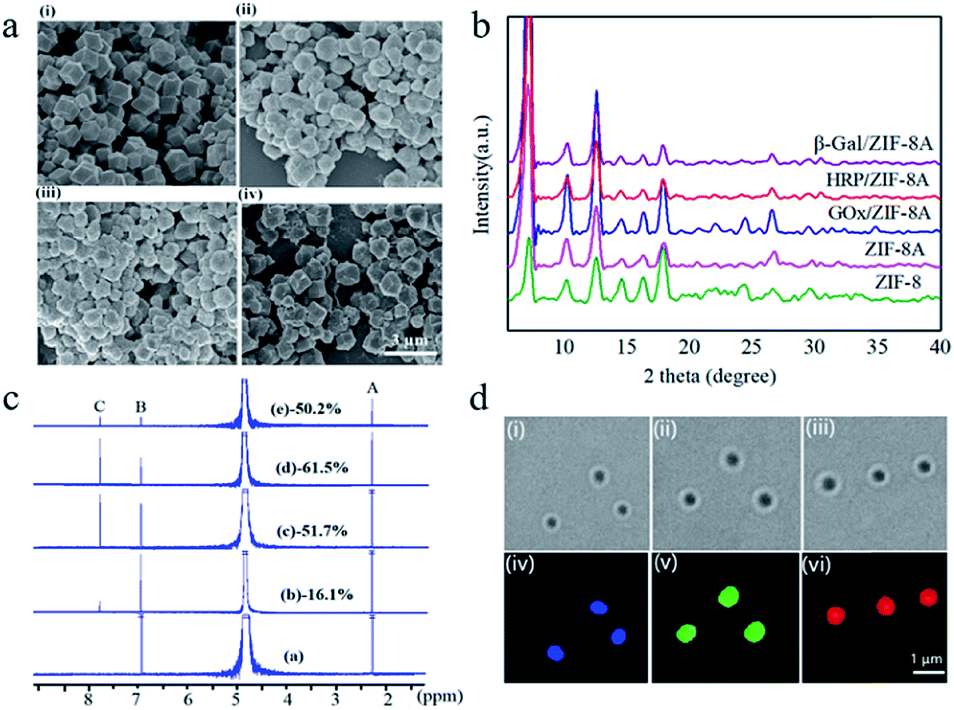 | ||
| Fig. 1 Characterization of enzyme/ZIF-8A: (a) SEM images, ZIF-8A (i), GOx/ZIF-8A (ii), HRP/ZIF-8A (iii), and β-gal/ZIF-8A (iv); (b) PXRD patterns; (c) 1H NMR spectra, ZIF-8 (curve a), ZIF-8A (curve b), GOx/ZIF-8A (curve c), HRP/ZIF-8A (curve d), and β-gal/ZIF-8A (curve e); (d) bright-field and CLSM images of GOx/ZIF-8A (blue) (i and iv), HRP/ZIF-8A (green) (ii and v), and β-gal/ZIF-8A (red) (iii and vi), respectively. | ||
The FTIR spectra (Fig. S2b†) of ZIF-8A and enzyme/ZIF-8A confirmed the signal peak of C–NH2 at 1580 cm−1, indicating that the amine group was functionalized on ZIF-8 and enzyme/ZIF-8. The 1H NMR spectra of ZIF-8 exhibited peaks at 2.4 (A) and 6.9 ppm (B), which correspond to methyl and methine groups of HmIm, respectively (Fig. 1c). Atz exchange resulted in a new peak at 7.8 ppm (C), which corresponds to the methine peak of Atz. The conversion reaction of Atz in ZIF-8A was determined using the intensity ratio of the methine peaks between B and C in the 1H NMR spectra47 (Fig. 1c), yielding ZIF-8A, GOx/ZIF-8A, HRP/ZIF-8A and β-gal/ZIF-8A with 16.1%, 51.7%, 61.5% and 50.2% Atz conversion, respectively.
