
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring NAD(P)H by an ultrasensitive fluorescent probe to reveal reductive stress induced by natural antioxidants in HepG2 cells under hypoxia†
Xiaohong
Pan
 ab,
Yuehui
Zhao
a,
Tingting
Cheng
b,
Aishan
Zheng
a,
Anbin
Ge
b,
Lixin
Zang
a,
Kehua
Xu
*a and
Bo
Tang
*a
ab,
Yuehui
Zhao
a,
Tingting
Cheng
b,
Aishan
Zheng
a,
Anbin
Ge
b,
Lixin
Zang
a,
Kehua
Xu
*a and
Bo
Tang
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Shandong Normal University, Jinan 250014, P. R. China. E-mail: tangb@sdnu.edu.cn; xukehua@sdnu.edu.cn
bDepartment of Pharmaceutical Sciences, Binzhou Medical University, Yantai 264003, P. R. China
First published on 19th July 2019
Abstract
Reductive stress, the opposite of oxidative stress, represents a disorder in the redox balance state which is harmful to biological systems. For decades, the role of oxidative stress in tumor therapy has been the focus of attention, while the effects of reductive stress have been rarely studied. Here, we report the anti-cancer effects of reductive stress induced by three natural antioxidants (resveratrol, curcumin and celastrol). Considering the fact that the solid tumor microenvironment suffers from hypoxia, we performed cell experiments under hypoxic conditions. In order to observe the reductive stress, we first developed an ultrasensitive fluorescent probe (TCF-MQ) for specifically imaging NAD(P)H which is a marker of reductive stress. TCF-MQ responded to NAD(P)H rapidly and exhibited high sensitivity with a detection limit of 6 nM. With the help of TCF-MQ, we found that upon the treatment of HepG2 cells with pharmacological doses of three natural antioxidants under hypoxic conditions, high levels of NAD(P)H were produced before cell death. The excess NAD(P)H resulted in reductive stress instead of oxidative stress. In contrast, under normoxic conditions, there was no reductive stress involved in the process of cell death induced by three natural antioxidants. Therefore, we hypothesize that the mechanism of cancer cell death induced by natural antioxidants under hypoxia should be attributed to the reductive stress.
Introduction
A natural antioxidant, such as resveratrol, curcumin or celastrol, is a self-protective substance produced by organisms to resist poor environments and reactive oxygen species (ROS). They mainly exist in fruits, vegetables and medicinal plants. Due to the antioxidant and free radical scavenging effects, natural antioxidants are traditionally used for food protection and disease prevention. Recently, several natural antioxidants have been reported to possess anti-tumor effects,1–3 but the mechanisms underlying their anticancer functions are debated. Most studies suggest that some natural antioxidants can stimulate the production of ROS to cause oxidative stress, which leads to cell apoptosis.4–6 However, these results are contradictory to their antioxidant effects. We found that there is a common feature in the above studies, that is, the tumor cells used in the experiments are all cultured under normoxic conditions (20% O2), there is abundant oxygen around the cells, which provides conditions for the production of reactive oxygen species.7 But this condition is obviously contrary to the hypoxic microenvironment of solid tumors.8,9 Hypoxia is a common feature in solid tumors. In hepatocellular carcinoma (HCC), the O2 values in most areas is in the range of 0–10 mm Hg (0–1.32%).10 Therefore, in this study, we revealed a new anti-cancer mechanism of three natural antioxidants (resveratrol, curcumin and celastrol) under simulated tumor hypoxic conditions.The redox balance in the body is fundamental to sustain life. Once redox imbalanced, cell damage and apoptosis are inevitable.11–13 For decades, studies of the anti-tumor effects of natural antioxidants have been focused on oxidative stress. However, reductive stress, another way of redox imbalance, is rarely noticed. Similar to oxidative stress, reductive stress also represents a disturbance in the redox state which can cause body damage and lead to cell death.14–16 Recently, reductive stress was defined as an excessive amount of reducing equivalents, and excess NAD(P)H and/or GSH is considered a marker of reductive stress.14,17,18
NAD(P)H, the general term of the nicotinamide adenine dinucleotide (NAD) derivatives NADH and NADPH,19 is a universal electron carrier in cellular processes. NAD(P)H and NAD(P)+ undergo reversible oxidation–reduction in many biological reactions,20 and the level of NAD(P)H is tightly linked to the redox environment in cells. Insufficient NAD(P)H induces oxidative stress due to increased ROS production, while excessive NAD(P)H leads to reductive stress because it cannot be properly oxidized.20 Besides, NAD(P)H is a highly reducible dynamic molecule with very high reactivity and instability which cannot be easily detected in cell and animal models. Therefore, an ultrasensitive and rapid method to selectively determine NAD(P)H in vivo must be developed to investigate the reductive stress.
Fluorescent probes are a powerful tool for direct visualization of biomolecules in vivo with high sensitivity and short response times.21,22 To date, several detection methods for reductive stress have been reported,23–25 and in particular, several small-molecule fluorescent probes for detecting NAD(P)H have been developed.26–31 Louie et al. synthesized a NADH-sensitive multimodal magnetic-resonance/optical-imaging contrast agent and successfully applied it in cells.27 Komatsu reported a ubiquinone-rhodol-derived fluorescent probe for imaging intracellular NAD(P)H.29 Recently, Chang et al. developed a boronic acid-containing fluorescent probe for NADH based on resazurin, which shows dramatically improved sensitivity (detection limit: 0.084 μM) by introducing boronic acid to bind with NADH.31 In addition, our group previously developed a near-infrared small-molecule fluorescent probe (DCI-MQ) based on dicyanoisophorone for monitoring of NAD(P)H in cells.32 This probe was successfully used to selectively detect endogenous NAD(P)H in cells with a detection limit of 12 nM. Although the above methods for detecting NAD(P)H are suitable for cell use, these methods are not suitable for monitoring the reductive stress due to their lower sensitivity.
Here, we designed and synthesized an ultrasensitive small-molecule fluorescent probe (TCF-MQ) for better detecting NAD(P)H. TCF-MQ uses a more electron-absorbing tricyanofuran as the chromophore which endows the probe with good sensitivity for NAD(P)H with a limit of detection (LOD) of 6 nM. This probe was successfully used to distinguish different levels of NAD(P)H in HepG2 cells under normoxic and hypoxic conditions. Furthermore, with the help of TCF-MQ, we found that high levels of NAD(P)H were produced before cell death after the treatment of HepG2 cells with pharmacological doses of natural antioxidants (resveratrol, curcumin and celastrol) under hypoxic conditions. The excess NAD(P)H resulted in reductive stress which might induce cell death.
