
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence isomerism-dependent self-assembly of glycopeptide mimetics with switchable antibiofilm properties†
Limin
Chen
 ab,
Jie
Feng
b,
Dan
Yang
a,
Falin
Tian
b,
Xiaomin
Ye
b,
Qiuping
Qian
b,
Shuai
Wei
*c and
Yunlong
Zhou
*ab
ab,
Jie
Feng
b,
Dan
Yang
a,
Falin
Tian
b,
Xiaomin
Ye
b,
Qiuping
Qian
b,
Shuai
Wei
*c and
Yunlong
Zhou
*ab
aSchool of Ophthalmology and Optometry, Eye Hospital, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou 325000, P. R. China. E-mail: zhouyl@wibe.ac.cn
bEngineering Research Center of Clinical Functional Materials and Diagnosis & Treatment Devices of Zhejiang Province, Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou 325000, P. R. China
cDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: shuaiwei@umich.edu
First published on 11th July 2019
Abstract
In biological systems, diverse amino acid sequences and functional decorations endow proteins with specific functions. Functionally modified oligopeptides are attractive building blocks to assemble stimuli-responsive biomimetic superstructures for mimicking soft structures in nature and biomaterial applications. In this work, we selectively synthesized the structurally simplest isomeric tripeptides (i.e., Ala–Gly–Gly–OH, Gly–Ala–Gly–OH and Gly–Gly–Ala–OH) to demonstrate how the subtlest change in sequence isomerism influences the self-assembly of glycopeptides. To impart self-assembly capability and stimuli-responsiveness, the isomeric tripeptides were modified with a hydrophobic n-butylazobenzene tail at the N-terminal. We observed three different self-assembled 1-D morphologies (i.e., nanotwists, nanoribbons and nanofibers) from the azobenzene-glycopeptides (AGPs) under the same conditions when the position of the Ala residue was switched. Experimental methods including transmission electron microscopy (TEM), atomic force microscopy (AFM), X-ray diffraction (XRD), Fourier transform infrared (FT-IR) spectroscopy and circular dichroism (CD) spectroscopy were used to characterize the structural details of glycopeptide mimetic assemblies. Martini coarse-grained molecular dynamics (MD) simulations confirmed such structural observations and investigated the differences in assembly mechanisms. Furthermore, the glycopeptide mimetic assemblies showed a reversible disassembly–assembly process in response to temperature, light or host–guest chemistry, and can be used as switchable antibiofilm nanoagents.
Introduction
Oligopeptides' capability of assembling into stimuli-responsive biomimetic superstructures has attracted great attention due to their interesting applications as biomaterials.1–5 The self-assembled superstructures are able to reversibly respond to external stimuli or environmental changes,6–10 which can be used to mimic soft structures in nature such as protein assemblies.11 A classic molecular model has been developed for constructing oligopeptide superstructures.12–16 Typically, an oligopeptide is anchored with a hydrophobic group at its N-terminal.17–22 The hydrophobic tails could be aliphatic or aromatic groups that enhance the self-assembly propensity of oligopeptides. Recent research has revealed that the variation of amino acid types or orders in the oligopeptides can result in different self-assembled morphologies and functional bioactivities.23–29 In addition, coarse-grain and full-atomistic molecular dynamics (MD) have been used to simulate the self-assembly behavior of peptides.30–35 However, the generalized self-assembly process and specific functions of peptide assemblies need to be further explored using sequence designs, MD simulations and functional modifications.36Peptides and saccharides are important molecular partners in many biological systems and often covalently bind to each other forming glycopeptides.37–39 In biological systems, the switching of amino acid sequences could lead to biological malfunction of proteins, for example, the positions of a serine and phosphoserine residue dictate the promotion or prevention of lipid hydrolysis functions of hormone-sensitive lipase.40 To demonstrate how the subtlest change in sequence isomerism could influence the self-assembly of glycopeptide molecules into highly ordered nanostructures, the structurally simplest isomeric tripeptides (i.e., Ala–Gly–Gly–OH, Gly–Ala–Gly–OH and Gly–Gly–Ala–OH) were selected. The N-terminal and C-terminal of the tripeptides were modified with a hydrophobic n-butylazobenzene tail and a hydrophilic N-methyl-D-glucamine residue (amino sugar alcohol derived from glucose), respectively. The azobenzene motif was anticipated to endow the azobenzene-glycopeptide (AGP) (Fig. 1) assemblies with stimuli-responsiveness under photoirradiation (i.e., UV and visible light) and host–guest interactions (i.e., β-cyclodextrin and adamantane).41 Importantly, considering that Ala and Gly are only different in the side-chain of a methyl group, we studied the morphology control of glycopeptide mimetic assemblies by switching the order of one Ala and two Gly residues. Moreover, we also evaluated the reversible disassembly–assembly process and potential biofunctions of the glycopeptide mimetic assemblies.