The incorporation efficiencies of GOx, HRP and β-gal in the ZIF-8A were calculated to be 50.6%, 42.7% and 78.3%, respectively, determined by using fluorescently labelled proteins (Fig. S3a†). To exclude the possible presence of enzyme on the surface of ZIF-8A, the GOx/ZIF-8 was treated with free HRP, HRP/ZIF-8A was treated with free GOx, and GOx/ZIF-8 and HRP/ZIF-8 was treated with free β-gal. However, after the PSM, the resulting enzyme/ZIF-8A did not show any cascade activity, implying that no enzyme was present on the ZIF-8A surface (Fig. S3b†). From the confocal laser scanning microscopy (CLSM) images (Fig. 1d), it can be observed that Alexa Fluor 350-labelled GOx (blue), FITC-labelled HRP (green) and Atto 647N NHS ester-labelled β-gal (red) were homogenously distributed in ZIF-8A nanocrystals.
In the two-enzyme cascade reaction, the catalytic efficiency was evaluated by the addition of glucose and using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) as a chromogenic substrate. GOx converts glucose into gluconic acid and generates H2O2 which is the substrate for HRP to oxidize ABTS2− to form ABTS−˙, and ABTS−˙ could be detected at 415 nm using ultraviolet-visible spectroscopy (UV-Vis).17 In the three-enzyme cascade reaction, lactose was used as a substrate. β-gal hydrolyzes lactose into glucose and galactose, followed by the cascade reaction by GOx and HRP (Scheme 1). Previous studies by us and other groups have proved that molecules larger than the window size of ZIF-8 can still flow into the crystals due to the inherited flexibility of ZIF-8 and the larger defects induced as a result of enzyme loading (Fig. S4†).10,48–50 The enhanced absorbance kinetics of the two-enzyme system (free HRP and GOx/ZIF-8A, and free GOx and HRP/ZIF-8A) and three-enzyme system (free GOx, HRP and β-gal/ZIF-8A) indicated that the activity of GOx, HRP and β-gal was well retained in ZIF-8A (Fig. S5a and b†). In contrast, the GOx/ZIF-8A and HRP/ZIF-8A particle mixture showed significantly reduced enzymatic activity (the three enzyme/ZIF-8A particle mixture showed negligible activity), suggesting that the substrate diffusion limitation rather than the intrinsic enzyme catalytic activity is a major contributing factor in the non-peptide functionalized enzyme/ZIF-8A particle mixture (Fig. S4a and b†). In addition, control experiments indicated that the single enzyme/ZIF-8A cannot trigger the catalytic cascade reaction (Fig. S6†).
To provide a site for peptide attachment, a bifunctional linker (SM(PEG)2) was coupled to the enzyme/ZIF-8A's amine group, allowing for covalent attachment at position 22 (i.e. cysteine residue) located in the loop region of the peptides via thiol-maleimide chemistry (Fig. 2a). 42-residue polypeptides JR2EC and JR2KC (helix-loop-helix motifs) were used.46,51 As shown by circular dichroism (CD) spectroscopy (Fig. 2b), peptides self-assembled into a helical conformation in PBS solution (pH 7.4). To super-assemble the enzyme/ZIF-8A particles, an aqueous suspension containing 1200 μg mL−1 of enzyme/ZIF-8A was mixed with an SM(PEG)2 linker and incubated for 30 min followed by removal of the unbound SM(PEG)2 by washing and centrifugation. JR2EC or JR2KC peptide was pre-treated with TCEP for 60 min prior to addition into the corresponding enzyme/ZIF-8A/linker system (JR2EC to GOx/ZIF-8A/SM(PEG)2, JR2KC to HRP/ZIF-8A/SM(PEG)2 and β-gal/ZIF-8A/SM(PEG)2). The final peptide-conjugated enzyme/ZIF-8A was obtained after 30 min incubation followed by centrifugation and washing steps. The peptide conjugation was confirmed by a shift in the zeta potential value of GOx/ZIF-8 from −11.9 mV to −22.0 mV due to the presence of negatively charged residues on JR2EC, and HRP/ZIF-8 and β-gal/ZIF-8 from −25.7 mV and −15.3 mV to −13.2 mV and −11.8 mV, respectively, due to the presence of positively charged residues on JR2KC (Fig. S7a and b†). The 1H NMR spectra of peptide-functionalized enzyme/ZIF-8A exhibited peaks at 2.05 ppm (a), which corresponds to the thiol group of cysteine in the peptide, further confirming the conjugation between the peptide and enzyme/ZIF-8A (Fig. S8†). After mixing the different peptide-functionalized enzyme/ZIF-8A particles, SEM analysis presents the super-assembly of two or