Results and discussion
Design and synthesis of TCF-MQ
2-Dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF) as a strong electron-withdrawing group has been widely used in non-linear optical materials.33,34 TCF-based fluorophores typically have a donor–π–acceptor (D–π–A) structure with long emission wavelengths, and many fluorescent probes have been developed.35 Here, we designed and synthesized a TCF-based fluorescent probe (TCF-MQ) for the detection of NAD(P)H (Scheme 1). TCF-MQ and all intermediate structures were fully characterized by 1H NMR, 13C NMR, and HRMS (see the Experimental section and the ESI†).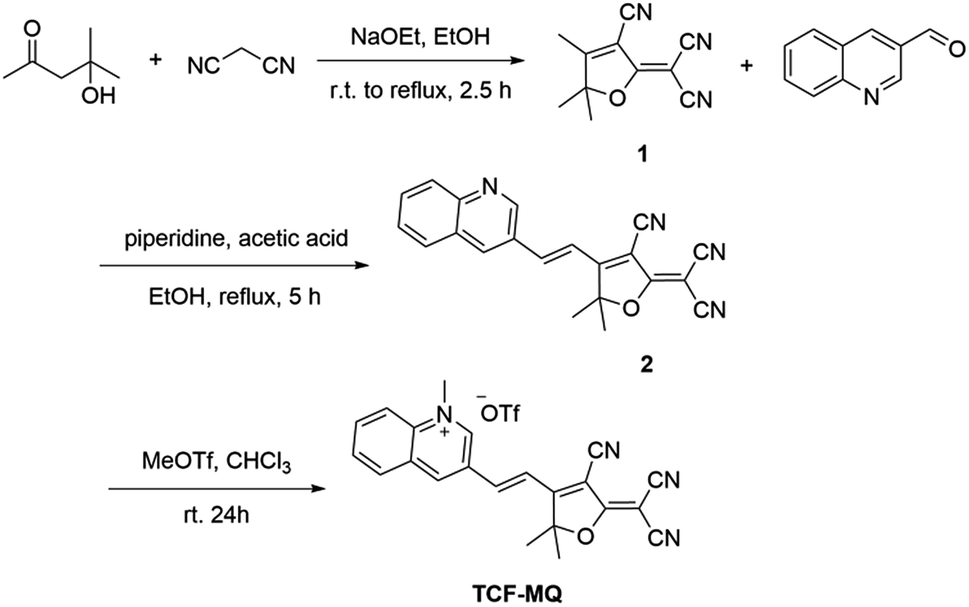 | ||
| Scheme 1 Synthesis of TCF-MQ. | ||
The proposed recognition mechanism of TCF-MQ toward NAD(P)H is shown in Scheme 2. The active hydrogen atom of NAD(P)H was added to the quinolinium moiety of the probe to form an adduct. To verify the sensing mechanism of TCF-MQ for NADH, the probe TCF-MQ and the product of TCF-MQ with NADH were analyzed by ESI-HRMS and 1H NMR spectroscopy. Once the reaction of TCF-MQ with NADH was completed, a mass peak at 353.1477 (m/z calcd = 353.1396) belonging to the reaction product was observed (Scheme 2 and Fig. S1†). From the 1H NMR analysis, after the reaction of TCF-MQ with NADH, the N–CH3 protons of TCF-MQ shifted upfield from 4.68 to 3.54 ppm, and a new singlet at 3.87 ppm emerged (Fig. S2†). These data confirmed our proposed recognition mechanism.
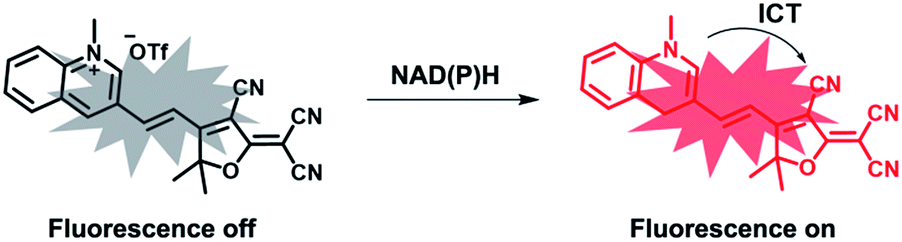 | ||
| Scheme 2 TCF-MQ for fluorescence detection of NAD(P)H. | ||
Fluorescence sensing performance analysis of TCF-MQ
The spectral properties of the probe with or without NADH were studied in PBS buffer solution (0.5% DMSO, 10 mM PBS, pH = 7.4). As shown in Fig. 1a, the probe displayed a weaker fluorescence peak at 604 nm (λex = 576 nm). As expected, after the reaction of the probe with NADH, a significant fluorescence enhancement at 610 nm (λex = 582 nm) was observed (quantum yield = 0.064). Meanwhile, the fluorescence intensity at 610 nm was enhanced gradually and reached a plateau within 25 min (Fig. 1b). The effect of pH on the fluorescence detection of NADH with TCF-MQ was also investigated, which indicated that TCF-MQ responded to NADH well under physiological conditions (at 37 °C and pH = 7.4) (Fig. S3†).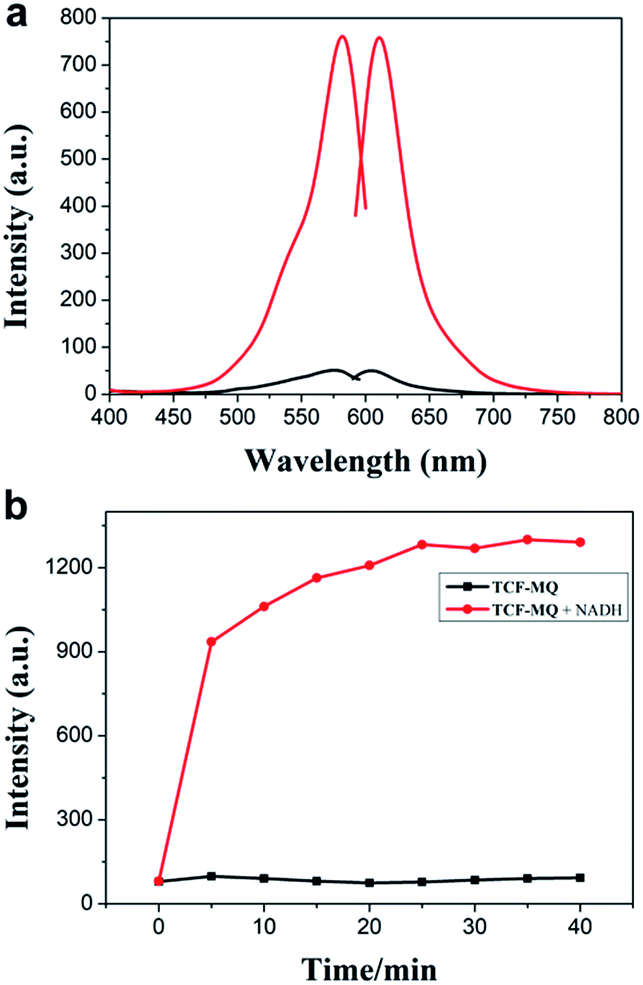 | ||
| Fig. 1 (a) Excitation and emission spectra of TCF-MQ (black) and the product of TCF-MQ with NADH (red). (b) Time-dependent fluorescence changes of TCF-MQ (10 μM) upon the addition of NADH (50 μM). | ||
Next, we investigated the sensitivity of the probe to NADH. The spectra of the solution of TCF-MQ treated with different concentrations of NADH (0 to 50 μM) were recorded. Upon treatment with NADH, the fluorescence intensities at 610 nm gradually increased with increasing concentrations of NADH (Fig. S4†), and there was a good linearity between the fluorescence intensities and NADH concentrations in the range of 0–3 μM (Fig. 2a). The regression equation is F = 83.22 + 238.42 [NADH], with a linear coefficient of 0.9819. The limit of detection (LOD) was calculated to be as low as 6 nM on the basis of S/N = 3, which indicates that TCF-MQ has excellent sensitivity to NADH and is more suitable for detecting NADH in living systems than DCI-MQ (LOD = 12 nM).26