 | ||
| Fig. 1 Molecular structures of glycopeptide mimetics denoted as AGP1, AGP2 and AGP3. | ||
Results and discussion
The AGP molecules were synthesized according to Scheme S1 (see the ESI† for details of synthetic procedures and compound characterization). AGP assemblies and hydrogels were prepared by fully dissolving the powders in water at the desired concentration at 70 °C before being cooled to room temperature for self-assembly. We performed rheological experiments on AGP hydrogels (30 mM). As shown in Fig. S1,† strain sweeps and frequency sweeps confirmed the formation of hydrogels for AGP molecules because G′ is greater than G′′. Furthermore, gel kinetics revealed a fast gelation process upon cooling from 70 °C to room temperature. Transmission electron microscopy (TEM) and atomic force microscopy (AFM) were used to characterize the morphology and dimensions of the self-assembled AGP nanostructures. The size of dried AGP nanostructures measured by TEM and AFM is potentially different relative to that of the hydrated forms.42 However, given the similarity across AGP1, AGP2 and AGP3, it is fair to assume that the same deviation between dried and hydrated nanostructures applies to all three, so it is reasonable to do a comparison of the dimensions of the dried samples. The AFM technique has a very good z resolution and a poor x–y resolution, while the TEM technique has a much higher x–y resolution. Therefore, we measured the width (i.e., x–y resolution) and height (i.e., z resolution) of AGP nanostructures using TEM and AFM imaging, respectively. Left-handed nanotwists with a helical pitch of 202 ± 12 nm were observed for AGP1 (Fig. 2A and B). The width of AGP1 nanotwists was measured to be 17.5 ± 2.9 nm according to TEM imaging (Fig. S2†), and the height was 7.3 nm ± 1.2 nm according to AFM imaging (Fig. S3†). In the case of either AGP2 or AGP3, untwisted nanofibers were observed (Fig. 2C–F). The width and height of AGP2 nanofibers were measured to be 18.5 ± 3.1 nm and 7.1 ± 0.8 nm according to TEM (Fig. S2†) and AFM (Fig. S3†) imaging respectively, indicating the formation of planar nanoribbons for AGP2. However, the width and height of AGP3 nanofibers were 6.9 ± 1.1 nm and 6.3 ± 1.5 according to TEM (Fig. S2†) and AFM imaging (Fig. S3†) respectively, indicating that AGP3 self-assembled into cylindrical nanofibers. Three different 1-D morphologies were observed when the position of the Ala residue was switched. We assume that peptide side chain interactions could be slightly varied when the position of the Ala residue was altered in the isomeric tripeptides of one Ala and two Gly residues. Slightly different peptide side chain interactions due to the sequence isomerism seem to have a non-negligible effect on the internal packing of AGP molecules within 1-D assemblies, and are ultimately reflected in the observed morphological transitions. Actually, the significance of peptide side chain interactions in determining the architectures of supramolecular assemblies was demonstrated by using isomeric tetrapeptide amphiphiles consisting of two glutamic acids and two valine residues.23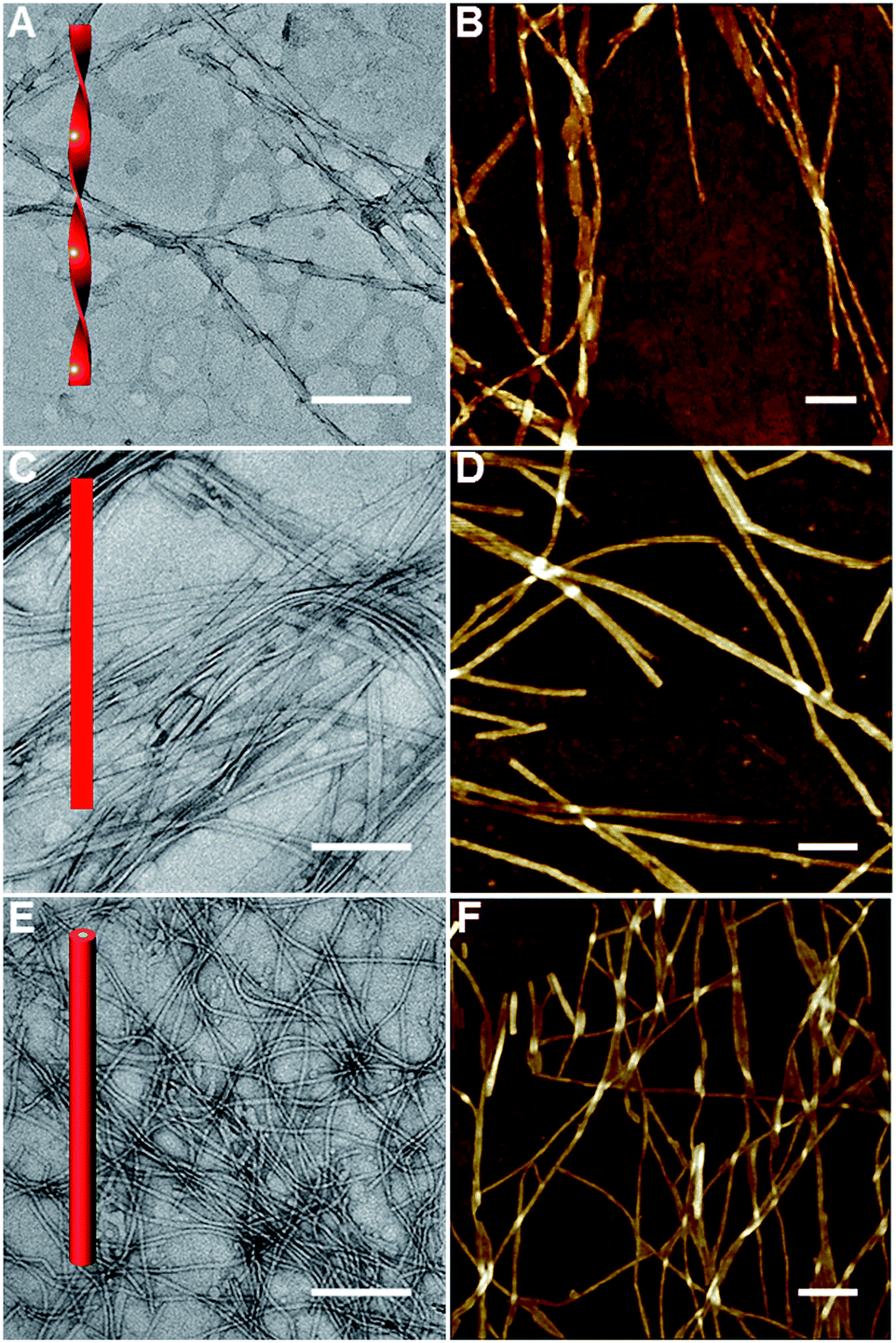 | ||
| Fig. 2 TEM and AFM imaging of AGP1 (A and B), AGP2 (C and D) and AGP3 (E and F) assemblies (0.6 mM) dried in air at room temperature. The scale bars represent 200 nm. | ||
To understand the difference of how AGP molecules are packed within the nanotwists, nanoribbons and nanofibers, we performed X-ray diffraction (XRD) experiments on their air-dried samples. According to the XRD patterns shown in Fig. 3A, AGP1 shows clear periodic diffraction peaks suggesting the formation of layered structures, which corresponds to layer spacings of 6.22 nm, 3.29 nm and 2.22 nm based on the Bragg equation (2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ = nλ, λ = 1.5418 Å). AGP2 also exhibits a lamellar structure (i.e., 6.18 nm, 3.12 nm, 2.06 nm), and shows a long layer spacing of 6.18 nm. However, AGP3 does not exhibit obvious periodic diffraction peaks, and shows a d-spacing of 5.56 nm. The d-spacing is larger than the extended molecular length (∼3.9 nm estimated by the CPK molecular model shown in Fig. S4†) but smaller than twice the length, suggesting the formation of interdigitated bilayer structures regardless of whether nanotwists, nanoribbons or nanofibers were formed.43 A slight increase of d-spacing for AGP1 relative to AGP2 can be associated with the twist of nanoribbons. The smallest d-spacing for AGP3 implied a much more closely packed bilayer structure relative to AGP1 and AGP2. The differences of internal packing of AGP molecules within 1-D assemblies according to the XRD patterns are responsible for the observed morphological transitions for AGP1, AGP2 and AGP3. To analyze the conformations of tripeptides within the nanotwists, nanoribbons and nanofibers, we performed Fourier transform infrared (FT-IR) spectroscopy experiments on AGP hydrogels in D2O. The signal in the amide I region (∼1600–1700 cm−1) (Fig. S5†) is broad, suggesting that multiple peaks could be hidden there, such as α-helices, β-sheets and random coils. Circular dichroism (CD) spectroscopy was used to investigate the transfer of chiral information from the chiral center (Ala) to chromophore (azobenzene) originating from the self-assembly