three enzyme/ZIF-8A particles (Fig. 2c(i) and (iii)). CLSM images further confirmed that GOx/ZIF-8A and HRP/ZIF-8A were bound together in the two enzyme/ZIF-8A system, and β-gal/ZIF8A, GOx/ZIF8A, and HRP/ZIF8A were conjugated in the three enzyme/ZIF-8A system (Fig. 2c(ii), (iv), and S9†). The 2D assemblies over 3D assemblies are likely due to the low grafting concentration of peptides on the MOF particles, and the relatively large, near-micrometer scale particle size. Previous studies suggested that the interactions between self-assembled micro-meter sized particles consist of an attractive force (resulting from the assembly) due to interparticle molecular hybridization, a steric and/or electrostatic repulsive force, and a strong attractive van der Waals interaction at short distances.52 All these factors could be responsible for the preference of 2D assemblies over 3D assemblies. To exclude the possibility of the random conjugation of enzyme/ZIF-8A, the conjugation of enzyme/ZIF-8A functionalized with the same peptide (JR2EC only or JR2KC only) was investigated. After mixing, only individual enzyme/ZIF-8A was presented and no significant particle super-assembly was observed (Fig. S10†), which strongly justifies the notion that the super-assembly is induced by the complementary coiled-coil peptide structures on the MOF surface (Fig. 2c and S11†). Analysis of the super-assembled enzyme/ZIF-8A structure specificity via CLSM is presented in Table S2.† It can be expected that regardless of the specificity of the super-assembled structures, the enzyme cascade reaction will be accelerated to some extent as compared to that of unassembled particles. Further particle functionalization by different complementary peptides is expected to improve the specificity of the super-assembled MOF particles in the three enzyme/ZIF-8A system.
 | ||
| Fig. 2 (a) Polypeptides (JR2EC or JR2KC) conjugated to ZIF-8A via a bifunctional linker SM(PEG)2. (b) CD spectra of self-assembled JR2EC (0.05 mg mL−1) and JR2KC (0.05 mg mL−1) confirming coiled-coil formation. (c) SEM and CLSM images of the super-assembled GOx/ZIF-8A (blue) and HRP/ZIF-8A (green) (i and ii) and the super-assembled GOx/ZIF-8A (blue), HRP/ZIF-8A (green) and β-gal/ZIF-8A (red) (iii and iv). The concentration of peptides is 0.1 mg mL−1. | ||
Next, the efficiency of the peptide-induced super-assembly for multi enzyme/ZIF-8A cascade reactions was studied. Control of the cascade efficiency was achieved by tuning the amounts of the linker (SM(PEG)2) and peptides on the enzyme/ZIF-8A particles (Fig. S10†). An insignificant difference was observed when using peptide concentrations from 0.1 mg mL−1 to 1.0 mg mL−1, suggesting 0.1 mg mL−1 polypeptide concentration was sufficient to super-assemble the enzyme/ZIF-8A. However, the kinetics in absorbance increased with increasing the linker (SM(PEG)2) concentration from 0.001 mM to 1 mM. Fig. 3 shows the “switch-on” cascade reaction detected by measuring the absorbance kinetics of the ABTS−˙ generated by the two enzyme/ZIF-8A and three enzyme/ZIF-8A cascade reactions. The activity of the super-assembled two enzyme/ZIF-8A particles exhibited a remarkable 7.3-fold enhancement compared with that of the unassembled GOx/ZIF-8A and HRP/ZIF-8A mixture (Fig. 3a); whilst the three-enzyme cascade reaction in the super-assembled enzyme/ZIF-8A showed a 4.4-fold enhancement in activity compared with the unassembled β-gal/ZIF-8A, GOx/ZIF-8A and HRP/ZIF-8A mixture (Fig. 3b). For the control, super-assembled pure ZIF-8A particles lacking the enzymes were used to react with ABTS and glucose/lactose. The results indicated that the peptides on the ZIF-8A surface did not promote ABTS conversion in the absence of the enzyme (Fig. S12†). The enhanced catalytic performance in the super-assembled enzyme/ZIF-8A particles could be attributed to the peptide induced inter-particle organization, which brings the different single enzyme/ZIF-8A particles to close proximity, where the high local concentration of the products generated by one of the enzymes is fed into the subsequent biocatalyst. The enzyme/ZIF-8A retained the crystal structure after the cascade reaction (Fig. S13†), further suggesting the superior stability of enzyme/ZIF-8A.