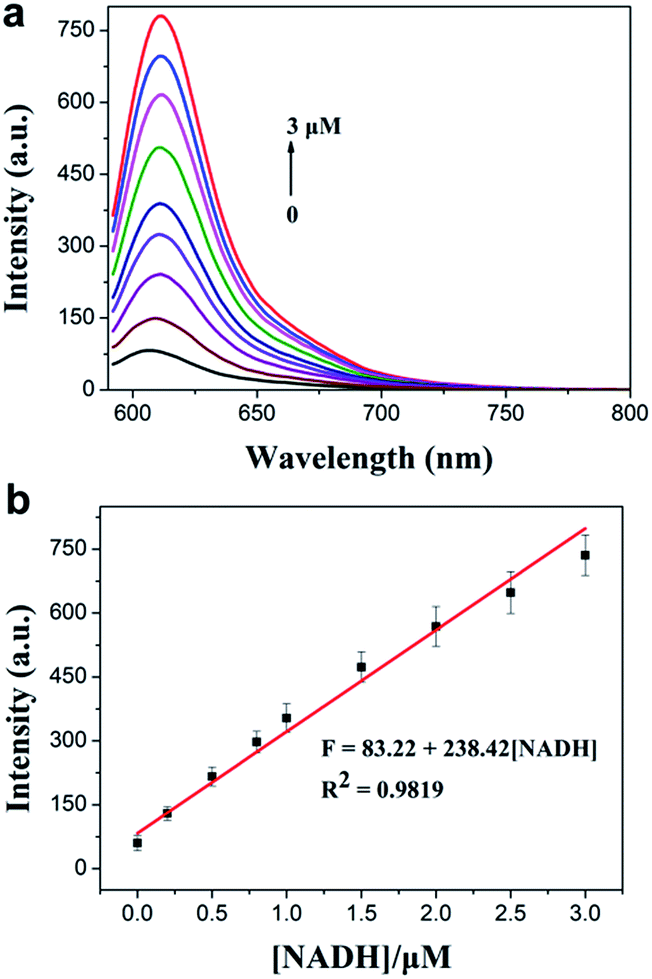 | ||
| Fig. 2 (a) Fluorescence intensity changes of TCF-MQ (10 μM) upon the addition of different concentrations of NADH (0–3 μM) in PBS (10 mM, pH 7.4, 0.5% DMSO as a cosolvent) at 37 °C for 30 min. (b) The linear correlation between emission intensities and concentrations of NADH. λex = 582 nm, λem = 610 nm, slit width: 5 nm/5 nm. | ||
Selective recognition of NAD(P)H by TCF-MQ
To illustrate the good selectivity of TCF-MQ toward NAD(P)H, a series of biologically relevant species including inorganic salts, amino acids and reducing agents were evaluated. As shown in Fig. 3, compared with NADPH and NADH, inorganic salts (K+, Na+, Ca2+, Mg2+, Zn2+, Fe3+, and Fe2+), amino acids (glycine, lysine, methionine, serine, cysteine, and homocysteine), reducing agents (glutathione, vitamin C, dithiothreitol, H2S2, and SO32−), NAD+, NADP+ and FADH2 did not cause obvious changes of the fluorescence intensity indicating that these do not interfere with the detection of NAD(P)H. In addition, the reactivity of TCF-MQ toward ROS was also tested. The results showed that biologically relevant ROS, including O2˙−, H2O2, NaClO, t-BuOOH and NO, did not trigger any fluorescence changes in the probe solution (Fig. S5†). These results demonstrate that TCF-MQ can be employed for the specific recognition of NAD(P)H with high selectivity.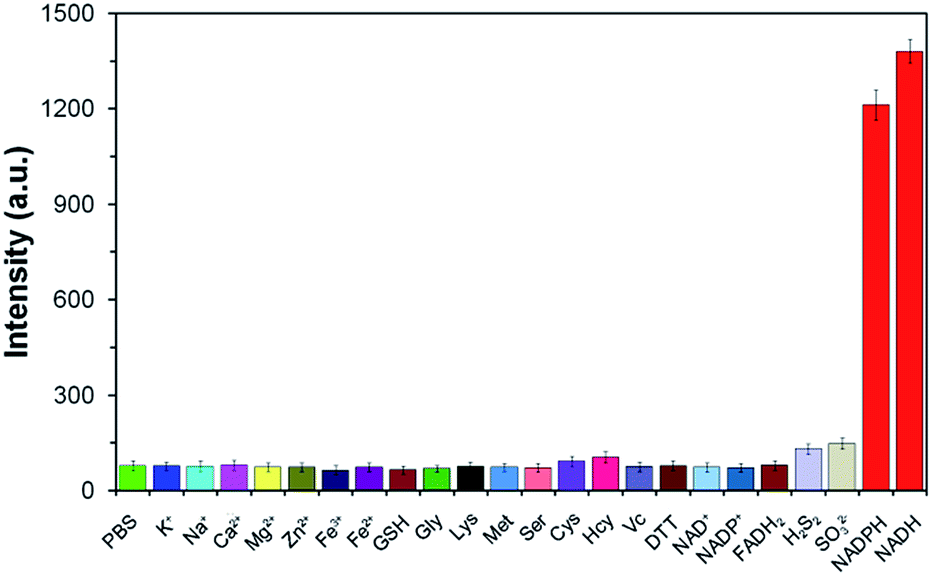 | ||
| Fig. 3 Fluorescence responses of the probe TCF-MQ (10 μM) for various analytes: 1 mM K+, Na+, Ca2+, Mg2+, Zn2+, Fe3+, Fe2+, GSH, Gly, Lys, Met, or Ser; 100 μM Cys, Hcy, VC, or DTT; 5 μM H2S2 or SO32−; and 50 μM NAD+, NADP+, FADH2, NADPH, or NADH. | ||
Bioimaging of NAD(P)H in living cells
After confirming the high sensitivity and selectivity of TCF-MQ for NAD(P)H, we explored its application to imaging endogenous NAD(P)H in living cells. Before being applied in cell imaging, we first tested the cytotoxicity of this probe by an MTT assay. The result showed that the TCF-MQ probe exhibited negligible cytotoxicity when HepG2 cells were treated with 0–50 μM probe for 18 h (Fig. S6†). The resistance to photobleaching experiments showed that no obvious fluorescence decrease was observed when cells were exposed to laser radiation for 600 s (Fig. S7†), suggesting that the probe is stable and can be used as a viable probe in living cells.Previous studies have shown that hypoxia can enhance NAD(P)H levels.36,37 Here, we investigated the imaging ability of TCF-MQ for NAD(P)H in HepG2 cells under hypoxia (1% O2) and normoxia (20% O2), respectively. HepG2 cells were cultured under hypoxic or normoxic conditions for 12 h, and then incubated with 10 μM TCF-MQ for 5–25 min. As shown in Fig. 4, the fluorescence of TCF-MQ became the brightest within 15 min, and the fluorescence intensity under hypoxia was higher than that under normoxia, indicating that more NAD(P)H was generated in cells under hypoxia. This result indicates that the probe can be used to monitor the levels of NAD(P)H in living cells.