of AGP molecules.44 As expected, fully dissolved non-assembling AGP solution at 70 °C did not give any CD signal of azobenzene due to the free state of azobenzene in solution (Fig. 3B and S7†). Upon the solution being cooled to 20 °C for self-assembly, a negative band at ∼331 nm and a positive band at ∼306 nm with a crossover at around 318 nm were observed for AGP1 (Fig. 3C). The CD bands of opposite sign centered at 318 nm are related to the π–π* transition of azobenzene, indicating that the chirality of Ala was transferred to azobenzene upon self-assembly. In the case of AGP2, a positive band at ∼374 nm and a negative band at ∼336 nm with a crossover at around 353 nm were found (Fig. 3E). As for AGP3, a positive band at ∼352 nm and a negative band at ∼312 nm with a crossover at around 326 nm were observed (Fig. 3G). The CD spectra variation for AGP1, AGP2 and AGP3 can be directly associated with the variation of chiral centers at odd and even positions.45 Furthermore, the CD spectra variation was also strongly dependent on the intensity and types of intermolecular or intramolecular interactions.46 To compare the intensity of intermolecular interactions for AGP1, AGP2 and AGP3, the critical aggregation concentration (CAC) values were determined (Fig. S6†) (see the ESI† for the detailed characterization). Given the high structural similarity for AGP1, AGP2 and AGP3, the CAC values for AGP2 (0.010 mM) < AGP1 (0.062 mM) < AGP3 (0.215 mM) can be directly associated with the different intensities of intermolecular interactions. Stronger intermolecular interactions (e.g., H-bonding and π–π stacking) for AGP2 > AGP1 > AGP3 will be more favorable for the transfer of chirality from Ala to azobenzene, and ultimately reflected in the CD spectra intensity variation. In addition, the different packing intensities for AGP1, AGP2 and AGP3 can be related to the observed morphological transitions.
sinθ = nλ, λ = 1.5418 Å). AGP2 also exhibits a lamellar structure (i.e., 6.18 nm, 3.12 nm, 2.06 nm), and shows a long layer spacing of 6.18 nm. However, AGP3 does not exhibit obvious periodic diffraction peaks, and shows a d-spacing of 5.56 nm. The d-spacing is larger than the extended molecular length (∼3.9 nm estimated by the CPK molecular model shown in Fig. S4†) but smaller than twice the length, suggesting the formation of interdigitated bilayer structures regardless of whether nanotwists, nanoribbons or nanofibers were formed.43 A slight increase of d-spacing for AGP1 relative to AGP2 can be associated with the twist of nanoribbons. The smallest d-spacing for AGP3 implied a much more closely packed bilayer structure relative to AGP1 and AGP2. The differences of internal packing of AGP molecules within 1-D assemblies according to the XRD patterns are responsible for the observed morphological transitions for AGP1, AGP2 and AGP3. To analyze the conformations of tripeptides within the nanotwists, nanoribbons and nanofibers, we performed Fourier transform infrared (FT-IR) spectroscopy experiments on AGP hydrogels in D2O. The signal in the amide I region (∼1600–1700 cm−1) (Fig. S5†) is broad, suggesting that multiple peaks could be hidden there, such as α-helices, β-sheets and random coils. Circular dichroism (CD) spectroscopy was used to investigate the transfer of chiral information from the chiral center (Ala) to chromophore (azobenzene) originating from the self-assembly of AGP molecules.44 As expected, fully dissolved non-assembling AGP solution at 70 °C did not give any CD signal of azobenzene due to the free state of azobenzene in solution (Fig. 3B and S7†). Upon the solution being cooled to 20 °C for self-assembly, a negative band at ∼331 nm and a positive band at ∼306 nm with a crossover at around 318 nm were observed for AGP1 (Fig. 3C). The CD bands of opposite sign centered at 318 nm are related to the π–π* transition of azobenzene, indicating that the chirality of Ala was transferred to azobenzene upon self-assembly. In the case of AGP2, a positive band at ∼374 nm and a negative band at ∼336 nm with a crossover at around 353 nm were found (Fig. 3E). As for AGP3, a positive band at ∼352 nm and a negative band at ∼312 nm with a crossover at around 326 nm were observed (Fig. 3G). The CD spectra variation for AGP1, AGP2 and AGP3 can be directly associated with the variation of chiral centers at odd and even positions.45 Furthermore, the CD spectra variation was also strongly dependent on the intensity and types of intermolecular or intramolecular interactions.46 To compare the intensity of intermolecular interactions for AGP1, AGP2 and AGP3, the critical aggregation concentration (CAC) values were determined (Fig. S6†) (see the ESI† for the detailed characterization). Given the high structural similarity for AGP1, AGP2 and AGP3, the CAC values for AGP2 (0.010 mM) < AGP1 (0.062 mM) < AGP3 (0.215 mM) can be directly associated with the different intensities of intermolecular interactions. Stronger intermolecular interactions (e.g., H-bonding and π–π stacking) for AGP2 > AGP1 > AGP3 will be more favorable for the transfer of chirality from Ala to azobenzene, and ultimately reflected in the CD spectra intensity variation. In addition, the different packing intensities for AGP1, AGP2 and AGP3 can be related to the observed morphological transitions.