 | ||
| Fig. 3 (a) Time-dependent absorbance changes observed by the reaction of two-enzyme and (b) three enzyme systems with or without peptide functionalization (0.1 mg mL−1 peptide and 0.1 mM linker). (c) Stimuli-responsive catalytic characteristics of the super-assembled GOx/ZIF-8A and HRP/ZIF-8A and (d) the super-assembled GOx/ZIF-8A, HRP/ZIF-8A, and β-gal/ZIF-8A with and without protease. | ||
To demonstrate the on demand “switching off” of the enzyme cascade reaction, the two and three enzyme/ZIF-8A super-assembled systems were incubated with protease, a peptide digestion cocktail (Fig. 3c and d). The presence of protease resulted in a significant decrease in the activity kinetics, suggesting the reaction was “switched off” due to the cleavage of JR2EC and JR2KC within the coiled-coil crosslinks. For the control, the activity of the enzyme/ZIF-8A particles without peptides on their surface against protease digestion was investigated. For the two-enzyme system, protease presented little effect on the cascade reaction, while the three-enzyme system showed no influence. This indicated that enzymatic activity was maintained in the ZIF-8A as a result of the protection provided by the encapsulation (Fig. S14†). SEM (Fig. S15(i) and (iii)†) and confocal microscopy (Fig. S15(ii) and (iv)†) images further confirmed that the super-assembled particle structures were disassembled after the addition of protease into the two enzyme/ZIF-8A and three enzyme/ZIF-8A system. The results highlight that these super-assembled MOFs, stabilized through JR2EC/JR2KC coiled-coils, have the potential to be further “switched off” upon exposure to chemical triggers.
Experimental section
Synthesis of the ZIF-8, GOx/ZIF-8, HRP/ZIF-8 and β-gal/ZIF-8 composite
An aqueous solution (0.4 mL) of enzyme (5 mg mL−1 GOx, 5 mg mL−1 HRP, or 5 mg mL−1 β-gal) and 2-methylimidazole water solution (0.2830 g, 4 mL) were first mixed before adding an aqueous solution of Zn(NO3)2 (0.0135 g, 1 mL) under stirring at room temperature. The mixture turned milky almost instantly after mixing. The reaction was allowed to proceed for 30 min, followed by 3 cycles of centrifugation at 6000 rpm for 10 min, and washing. The final product was suspended in 1 mL DI water. ZIF-8 nanocrystals were synthesized following the same process without adding enzyme. The effect of pH during enzyme/ZIF-8A synthesis on enzyme activity was evaluated using DI water and PBS (pH 7.4, 8.6 and 9.6).Synthesis of ZIF-8A, GOx/ZIF-8A, HRP/ZIF-8A and β-gal/ZIF-8A via a post-synthetic ligand exchange method (PSM)
Atz (24 mg, 0.28 mmol) was added to the ZIF-8 (or GOx/ZIF-8, HRP/ZIF-8 and β-gal/ZIF-8) suspension. The PSM reaction was performed at 50 °C for 2 h. The product was collected after 3 cycles of centrifugation at 6000 rpm for 10 min and washing. The final product was suspended in 1 mL DI water.Characterization of enzyme/ZIF-8 and enzyme/ZIF-8A
SEM images of samples were taken on a NanoSEM 450 field-emission scanning electron microscope (FE-SEM). Powder X-ray diffraction patterns (PXRD) were recorded using an Empyrean XRD Xpert Materials Research diffractometer (MRD) system with a Cu Kα anode (λ = 0.15406 nm) at 40 kV and 40 mA. FTIR (Alpha-FTIR) was used to determine the amine group functionalized on the enzyme/ZIF-8. NMR analysis was used for determining the Atz conversion. 1H NMR spectra were acquired by dissolving the MOFs in diluted HCl with D2O (10/90 v/v) at 25 °C on a Bruker Avance III 400 MHz NMR (Rabi).Determination of the enzyme incorporation efficiency by a fluorescent protein labeling method