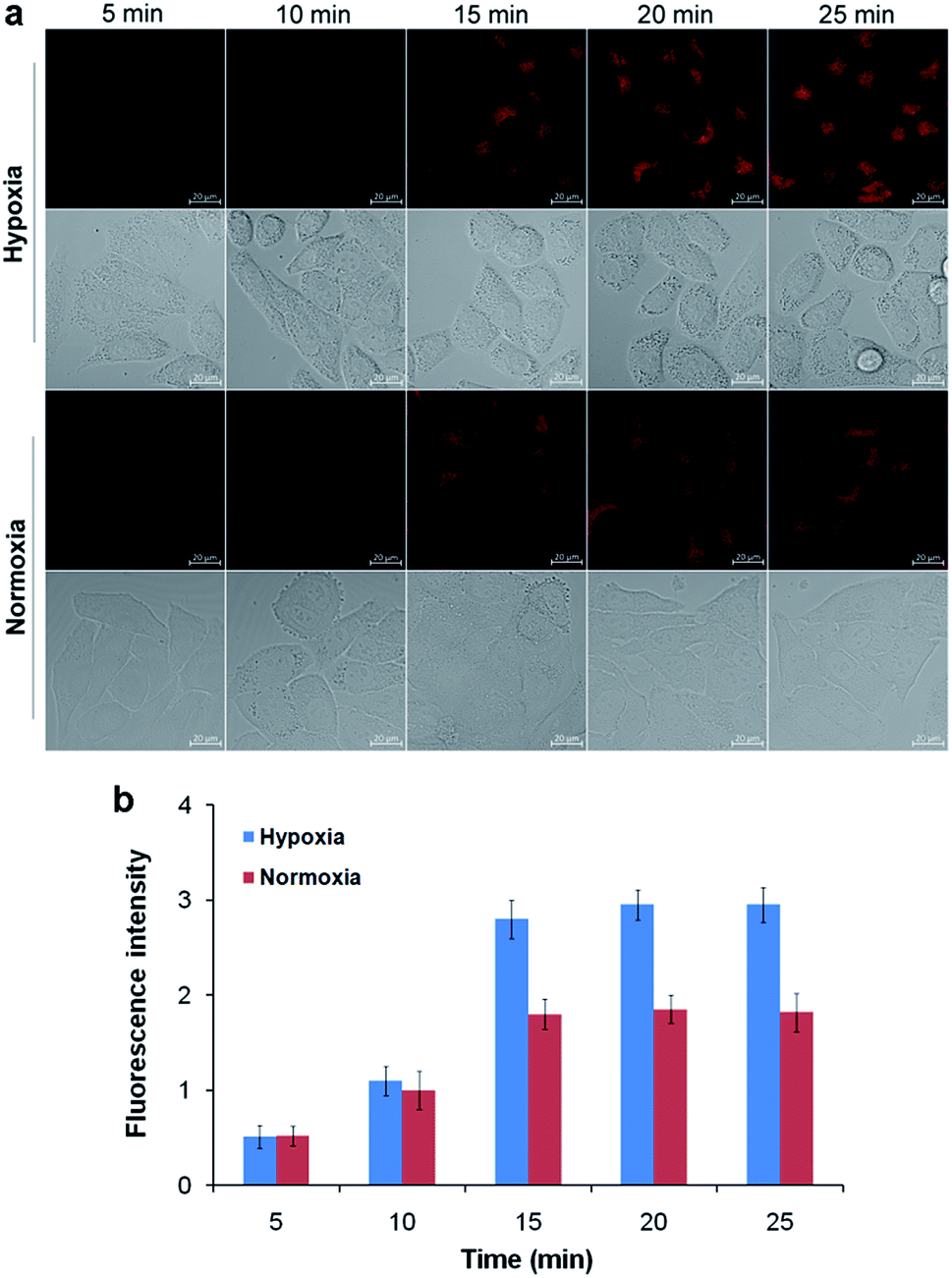 | ||
| Fig. 4 Fluorescence imaging of NAD(P)H in living cells. HepG2 cells were cultured under hypoxic (1% O2) or normoxic (20% O2) conditions for 12 h, and then incubated with 10 μM TCF-MQ probe for 5–25 min. (a) The fluorescence images were obtained using confocal microscopy. (b) The fluorescence intensity was quantified based on the results of the relative fluorescence intensity per cell in the scanned area. The scale bar in all fluorescence images of cells is 20 μm. | ||
Revealing reductive stress induced by natural antioxidants
Next, we used the TCF-MQ probe to observe the NAD(P)H levels in the process of liver cancer cell death induced by natural antioxidants (resveratrol, curcumin and celastrol). HepG2 cells were exposed to different concentrations of resveratrol, curcumin and celastrol for different periods of time under hypoxic and normoxic conditions, respectively. After treatment, the cells were loaded with 10 μM TCF-MQ for 20 min, and then, the fluorescence was detected by confocal microscopy. Under hypoxic conditions, the HepG2 cells treated with 0–10 μg mL−1 resveratrol for 6 h had a concentration-dependent increase in fluorescence. However, when the resveratrol concentration reached 20 μg mL−1, the fluorescence intensity decreased due to cell death (Fig. 5a, d and g). Additionally, after the HepG2 cells were treated with 10 μg mL−1 resveratrol for 0–9 h, the fluorescence intensity was also enhanced in a time-dependent manner (Fig. S8†). In contrast, under normoxic conditions, after the HepG2 cells were exposed to 0–20 μg mL−1 resveratrol for 6 h or 10 μg mL−1 resveratrol for 0–9 h, the cells exhibited much lower fluorescence compared with that under hypoxic conditions, and there was no enhancement of fluorescence in the process of cell death (Fig. 5a, d, g and S8†). Curcumin and celastrol showed similar results to resveratrol. When HepG2 cells were treated with curcumin or celastrol under hypoxic conditions, the fluorescence intensity of NAD(P)H was increased in a concentration- and time-dependent manner, but decreased with cell death (Fig. 5b–i). However, under normoxic conditions, only intracellular background fluorescence of TCF-MQ was observed during cell death induced by curcumin and celastrol (Fig. S9 and S10†). These results indicate that reductive stress is involved in the process of HepG2 cell death induced by resveratrol, curcumin and celastrol, and cell death may be attributed to the reductive stress.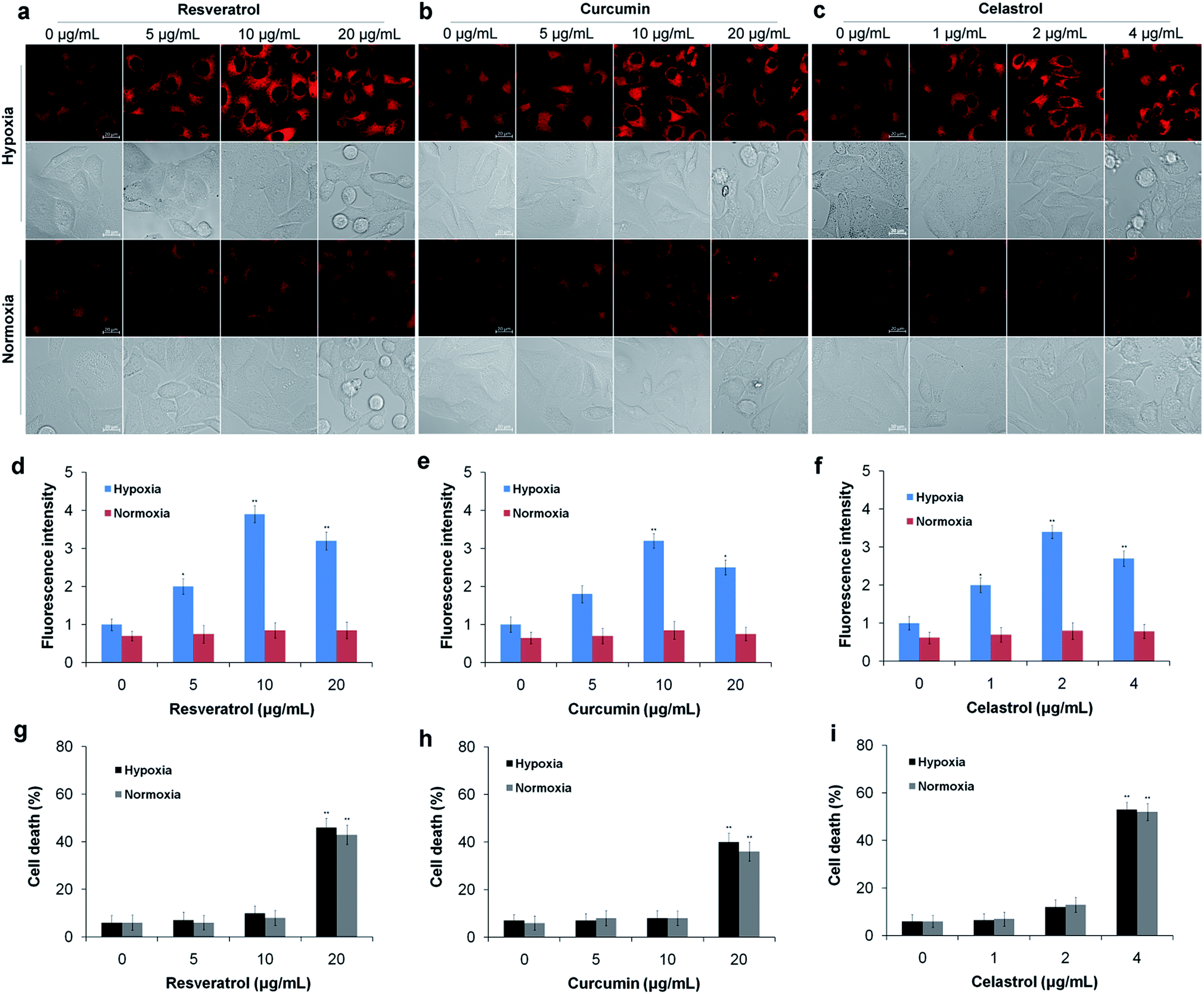 | ||