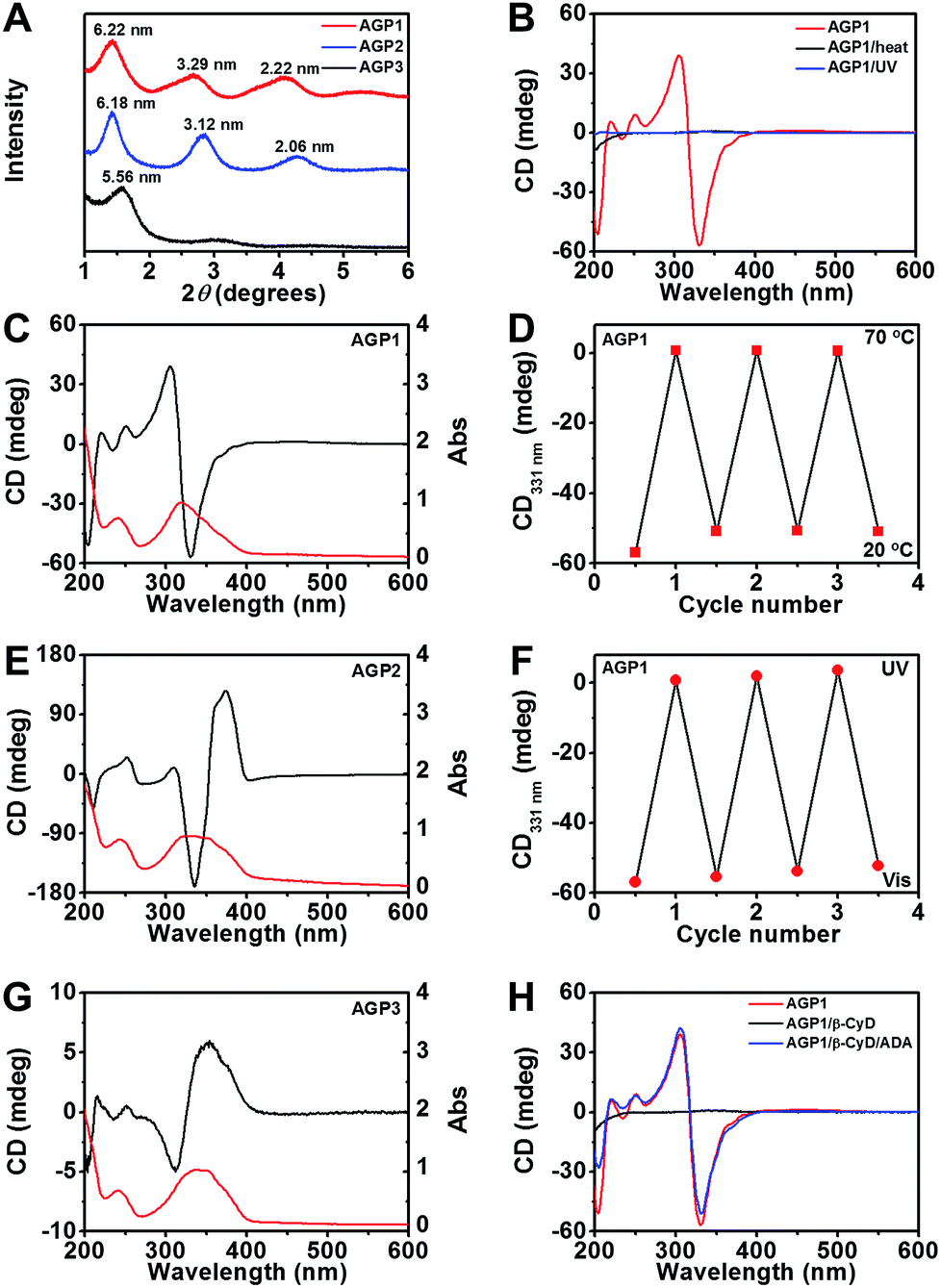 | ||
| Fig. 3 (A) XRD patterns of air-dried AGP hydrogels (30 mM). (B) CD spectra of AGP1 assemblies (0.6 mM) in response to heat (70 °C) or UV irradiation (λ365 nm, 3.7 mW cm−2, 10 min). (C, E and G) CD/UV-Vis (black/red traces) spectra of AGP1, AGP2 or AGP3 assemblies (0.6 mM). (D and F) Switchable CD signals at 331 nm from AGP1 assemblies (0.6 mM) in response to temperature changes or alternating UV/Vis illumination. (H) CD spectra of AGP1 assemblies (0.6 mM) in response to host–guest chemistry by β-CyD (nβ-CyD:nAGP1 = 8:1) and ADA (nADA:nβ-CyD = 1:1). | ||
We next assessed a reversible disassembly–assembly process of AGP assemblies in response to different stimuli (heat, light or host–guest chemistry) using CD spectroscopy. Upon heating to 70 °C, the CD signal of azobenzene disappeared (Fig. 3B, D and S7†). Upon cooling to 20 °C, the CD signal almost recovered (Fig. 3D and S7†). This reversible process can be repeated for at least 3 cycles. It can be ascribed to the weak intermolecular interactions that can be reversibly disrupted and recovered at 70 °C and 20 °C, respectively. After UV irradiation for 10 min, no CD signal of azobenzene was detected (Fig. 3B, F and S7†). After visible light irradiation for 60 min, the CD signal almost recovered (Fig. 3F and S7†). This reversible process can be repeated for at least 3 cycles. The UV-triggered cis-azobenzene is more hydrophilic than its trans-form owing to the enhancement of the dipole moment, and the altered molecular geometry of cis-azobenzene may restrict the close packing of the AGP molecules within assemblies.47,48 TEM imaging confirmed the disassembly of AGP1 nanotwists upon UV irradiation (Fig. S8A†). Under visible light irradiation, the azobenzene unit gradually recovered its trans-form, thus resulting in the reappearance of the CD signal. TEM imaging revealed the recovered AGP1 nanotwists (Fig. S8B†). Azobenzene trans–cis photoisomerization was also observed using UV-Vis spectroscopy (Fig. S7†) as determined by a diminished intensity of the peak at 350 nm corresponding to the π–π* transition and an increase in the intensity of the peak at 440 nm corresponding to the n–π* transition. Besides the thermal and optical stimuli, CD spectra of AGP assemblies were also switched off and on by host–guest interactions. It is expected that β-cyclodextrin (β-CyD) that possesses a hydrophilic exterior and hydrophobic cavity can encapsulate the n-butylazobenzene tail of AGP molecules. When β-CyD was added to AGP assemblies (0.6 mM) at room temperature, the CD signal of azobenzene disappeared in a few minutes (Fig. 3H and S7†), suggesting a fast host–guest complexation process. The AGP molecules have characteristic CAC values (AGP1: 0.062 mM; AGP2: 0.010 mM; AGP3: 0.215 mM), suggesting the existence of non-assembling AGP molecules in self-assembly solution. Moreover, supramolecular assemblies that are held together by weak intermolecular noncovalent interactions show dynamic and reversible assembly-to-disassembly properties. Thus, we speculate that the host–guest complexation between β-CyD and non-assembling AGP molecules would gradually promote the disassembly of AGP-aggregates. The disassembly of AGP1 nanotwists after host–guest complexation by β-CyD was confirmed by TEM imaging (Fig. S8C†). After adding 2-adamantanamine hydrochloride (ADA), the CD spectra almost recovered (Fig. 3H and S7†). Following competitive replacement by ADA, the released AGP1 reassembled into nanotwists as confirmed by TEM imaging (Fig. S8D†). These results demonstrated multiple stimuli-responsive properties of AGP assemblies.