10 μL of Alexa Fluor 350 (λex 346 nm; λem 442 nm), fluorescein isothiocyanate (FITC; λex 492 nm; λem 518 nm) and Atto 647N NHS ester (λex 647 nm; λem 661 nm) dissolved in water (2 mg mL−1) were slowly added to 1 mL of GOx, HRP and β-gal solution (5 mg mL−1 of enzyme in 0.5 M carbonate buffer, pH 8.3), respectively. After 2 h, excess non-reacted dyes were removed via dialysis against DI water. GOx, HRP and β-gal were labeled with Alexa Fluor 350 NHS ester, fluorescein isothiocyanate (FITC) and Atto 647N NHS ester, respectively, and subjected to the same procedure to synthesize the enzyme/ZIF-8A composite. The fluorescent protein labeling method was used for quantitative analysis of the enzyme incorporation efficiency of enzyme/ZIF-8A using a SpectraMax M5 microplate reader (Molecular Devices). Confocal laser scanning microscopy (CLSM) images of the fluorescently labeled enzyme/ZIF-8A particles were taken with an Olympus IX53 CLSM equipped with the corresponding filter sets and a 60× oil immersion objective.Evaluation of the activities of GOx, HRP and β-gal incorporated in the ZIF-8A
To evaluate the biocatalytic activity of individual enzyme/ZIF-8A particles, 5 μg mL−1 free HRP instead of 60 μg mL−1 HRP/ZIF-8A was added to in the two enzyme/ZIF-8A cascade system to evaluate the activity of GOx/ZIF-8A; however 5 μg mL−1 free GOx instead of GOx/ZIF-8A was added to evaluate the activity of HRP/ZIF-8A. In the three enzyme/ZIF-8A cascade system, 5 μg mL−1 free GOx and 5 μg mL−1 free HRP instead of GOx/ZIF-8A and HRP/ZIF-8A were added to evaluate the activity of β-gal/ZIF-8A. ABTS was used as a chromogenic substrate. The two-enzyme catalytic reaction was initiated in 220 μL PBS solution containing 314 mM ABTS, 1.2 mM glucose, 60 μg mL−1 GOx/ZIF-8A and 60 μg mL−1 HRP/ZIF-8A. And the three-enzyme catalytic reaction was initiated in 270 μL PBS solution containing 532 mM ABTS, 29 mM lactose, 60 μg mL−1 GOx/ZIF-8A, 60 μg mL−1 HRP/ZIF-8A and 60 μg mL−1 β-gal/ZIF-8A. The absorbance at 415 nm was recorded using a SpectraMax M5 microplate reader (Molecular Devices). To ensure that no ABTS conversion takes place when only one enzyme is present, we evaluated the absorbance of 60 μg mL−1 GOx/ZIF-8A or 60 μg mL−1 HRP/ZIF-8A in the presence of 314 mM ABTS and 1.2 mM glucose (two-enzyme system) and 60 μg mL−1 β-gal/ZIF-8A in the presence of 532 mM ABTS and 29 mM lactose for β-gal/ZIF-8A (three-enzyme system).Peptide conjugation to enzyme/ZIF-8A
GOx/ZIF-8A, HRP/ZIF-8A and β-gal/ZIF-8A were diluted 10 times in DI water prior to use. Enzyme/ZIF-8A (1200 μg mL−1) and linker SM(PEG)2 (4 μL, 0.1 mM in PBS, pH 7.4) were mixed and incubated for 30 min under constant shaking (500 rpm) at 37 °C, followed by removal of the unbound SM(PEG)2 by centrifugation (6000 rcf, 4 °C, 10 min). Next, JR2EC or JR2KC peptides (25 mg mL−1) were pre-treated with TCEP (0.1 M in 20 mM MOPS buffer) for 60 min at 37 °C under constant shaking. TCEP-treated peptides (20 μL, 0.1 mg mL−1) was then added into the corresponding enzyme/ZIF-8A/linker system (JR2EC to GOx/ZIF-8/SM(PEG)2, and JR2KC to HRP/ZIF-8/SM(PEG)2 and β-gal/ZIF-8/SM(PEG)2) and incubated for 30 min under constant shaking (500 rpm) at 37 °C. The resulting products were centrifuged at 6000 rcf, 4 °C, 10 min, to remove unbound peptides. A control experiment of ZIF-8A without enzyme in the presence of 314 mM ABTS and 1.2 mM glucose or 532 mM ABTS and 29 mM lactose was conducted to exclude the possibility that peptides might trigger an increased ABTS conversion even in the absence of the enzyme.Unbound peptide concentration determination in the supernatant via Bradford assay