| Fig. 5 Detection of NAD(P)H levels induced by natural antioxidants in living cells. (a) HepG2 cells were exposed to 0–20 μg mL−1 resveratrol for 6 h under hypoxic conditions (1% O2) and normoxic conditions (20% O2), and then the cells were incubated with 10 μM TCF-MQ probe for 20 min before the fluorescence images were obtained using confocal microscopy. (b) HepG2 cells were exposed to 0–20 μg mL−1 curcumin for 6 h under hypoxic conditions (1% O2) and normoxic conditions (20% O2), and then the cells were treated using the method described in (a). (c) HepG2 cells were exposed to 0–4 μg mL−1 celastrol for 6 h under hypoxic conditions (1% O2) and normoxic conditions (20% O2), and then the cells were treated using the method described in (a). (d–f) The fluorescence intensity in the experiments in (a–c) was quantified based on the results of the relative fluorescence intensity per cell in the scanned area. (g) HepG2 cells were exposed to 0–20 μg mL−1 resveratrol for 6 h under hypoxic conditions (1% O2) and normoxic conditions (20% O2), and then the cells were collected and stained with trypan blue. (h) HepG2 cells were exposed to 0–20 μg mL−1 curcumin for 6 h under hypoxic conditions (1% O2) and normoxic conditions (20% O2), and then the cells were collected and stained with trypan blue. (i) HepG2 cells were exposed to 0–4 μg mL−1 celastrol for 6 h under hypoxic conditions (1% O2) and normoxic conditions (20% O2), and then the cells were collected and stained with trypan blue (*p < 0.05, **p < 0.01, t test). The scale bar in all fluorescence images of cells is 20 μm. | ||
Because previous studies have shown that natural antioxidants (resveratrol, curcumin and celastrol) can stimulate the production of ROS in tumor cells cultured in vitro,4–6 we assessed whether oxidative stress was induced as HepG2 cells were treated with the above three natural antioxidants under hypoxic conditions. In this experiment, a previously described H2O2 probe was employed.38 HepG2 cells were exposed to resveratrol (10 μg mL−1), curcumin (10 μg mL−1) and celastrol (2 μg mL−1) for 0–9 h under hypoxic conditions, respectively. Then, the cells were incubated with the H2O2 probe for 15 min. As a result, there was no obvious increase in H2O2 levels in the three antioxidant treatment groups compared with the control group (Fig. S11–S13†). These results indicate that the three antioxidants do not cause oxidative stress under hypoxic conditions.