To elucidate the molecular details of the self-assembled 1-D morphologies of AGP1, AGP2 and AGP3, we performed Martini coarse-grained MD simulations in each case.30–35,49 Under the same simulation conditions (as detailed in the ESI†), we noticed a dramatic difference in the self-assembled morphologies of the three molecules (Fig. 4). In each case, we demonstrated the snapshots of the resulting structure from its face, side, and end perspectives. The face perspectives are the same as presented in the TEM images. The ends represent the potential self-assembly propagation direction, where structures are repeatedly connected to each other to form the fibers. The way we define the ends versus sides is based on the greater exposure of the hydrophobic inner part of the fiber to the solvent, where continuous assembly would occur. Simulation sizes were limited to around 800 units in each case with fully solvated environments for reasonably long timescales. As shown in Fig. 4A and B, the AGP1 and AGP2 assemblies showed similar ribbon morphologies from the face perspective. However, from the end perspective, the AGP1 showed an obvious curved edge compared to the straight-line edge of the AGP2. These results are consistent with the observations from the TEM images: both AGP1 and AGP2 self-assembled into long ribbon-like fibers while the AGP1 fibers formed a helical structure in a large length scale but the AGP2 fibers did not. Rather differently, ∼800 AGP3 assembled into ∼2 perfect circular subunits according to the end perspective as shown in Fig. 4C. According to the face perspective, the repeating circular subunits stacked end to end forming the unique cylindrical nanofiber, which is well consistent with the experimental results. To calculate the curvatures of the equilibrated assemblies of AGP1 and AGP3, we measured characteristic distances for each from their trajectories of 50 ns close to the end. Three distances between the atom pairs of (508-C1, 77-C1), (77-C1, 736-C1), and (736-C1, 508-C1) were measured for L2D2, and two distances between the atom pairs (G604-C1, G688-C1) and (G576-C1, G450-C1) were measured for L4D4. The histograms of each are plotted as in Fig. S11 and S12,† respectively. Based on the most populated distances, we calculated the curvatures to be 0.13 nm−1 for AGP1 and 0.37 nm−1 for AGP3.
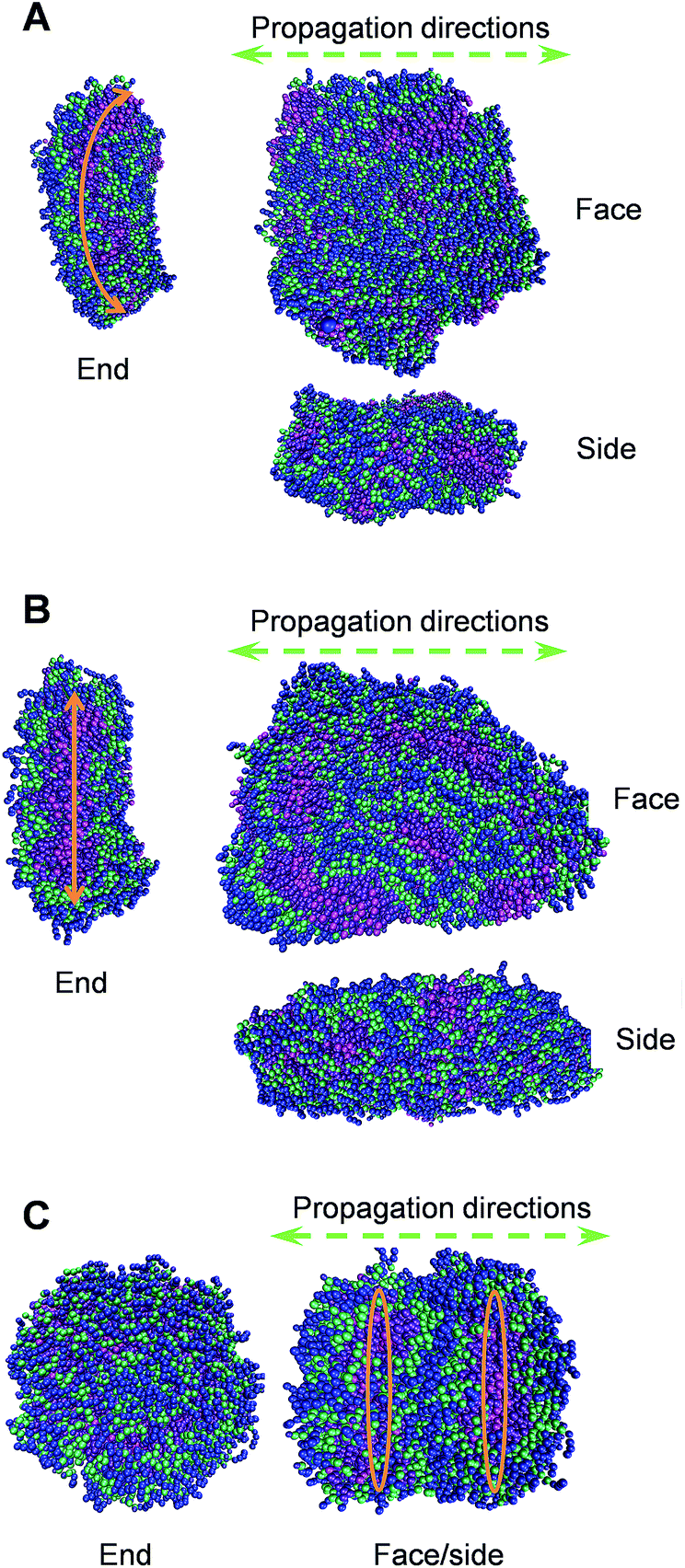 | ||
| Fig. 4 Representative conformations of the self-assembled morphologies of AGP1 (A), AGP2 (B), and AGP3 (C) from simulations of ∼800 units each. Each molecule is colored in such a way that the hydrophobic end is in magenta and violet, three amino acid residues are in green, and the hydrophilic end is in blue. | ||
To analyze the sequence isomerism-dependent self-assembled morphology difference in a large length scale, we take a close look at the structural details especially on the inner hydrophobic parts that formed as the scaffolds for the fibers as shown in Fig. 5. One reasonable assumption is that each small group of three molecules first self-assembled into different short-range assembly subunit cells, which further propagate to fibers with an observable morphology difference. Therefore, with the detailed structures of the scaffolds from simulations, we tried to identify the repeating assembly subunit cells that composed the large fiber morphology. As shown in Fig. 5, we clearly captured the different subunit cells. The AGP1 formed small hexagonal subunit cells that packed into a ribbon-like fiber with the curved edge (Fig. 5A). In contrast, the subunit cells of the AGP2 are parallelograms with sizes about twice larger than those of the AGP1 hexagons (Fig. 5B). As shown in Fig. 4, the parallelograms of the AGP2 fibers have straight-line edges with no curvature. As previously discussed, the AGP3 first formed circular subunit cells (Fig. 5C), which then packed into special cylindrical nanofibers. Smaller simulation systems with a lower number of molecules (less than 64) were used to understand the formation of subunit cells at the