Standard solutions of peptides were prepared in DI water. 100 μL Bradford reagent was mixed with 100 μL of each standard solution or 100 μL supernatant of peptide-conjugated enzyme/ZIF-8A in a 96-well plate. The mixture was incubated for 5 min and the absorbance was measured at 595 nm using a SpectraMax M5 microplate reader (Molecular Devices).Characterization of super-assembled enzyme/ZIF-8A structures
CD spectra were measured using an Applied Photophysics Chirascan Plus CD spectrometer. Samples were measured using a 0.5 mm path length cuvette. The spectra were recorded from 200 to 260 nm at 2 nm internals with a 1 nm bandwidth. CD spectra of the polypeptides indicate the formation of coiled-coil structures with minima at 206 and 222 nm. The zeta potential was measured using a Zetasizer Nano ZS (Malvern). NMR analysis was used to confirm the peptide conjugation. 1H NMR spectra were acquired in diluted HCl with D2O (10/90 v/v) at 25 °C on a Bruker Avance III 400 MHz NMR (Rabi). CLSM and SEM images of the super-assembled enzyme/ZIF-8A were also obtained as per the procedure described in the Characterization section.Switching off the cascade reaction by protease
0.5 μL protease from Bacillus licheniformis (2.4 U g−1) was added to 50 μL of enzyme/ZIF-8A super-assembled system and incubated for 30 min at 37 °C. The absorbance was recorded using a SpectraMax M5 microplate reader (Molecular Devices). A control experiment of the activity of the proposed enzyme/ZIF-8A particles without peptides against protease digestion was also performed.Conclusions
We report a peptide-induced super-assembly of enzyme-containing MOFs for programmed enzyme cascade reactions. Peptides with coiled-coils as crosslinkers allowed the super-assembly of two and three single enzyme-containing MOF particles, which dramatically accelerate (switch-on) the cascade reaction when compared to unassembled MOF nanoparticles. Further incubation in protease terminates the peptide crosslinking and leads to programmed “switch-off” of the cascade reaction. This super-assembly approach for programmed “on” and “off” multi-enzyme cascades is expected to facilitate further development of smart stimuli-responsive systems for advanced energy and biotechnology applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australia National Health and Medical Research Council (NHMRC APP1163786, K. L.), the Scientia Fellowship program at UNSW (R. C., K. L.), and the Australian Research Council (ARC) (DP190101008, K. L.). J. L. acknowledges the support from the China Scholarship Council. F. M. acknowledges the support from the Australian Government Research Training Program Scholarship. R. C. acknowledges the support from the Australian Research Council Discovery Early Career Researcher Award (ARC DECRA DE170100068).References
- E. García-Junceda, I. Lavandera, D. Rother and J. H. Schrittwieser, Nat. Nanotechnol., 2016, 11, 409–420 CrossRef PubMed.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- Z. Zhu, T. K. Tam, F. Sun, C. You and Y.-H. P. Zhang, Nat. Commun., 2014, 5, 3026 CrossRef PubMed.