To further confirm the reductive stress induced by natural antioxidants in HepG2 cells under hypoxic conditions, we conducted a similar experiment in vivo. Tumor bearing mice were treated with different concentrations of resveratrol, curcumin and celastrol via subcutaneous injection at the tumor site for 10 days. After treatment, the mice received a subcutaneous administration of the TCF-MQ probe (10 μM) into the tumor and were incubated for 30 min. Then, fluorescence images were obtained using an in vivo imaging system. The fluorescence signal of the probe obviously increased in a dose-dependent manner in the resveratrol treatment groups (Fig. 6a and d). Similar to the results for resveratrol, the fluorescence intensity of the probe was also enhanced in the curcumin and celastrol treatment groups (Fig. 6b, c, e and f), indicating that NAD(P)H levels were elevated during the treatment of liver cancer cells in mice with three natural antioxidants. These findings support the in vitro results obtained using the HepG2 cells under hypoxia showing that reductive stress was induced by treatment with natural antioxidants. The tumor volume measurement experiments showed that resveratrol could significantly inhibit tumor growth in a dose- and time-dependent manner (Fig. 6g). Curcumin and celastrol could also suppress the tumor growth in mice (Fig. 6h and i), indicating the reductive stress induced by three natural antioxidants can inhibit the growth of a liver cancer tumor.
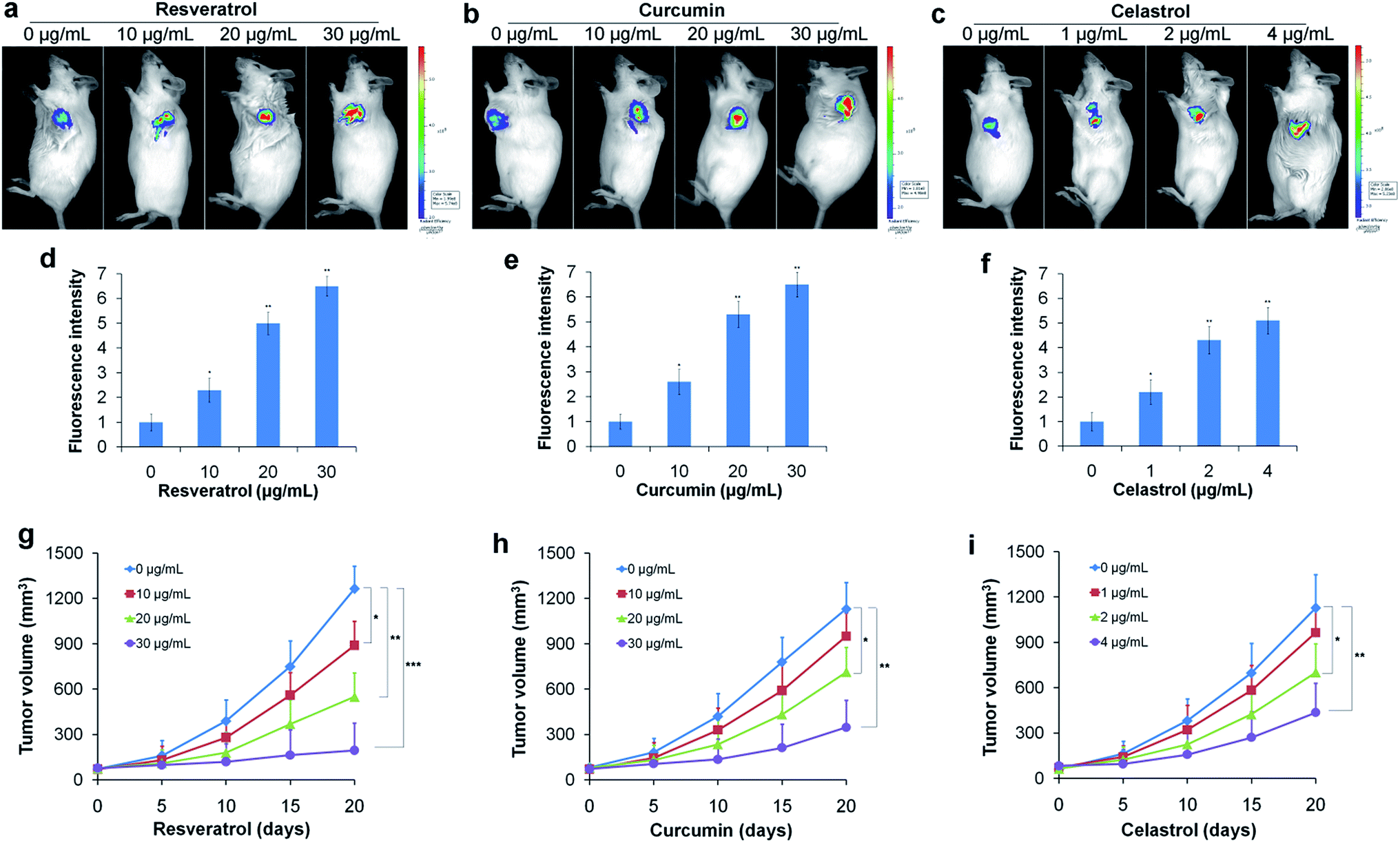 | ||
| Fig. 6 Detection of NAD(P)H levels induced by natural antioxidants in vivo. (a) Tumor-bearing mice were treated with 0–30 μg mL−1 resveratrol for 10 days, and then, 10 μM TCF-MQ probe was subcutaneously injected into the tumor for 30 min before the fluorescence was analyzed using an in vivo imaging system. (b) Tumor-bearing mice were treated with 0–30 μg mL−1 curcumin for 10 days, then the TCF-MQ probe was added using the method described in (a). (c) Tumor-bearing mice were treated with 0–4 μg mL−1 celastrol for 10 days, and then the TCF-MQ probe was added using the method described in (a). (d) The quantitative fluorescence intensity for (a). (e) The quantitative fluorescence intensity for (b). (f) The quantitative fluorescence intensity for (c). (g) Tumor-bearing mice were treated with 0–30 μg mL−1 resveratrol for different number of days, and the tumor volumes were measured using digital calipers. (h) Tumor-bearing mice were treated with 0–30 μg mL−1 curcumin for different numbers of days, and the tumor volumes were measured using digital calipers. (i) Tumor-bearing mice were treated with 0–4 μg mL−1 celastrol for different numbers of days, and the tumor volumes were measured using digital calipers (*p < 0.05, **p < 0.01, ***p < 0.001, t test). | ||
Conclusions
In summary, we developed a novel small-molecule fluorescent probe, TCF-MQ, for monitoring NAD(P)H and revealing the reductive stress. TCF-MQ can rapidly respond to NAD(P)H with good selectivity and exhibited very high sensitivity to NAD(P)H with a low detection limit of 6 nM. The probe was successfully used to image endogenous NAD(P)H in living cells and to distinguish the different levels of NAD(P)H in HepG2 cells under normoxic conditions and hypoxic conditions. Moreover, with the help of TCF-MQ, we found that upon the treatment of HepG2 cells with pharmacological doses of three natural antioxidants (resveratrol, curcumin and celastrol) under hypoxic conditions, high levels of NAD(P)H were produced before cell death. The excess NAD(P)H resulted in reductive stress instead of oxidative stress. In contrast, under normoxic conditions, there was no obvious increase in NAD(P)H content in the process of cell death induced by resveratrol, curcumin and celastrol. Thus, we speculate that natural antioxidants might induce tumor cell death via reductive stress under the tumor hypoxic microenvironment, whereas under a normoxic environment, natural antioxidants induced cell death through oxidative stress based on previous reports.4–6 The mechanism of reductive stress inducing tumor cell death under hypoxic conditions is undergoing further research. Our findings reveal a new anti-cancer mechanism of resveratrol, curcumin and celastrol and provide a new research area for natural antioxidants.Experimental
Materials and methods
3-Hydroxy-3-methyl-2-butanone, malononitrile, methyl trifluoromethanesulfonate and 3-quinolinecarboxaldehyde were purchased from Sun Chemical Technology (Shanghai, China). β-NADPH and β-NADH were obtained from Aladdin (Shanghai, China). Piperidine, sodium ethoxide, anhydrous ethanol, chloroform and acetonitrile were from Sinopharm Chemical Reagent (Shanghai, China). Resveratrol, curcumin and celastrol were from Dalian Meilun Biotechnology (Dalian, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphnyl-2H-tetrazolium bromide (MTT) was from Sigma-Aldrich (St. Louis, MO, USA). 1H NMR and 13C NMR spectra were recorded on a nuclear magnetic resonance spectrometer (Advance 400 MHz, Bruker, Switzerland). High-resolution mass spectroscopy (HRMS) was performed on a high resolution quadrupole-time of flight mass spectrometer (Bruker, Germany). Fluorescence spectra were recorded on a fluorescence spectrometer (Edinburgh Instruments, U.K.). The fluorescence images were obtained with a confocal microscope (LSM 880, Zeiss, Germany).Synthesis of the probe TCF-MQ