molecular level (as detailed in the ESI†). We first evaluated if our model could capture the bimolecule layer structure as assumed. In fully hydrated environments, the hydrophobic ends are expected to hide inside the assembled structure while leaving the hydrophilic end exposed to the solvent molecules, so that the hydration free energy could be minimized. As tested, for AGP1, AGP2 and AGP3, randomly oriented molecules interacted with each other forming the bimolecule layers in equilibrium that confirmed the XRD results. These results built the foundation for our large-scale simulation, where we initiated the simulation with two layers of scattered molecules with hydrophobic ends facing each other. It was a reasonable starting point based on the tested assumption that saved extensive computational time. With medium sizes (64 to 192) of simulation systems, we obtained a part of the unit cell for AGP3 (Fig. S7†), which is identical to the repeating structure observed in the larger fiber structure (Fig. 5) in terms of angles formed by branches. However, such particular unit cells were not obtained for AGP1 and AGP2. According to the observed numbers of repeating subunit cells in each assembled structures shown in Fig. 5, we estimated the unit cell size in a number of molecules to be around 130 (AGP3), 260 (AGP2), and 400 (AGP1). This means that the collaborative effects to form a subunit cell require a different number of molecules in each case. This could be the reason why it was easier to capture the subunit cells for the AGP3 than the others.
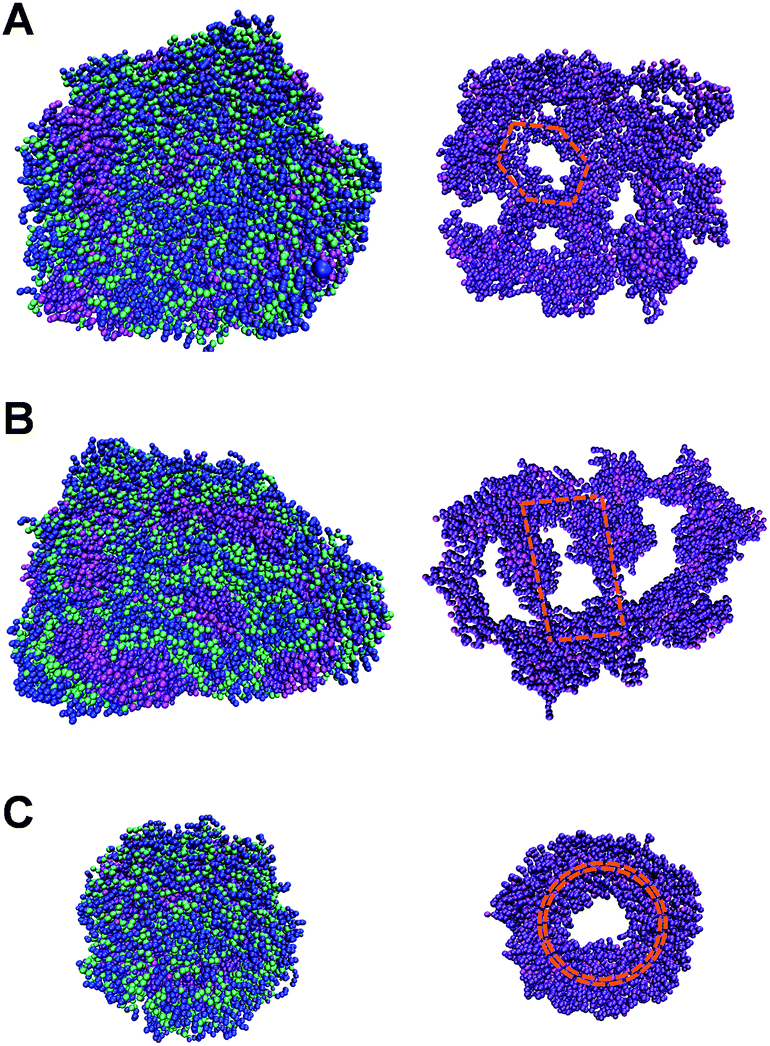 | ||
| Fig. 5 The reprint of the face snapshots of AGP1 (A) and AGP2 (B) and the end snapshot of AGP3 (C) on the top and the corresponding inner hydrophobic scaffolds at the bottom (in magenta and violet colors). | ||
A multivalent glycopeptide dendrimer inhibitor of the galactose-specific lectin LecA and of P. aeruginosa biofilms was reported.50 Here, we evaluated the antibiofilm activity of glycopeptide mimetic assemblies against methicillin-resistant S. aureus. Bacterial biofilms are microbial communities on a surface that are held together by an extracellular matrix composed of polysaccharides, proteins, DNA, lipids, etc.51 It is estimated that up to 80% of bacterial infections are accompanied by biofilm formation. Since bacteria in biofilms are more resistant to host immune responses and antibiotic therapies than bacteria in the planktonic state,52 biofilm-associated infections pose a major health threat, and there is a pressing need for antibiofilm agents. The AGP assemblies were screened for biofilm inhibition activity against S. aureus using a microtiter dish biofilm formation assay that involved recording the OD550 of crystal violet-stained biofilms (see the ESI† for the experimental procedures). Crystal violet assay of the formed S. aureus biofilms in the presence of AGP1, AGP2 and AGP3 assemblies revealed comparative biofilm inhibition activity (Fig. 6A). The formed biofilms in the presence of AGP assemblies were stained with SYTO9 and observed by fluorescence microscopy imaging. The imaging results (Fig. 6G and S13†) showed significantly reduced green fluorescence from the formed biofilms in the presence of AGP assemblies in comparison with blank control, also indicating the biofilm inhibition activity of AGP assemblies. Notably, after host–guest complexation by β-CyD, the biofilm inhibition activity of AGP1 assemblies disappeared, which was subsequently recovered after competitive replacement with ADA according to the crystal violet assay (Fig. 6D) and fluorescence microscopy imaging (Fig. 6H and I). To understand the inhibitory effect on biofilm formation, we also assessed biofilm elimination capability of AGP assemblies by 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-arboxanilide (XTT) (which characterizes living biofilm biomass) and crystal violet (which does not differentiate between live and dead biofilm bacterial cells) assays (see the ESI† for the experimental procedures). After