- Y. Zhang, M. A. Arugula, M. Wales, J. Wild and A. L. Simonian, Biosens. Bioelectron., 2015, 67, 287–295 CrossRef CAS PubMed.
- V. Sanz, S. de Marcos and J. Galbán, Biosens. Bioelectron., 2007, 22, 2876 CrossRef CAS PubMed.
- E. O'Reilly and C. Iglesias, Perspect. Sci., 2015, 4, 55–61 CrossRef.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694–1697 CrossRef CAS.
- K. Ariga, Q. Ji, T. Mori, M. Naito, Y. Yamauchi, H. Abe and J. P. Hill, Chem. Soc. Rev., 2013, 42, 6322–6345 RSC.
- D. N. Tran and K. J. Balkus Jr, ACS Catal., 2011, 1, 956–968 CrossRef CAS.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS.
- D. Feng, T. F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y. P. Chen, X. Wang, K. Wang, X. Lian, Z. Y. Gu, J. Park, X. Zou and H. C. Zhou, Nat. Commun., 2015, 6, 5979 CrossRef PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- S. Gao, J. Hou, Z. Deng, T. Wang, S. Beyer, A. Guilherme Buzanich, J. J. Richardson, A. Rawal, R. Seidel, M. Y. Zulkifli, W. Li, T. D. Bennett, A. K. Cheetham, K. Liang and V. Chen, Chem, 2019, 5, 1–12 Search PubMed.
- H. Ren, L. Zhang, J. An, T. Wang, L. Li, X. Si, L. He, X. Wu, C. Wang and Z. Su, Chem. Commun., 2014, 50, 1000–1002 RSC.
- A. Tiwari, A. Singh, N. Garg and J. K. Randhawa, Sci. Rep., 2017, 7, 12598 CrossRef PubMed.
- F. K. Shieh, S. C. Wang, C. I. Yen, C. C. Wu, S. Dutta, L. Y. Chou, J. V. Morabito, P. Hu, M. H. Hsu, K. C. Wu and C. K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed.
- W. H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nat. Catal., 2018, 1, 689–695 CrossRef CAS.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. J. C. C. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- S. Gao, J. Hou, J. Zeng, J. J. Richardson, Z. Gu, X. Gao, D. Li, M. Gao, D. W. Wang, P. Chen, V. Chen, K. Liang, D. Zhao and B. Kong, Adv. Funct. Mater., 2019, 29, 1808900 CrossRef.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. J. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed.
- X. Lian, Y. P. Chen, T. F. Liu and H. C. Zhou, Chem. Sci., 2016, 7, 6969–6973 RSC.
- X. Liu, W. Qi, Y. Wang, R. Su and Z. He, Nanoscale, 2017, 9, 17561–17570 RSC.
- Z. L. Xu, G. W. Xiao, H. F. Li, Y. Shen, J. Zhang, T. Pan, X. Y. Chen, B. Zheng, J. S. Wu, S. Li, W. N. Huang and F. W. Huo, Adv. Funct. Mater., 2018, 28, 1802479 CrossRef.
- H. Chen, S. Jia, J. Zhang, M. Jang, X. Chen, K. Koh and Z. Wang, Anal. Methods, 2015, 7, 8942–8946 RSC.
- F. Mazur, L. Liu, H. Li, J. Huang and R. Chandrawati, Sens. Actuators, B, 2018, 268, 182–187 CrossRef CAS.
- C. Jumeaux, E. Kim, P. D. Howes, H. Kim, R. Chandrawatia and M. M. Stevens, Nanoscale Adv., 2019, 1, 532–536 RSC.
- Y. Chu, J. Hou, C. Boyer, J. J. Richardson, K. Liang and J. Xu, Applied Materials Today, 2018, 10, 93–105 CrossRef.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. A. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288 RSC.