In vitro detection of NAD(P)H
The stock solution of TCF-MQ (2 × 10−3 M) was prepared in DMSO. Stock solutions of NAD(P)H and various interfering reagents (Na+, K+, Ca2+, Mg2+, Zn2+, Fe2+, Fe3+, glycine, lysine, serine, methionine, cysteine, homocysteine, glutathione, dithiothreitol, vitamin C, H2S2, SO32−, NAD+, NADP+, FADH2, H2O2, NaClO, t-BuOOH, and NO) were prepared in double-distilled water, but superoxide anion free radicals (O2−˙) were prepared from a solution of KO2 in DMSO. For spectral measurements, the stock solution of the TCF-MQ probe was diluted with PBS buffer to give a final concentration of 10 μM. NAD(P)H and various analytes were added to the solution of TCF-MQ (10 μM) in PBS buffer (10 mM, pH = 7.4, containing 0.5% DMSO). The fluorescence emission spectra were recorded in a range from 592–800 nm (λex = 582 nm, λem = 610 nm, slit width: 5 nm/5 nm).Cell culture
The cell culture was performed according to our previous paper.39 Briefly, HepG2 cells were cultured in DMEM (high glucose) supplemented with 10% FBS and 1% antibiotics (100 U mL−1 penicillin and 100 mg mL−1 streptomycin) at 37 °C in a humidified atmosphere of 95% air (20% O2) and 5% CO2. For simulating the tumor hypoxic microenvironment, HepG2 cells were cultured in a mixture containing 1% O2, 94% N2 and 5% CO2 using a low oxygen incubator (Bugbox M, Ruskinn, England).Fluorescence imaging in living cells
The fluorescence of NAD(P)H and H2O2 was detected by confocal microscopy. Briefly, HepG2 cells (5 × 104) were seeded on glass-bottom culture dishes (15 mm) for 24 h. Then, the cells were exposed to different concentrations of three natural antioxidants for different periods of time under hypoxic and normoxic conditions, respectively. After treatment, the cells cultured under hypoxia and normoxia were simultaneously incubated with 10 μM (1 mL) NAD(P)H probe for 20 min or 10 μM (1 mL) H2O2 probe for 15 min in FBS-free DMEM at 37 °C, and then, the cells were washed three times with PBS buffer and imaged immediately using a confocal microscope. The TCF-MQ probe was excited at 582 nm and the fluorescence emission range was set from 600–650 nm. The H2O2 probe was excited at 532 nm and the fluorescence emission range was set from 600–700 nm.Tumor model preparation and fluorescence imaging
All animal experiments were performed according to the Principles of Laboratory Animal Care (People's Republic of China) and the Guidelines of the Animal Investigation Committee, and approved by the local Animal Care and Use Committee. Kunming mice were purchased from Qingdao Daren Fucheng Animal Co., Ltd. Hepatocellular carcinoma H22 cells (1 × 107) derived from mice were injected into the abdominal cavity of mice, and ascites was formed after 5–7 days. After three passages, ascites was used. We selected 8 week-old mice and injected H22 cells subcutaneously into the axillary lateral of the left forelimb at a density of 1 × 107 cells per mL with 100 μL per mice. After 7 days, these mice were given different concentrations of natural antioxidants through subcutaneous injection at the tumor site for different periods of time. After treatment, the mice were subcutaneously injected with the TCF-MQ probe and incubated for 30 min. Then, fluorescence images were obtained using an in vivo imaging system (IVIS) with 582 nm excitation and 600–650 nm to detect NAD(P)H. In addition, the tumor volume was measured using digital calipers and calculated as volume = 0.5 × (length × width2).40Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21535004, 21575081, 21775091 and 91753111), the Key Research and Development Program of Shandong Province (2018YFJH0502), the Shandong Provincial Natural Science Foundation of China (ZR2014JL055) and the Key Research and Development Plan of Yantai City (2019XDHZ098).Notes and references
- D. Sinha, N. Sarkar, J. Biswas and A. Bishayee, Semin. Cancer Biol., 2016, 40–41, 209–232 CrossRef CAS PubMed.
- J. Y. Wang, X. Wang, X. J. Wang, B. Z. Zheng, Y. Wang, X. Wang and B. Liang, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 7492–7499 Search PubMed.
- S. Shrivastava, M. K. Jeengar, V. S. Reddy, G. B. Reddy and V. G. Naidu, Exp. Mol. Pathol., 2015, 98, 313–327 CrossRef CAS PubMed.
- D. J. Colin, E. Limagne, K. Ragot, G. Lizard, F. Ghiringhelli, É. Solary, B. Chauffert, N. Latruffe and D. Delmas, Cell Death Dis., 2014, 5, e1533 CrossRef CAS PubMed.