being treated with the AGP assemblies, the amount of the preformed S. aureus biofilms significantly reduced as determined by the XTT assay (∼90% removal efficiency) (Fig. 6B) and crystal violet assay (∼80% removal efficiency) (Fig. 6C). To visualize the reduction of biofilm biomass, the preformed biofilms after treatment with the AGP assemblies were stained with LIVE/DEAD dyes and observed by fluorescence microscopy imaging. The imaging results (Fig. S14A–D†) were consistent with the XTT and crystal violet assay results, indicating the biofilm removal efficacy of the AGP assemblies. Because the bacterial cell wall usually consists of a large number of glycosyl groups in peptidoglycan, the bacteria have a strong ability to form hydrogen bonds with the molecules having similar structures. The self-assembled AGP nanostructures bearing multivalent tripeptide-glycosylamines are expected to ensure strong affinity to the bacterial cell wall.53–55 Therefore, it is reasonable to speculate that the affinity of the AGP assemblies with bacterial surfaces causes the gradual removal of the biofilms by weakening the attachment of the bacteria to the biofilm,56 and thus results in a reduction of the biofilm biomass. Upon host–guest complexation by β-CyD, the AGP nanostructures disassembled and the resulting AGP/β-CyD complex effectively switched off the biofilm removal efficacy (Fig. 6E, F and S14E†). Accordingly, we assume that the destruction of self-assembled multi-valency of AGP nanostructures results in much lower affinity with the bacterial cell wall, and thus turns off the bacterial cell detachment from the biofilm. Following a competitive replacement by ADA, the released AGP molecules reassembled and the biofilm removal efficiency was almost recovered (switch on) (Fig. 6E, F and S14F†). In a combination of biofilm removal results, it is rational to speculate that the affinity of the AGP assemblies with bacterial surfaces leads to disruption of the interactions of bacteria to the substrate surface, and interference with bacterial adhesion to the substrate surface during the biofilm formation,57 and ultimately exhibits the biofilm inhibition activity. Similarly, the switch off of the biofilm inhibition activity upon host–guest complexation by β-CyD was because of the much lower affinity of the AGP/β-CyD complex to the bacterial cell wall. Following competitive replacement by ADA, the switch on of the biofilm inhibition activity was due to the reassembly of the released AGP molecules. Taken together, the AGP assemblies showed antibiofilm (biofilm inhibition and elimination) activity. Based on a reversible disassembly–assembly process by host–guest chemistry, the antibiofilm activity of AGP assemblies could be reversibly switched off and on, which makes AGP assemblies promising intelligent antibacterial nanomaterials.
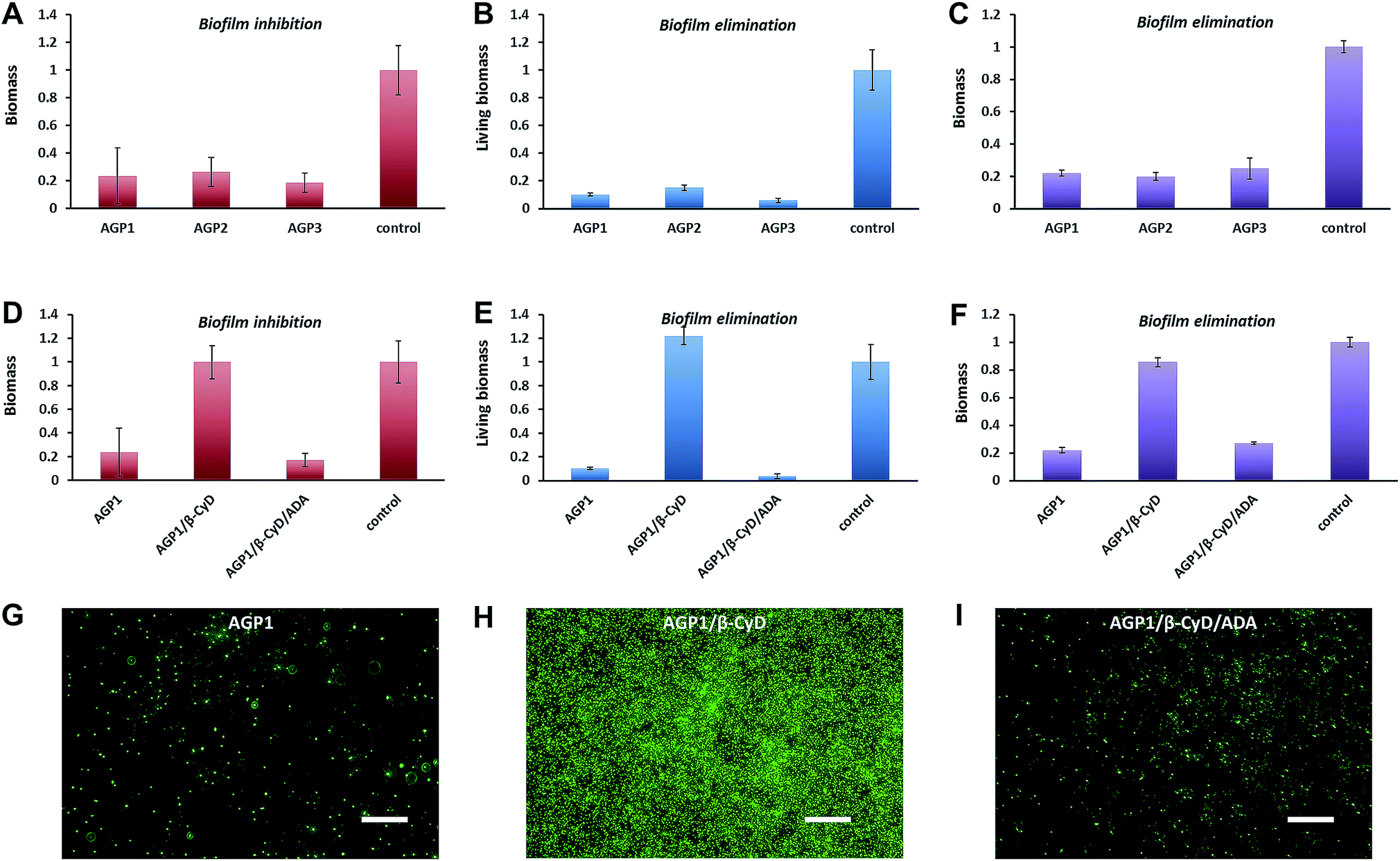 | ||
| Fig. 6 (A and D) The biomass of the formed S. aureus biofilms in the presence of 0.6 mM AGP1, AGP2, AGP3, AGP1/β-CyD or AGP1/β-CyD/ADA for 24 h at 37 °C as quantified by crystal violet assay. (B and E) The living biomass of the preformed S. aureus biofilms after treatment with 0.6 mM AGP1, AGP2, AGP3, AGP1/β-CyD or AGP1/β-CyD/ADA for 24 h at 37 °C as quantified by XTT assay. (C and F) The biomass of the preformed S. aureus biofilms after treatment with 0.6 mM AGP1, AGP2, AGP3, AGP1/β-CyD or AGP1/β-CyD/ADA for 24 h at 37 °C as quantified by crystal violet assay. (G, H and I) Fluorescence microscopy imaging of the formed S. aureus biofilms in the presence of 0.6 mM AGP1, AGP1/β-CyD or AGP1/β-CyD/ADA for 24 h at 37 °C after SYTO9-staining. The scale bars are 20 μm. | ||
Conclusions