- Q. Y. Lin, J. A. Mason, Z. Li, W. Zhou, M. N. O'Brien, K. A. Brown, M. R. Jones, S. Butun, B. Lee, V. P. Dravid, K. Aydin and C. A. Mirkin, Science, 2018, 359, 669–762 CrossRef CAS PubMed.
- B. S. Flavel, M. Nambiar and J. G. Shapter, Silicon, 2011, 3, 163–171 CrossRef CAS.
- R. Li, H. Huang, L. Huang, Z. Lin, L. Guo, B. Qiu and G. Chen, Electrochim. Acta, 2013, 109, 233–237 CrossRef CAS.
- M. Puiu and C. Bala, Peptide-based biosensors, Bioelectrochemistry, 2018, 120, 66–75 CrossRef CAS PubMed.
- Y. Loo, S. Zhang and C. A. E. Hauser, Biotechnol. Adv., 2012, 30, 593–603 CrossRef CAS PubMed.
- A. L. Boyle, E. H. C. Bromley, G. J. Bartlett, R. B. Sessions, T. H. Sharp, C. L. Williams, P. M. G. Curmi, N. R. Forde, H. Linke and D. N. J. Woolfson, J. Am. Chem. Soc., 2012, 134, 15457–15467 CrossRef CAS PubMed.
- R. Chandrawati and M. M. Stevens, Chem. Commun., 2014, 50, 5431 RSC.
- F. Mazur and R. Chandrawati, Adv. Biosyst., 2019, 3, 1800330 CrossRef.
- M. S. Mannoor, S. Zhang, A. J. Link and M. C. McAlpine, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19207–19212 CrossRef CAS PubMed.
- S. Fujita and K. Matsuura, Org. Biomol. Chem., 2017, 15, 5070 RSC.
- E. De Santis, H. Alkassem, B. Lamarre, N. Faruqui, A. Bella, J. E. Noble, N. Micale, S. Ray, J. R. Burns, A. R. Yon, B. W. Hoogenboom and M. G. Ryadnov, Nat. Commun., 2017, 8, 2263 CrossRef PubMed.
- M. Obana, B. R. Silverman and D. A. Tirrell, J. Am. Chem. Soc., 2017, 139, 14251–14256 CrossRef CAS PubMed.
- G. Indelicato, N. Wahome, P. Ringler, S. A. Müller, M. P. Nieh, P. Burkhard and R. Twarock, Biophys. J., 2016, 110, 646–660 CrossRef CAS PubMed.
- M. M. Stevens, N. T. Flynn, C. Wang, D. A. Tirrell and R. Langer, Adv. Mater., 2004, 16, 915–918 CrossRef CAS.
- M. G. Ryadnov, B. Ceyhan, C. M. Niemeyer and D. N. Woolfson, J. Am. Chem. Soc., 2003, 125, 9388–9394 CrossRef CAS PubMed.
- A. J. Gormley, R. Chandrawati, A. J. Christofferson, C. Loynachan, C. Jumeaux, A. Artzy-Schnirman, D. Aili, I. Yarovsky and M. M. Stevens, Chem. Mater., 2015, 27, 5820–5824 CrossRef CAS.
- K. Y. Cho, H. An, X. H. Do, K. Choi, H. G. Yoon, H. K. Jeong, J. S. Lee and K. Y. J. Baek, J. Mater. Chem. A, 2018, 6, 18912–18919 RSC.
- K. Liang, J. J. Richardson, C. J. Doonan, X. Mulet, Y. Ju, J. Cui, F. Caruso and P. Falcaro, Angew. Chem., Int. Ed., 2017, 56, 8510–8515 CrossRef CAS PubMed.
- K. Liang, C. J. Coghlan, S. G. Bell, C. Doonan and P. Falcaro, Chem. Commun., 2016, 52, 473–476 RSC.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- S. K. Lim, C. Sandén, R. Selegård, B. Liedberg and D. Aili, Sci. Rep., 2016, 6, 21123 CrossRef CAS PubMed.
- M. P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4225–4229 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02021g |
| This journal is © The Royal Society of Chemistry 2019 |