- F. Song, L. Zhang, H. X. Yu, R. R. Lu, J. D. Bao, C. Tan and Z. Sun, Food Chem., 2012, 132, 43–50 CrossRef CAS PubMed.
- J. H. Kim, J. O. Lee, S. K. Lee, N. Kim, G. Y. You, J. W. Moon, J. Sha, S. J. Kim, S. H. Park and H. S. Kim, Cell. Signalling, 2013, 25, 805–813 CrossRef CAS PubMed.
- L. J. Yan, J. Diabetes Res., 2014, 2014, 137919 Search PubMed.
- E. B. Rankin and A. J. Giaccia, Science, 2016, 352, 175–180 CrossRef CAS PubMed.
- N. C. Denko, Nat. Rev. Cancer, 2008, 8, 705–713 CrossRef CAS PubMed.
- X. X. Xiong, X. Y. Qiu, D. X. Hu and X. Q. Chen, Mol. Pharmacol., 2017, 92, 246–255 CrossRef CAS PubMed.
- X. X. Fan, H. D. Pan, Y. Li, R. J. Guo, E. L. Leung and L. Liu, Pharmacol. Ther., 2018, 191, 148–161 CrossRef CAS PubMed.
- M. Inoue, R. Nakashima, M. Enomoto, Y. Koike, X. Zhao, K. Yip, S. H. Huang, J. N. Waldron, M. Ikura, F. F. Liu and S. V. Bratman, Nat. Commun., 2018, 9, 5116 CrossRef PubMed.
- B. Petrova, K. Liu, C. Tian, M. Kitaoka, E. Freinkman, J. Yang and T. L. Orr-Weaver, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7978–E7986 CrossRef CAS PubMed.
- N. S. Rajasekaran, P. Connell, E. S. Christians, L. J. Yan, R. P. Taylor, A. Orosz, X. Q. Zhang, T. J. Stevenson, R. M. Peshock, J. A. Leopold, W. H. Barry, J. Loscalzo, S. J. Odelberg and I. J. Benjamin, Cell, 2007, 130, 427–439 CrossRef CAS PubMed.
- H. Zhang, P. Limphong, J. Pieper, Q. Liu, C. K. Rodesch, E. Christians and I. J. Benjamin, FASEB J., 2012, 26, 1442–1451 CrossRef CAS PubMed.
- D. E. Handy and J. Loscalzo, Free Radical Biol. Med., 2017, 109, 114–124 CrossRef CAS PubMed.
- S. Dimmeler and A. M. Zeiher, Cell, 2007, 130, 401–402 CrossRef CAS PubMed.
- A. C. Brewer, S. B. Mustafi, T. V. Murray, N. S. Rajasekaran and I. J. Benjamin, Antioxid. Redox Signaling, 2013, 18, 1114–1127 CrossRef CAS PubMed.
- H. Komatsu, Y. Shindo, K. Oka, J. P. Hill and K. Ariga, Angew. Chem., Int. Ed. Engl., 2014, 53, 3993–3995 CrossRef CAS PubMed.
- V. A. Aleshin, A. V. Artiukhov, H. Oppermann, A. V. Kazantsev, N. V. Lukashev and V. I. Bunik, Cells, 2015, 4, 427–451 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, K. B. Zheng, L. W. He and W. M. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- J. Yin, Y. Kwon, D. Kim, D. Lee, G. Kim, Y. Hu, J. H. Ryu and J. J. Yoon, J. Am. Chem. Soc., 2014, 136, 5351–5358 CrossRef CAS PubMed.
- X. Ao, S. A. Bright, N. C. Taylor and R. B. P Elmes, Org. Biomol. Chem., 2017, 15, 6104–6108 RSC.
- A. Tirla and P. Rivera-Fuentes, Angew. Chem., Int. Ed. Engl., 2016, 55, 14709–14712 CrossRef CAS PubMed.
- A. Tirla and P. Rivera-Fuentes, Chimia, 2018, 72, 241–244 CrossRef PubMed.
- C. A. Roeschlaub, N. L. Maidwell, M. R. Rezai and P. G. Sammes, Chem. Commun., 1999, 1637–1638 RSC.
- C. Tu, R. Nagao and A. Y. Louie, Angew. Chem., Int. Ed., 2009, 48, 6547–6551 CrossRef CAS PubMed.
- S. O. Jung, J. Y. Ahn and S. Kim, Tetrahedron Lett., 2010, 51, 3775–3778 CrossRef CAS.
- H. Komatsu, Y. Shindo, K. Oka, I. P. Hill and K. Ariga, Angew. Chem., Int. Ed., 2014, 53, 3993–3995 CrossRef CAS PubMed.
- M. A. Fomin, R. I. Dmitrev, J. Jenkins, D. B. Papkovsky, D. Heindl and B. Konig, ACS Sens., 2016, 1, 702–709 CrossRef CAS.
- L. Wang, J. Zhang, B. Kim, J. Peng, S. N. Berry, Y. Ni, D. Su, J. Lee, L. Yuan and Y. T. Chang, J. Am. Chem. Soc., 2016, 138, 10394–10397 CrossRef CAS PubMed.
- Y. Zhao, K. Wei, F. Kong, X. Gao, K. Xu and B. Tang, Anal. Chem., 2018, 91, 1368–1374 CrossRef PubMed.
- C. Zhang, L. R. Dalton, M.-C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043–3050 CrossRef CAS.
- W. Bentoumi, J. C. Mulatier, P. A. Bouit, O. Maury, A. Barsella, J. P. Vola, E. Chastaing, L. Divay, F. Soyer, P. Le Barny, Y. Bretonnière and C. Andraud, Chemistry, 2014, 20, 8909–8913 CAS.
- A. C. Sedgwick, H. H. Han, J. E. Gardiner, S. D. Bull, X. P. He and T. D. James, Chem. Commun., 2017, 53, 12822–12825 RSC.
- L. B. Jiang, L. Cao, Y. Q. Ma, Q. Chen, Y. Liang, F. L. Yuan, X. L. Li, J. Dong and N. Chen, Osteoarthr. Cartil., 2018, 26, 138–148 CrossRef PubMed.
- O. Polesskaya, A. Sun, G. Salahura, J. N. Silva, S. Dewhurst and K. Kasischke, J. Visualized Exp., 2012, 3466 Search PubMed.
- X. Liu, B. Hu, R. Cheng, F. Kong, X. Pan, K. Xu and B. Tang, Chem. Commun., 2016, 52, 6693–6696 RSC.
- X. H. Pan, X. X. Song, C. Wang, T. T. Cheng, D. R. Luan, K. H. Xu and B. Tang, Theranostics, 2019, 9, 1794–1808 CrossRef CAS.
- W. S. Henry, T. Laszewski, T. Tsang, F. Beca, A. H. Beck, S. S. McAllister and A. Toker, Cancer Res., 2017, 77, 790–801 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02020a |
| This journal is © The Royal Society of Chemistry 2019 |