In summary, we synthesized three isomeric glycopeptide mimetics by modifying Ala–Gly–Gly–OH (or Gly–Ala–Gly–OH, Gly–Gly–Ala–OH) with an n-butylazobenzene group at its N-terminal. Different self-assembled 1-D morphologies (i.e., nanotwists, nanoribbons and nanofibers) were observed under the same conditions when the position of the Ala residue was switched. Martini coarse-grained MD simulations confirmed the sequence isomerism-dependent self-assembled morphological transitions. The glycopeptide mimetic assemblies showed a reversible disassembly–assembly process in response to different stimuli (temperature, light and host–guest chemistry), and can be used as switchable antibiofilm nanoagents. The ability of this nanomaterial to reversibly turn-off and turn-on antibacterial activity is particularly interesting for combating bacterial resistance.58Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21773172, No. 21603166, No. 21573162), WIBEZD 2014001-02 and Wenzhou Public Welfare Science and Technology Project (S20170014).Notes and references
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef PubMed.
- Y. Xie, Y. Wang, W. Qi, R. Huang, R. Su and Z. He, Small, 2017, 13, 1700999 CrossRef PubMed.
- H. Wang, J. Shi, Z. Feng, R. Zhou, S. Wang, A. A. Rodal and B. Xu, Angew. Chem., Int. Ed., 2017, 56, 16297–16301 CrossRef CAS PubMed.
- C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS PubMed.
- C. H. Chen, L. C. Palmer and S. I. Stupp, Nano Lett., 2018, 18, 6832–6841 CrossRef CAS PubMed.
- W. Jeong, S. Hyun Kwon and Y. Lim, Adv. Funct. Mater., 2018, 28, 1803114 CrossRef.
- T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746–14752 CrossRef CAS PubMed.
- L. Qin, P. Duan, F. Xie, L. Zhang and M. Liu, Chem. Commun., 2013, 49, 10823–10825 RSC.
- H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740–750 CrossRef CAS.
- C. Zhang, R. Shafi, A. Lampel, D. MacPherson, C. G. Pappas, V. Narang, T. Wang, C. Maldarelli and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 14511–14515 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Álvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 6141, eaat6141 Search PubMed.
- N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem.–Eur. J., 2017, 23, 981–993 CrossRef CAS PubMed.
- J. Li, X. Du, S. Hashim, A. Shy and B. Xu, J. Am. Chem. Soc., 2017, 139, 71–74 CrossRef CAS PubMed.
- P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS PubMed.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS PubMed.
- M. Amit, S. Yuran, E. Gazit, M. Reches and N. Ashkenasy, Adv. Mater., 2018, 30, 1707083 CrossRef PubMed.
- S. Cavalli, F. Albericio and A. Kros, Chem. Soc. Rev., 2010, 39, 241–263 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- H. Wang, Z. Yang and D. J. Adams, Mater. Today, 2012, 15, 500–507 CrossRef CAS.
- H. Cui, A. G. Cheetham, E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 12461–12468 CrossRef CAS.
- Y. Hu, R. Lin, P. Zhang, J. Fern, A. G. Cheetham, K. Patel, R. Schulman, C. Kan and H. Cui, ACS Nano, 2016, 10, 880–888 CrossRef CAS PubMed.
- A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS.
- S. T. Wang, Y. Lin, R. K. Spencer, M. R. Thomas, A. I. Nguyen, N. Amdursky, E. Thomas Pashuck, S. C. Skaalure, C. Y. Song, P. A. Parmar, R. M. Morgan, P. Ercius, S. Aloni, R. N. Zuckermann and M. M. Stevens, ACS Nano, 2017, 11, 8579–8589 CrossRef CAS PubMed.
- Y. Wang, K. Kaur, S. J. Scannelli, R. Bitton and J. B. Matson, J. Am. Chem. Soc., 2018, 140, 14945–14951 CrossRef CAS PubMed.
- Q. Xing, J. Zhang, Y. Xie, Y. Wang, W. Qi, H. Rao, R. Su and Z. He, ACS Nano, 2018, 12, 12305–12314 CrossRef CAS.
- F. Zhao, H. Guo, Z. Zhang, J. Ye, L. Liu, C. X. Zhao and Z. Shao, J. Mater. Chem. B, 2017, 5, 5189–5195 RSC.
- N. Brown, J. Lei, C. Zhan, L. J. W. Shimon, L. Adler-Abramovich, G. Wei and E. Gazit, ACS Nano, 2018, 12, 3253–3262 CrossRef CAS PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862–1876 CAS.
- C. Guo, Z. A. Arnon, R. Qi, Q. Zhang, L. Adler-Abramovich, E. Gazit and G. Wei, ACS Nano, 2016, 10, 8316–8324 CrossRef CAS PubMed.
- O. S. Lee, S. I. Stupp and G. C. Schatz, J. Am. Chem. Soc., 2011, 133, 3677–3683 CrossRef CAS PubMed.
- X. Mu, K. M. Eckes, M. M. Nguyen, L. J. Suggs and P. Ren, Biomacromolecules, 2012, 13, 3562–3571 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- J. Wang and B. Xu, Chem, 2018, 4, 1765–1767 CAS.
- I. J. Edwards, Nat. Rev. Urol., 2012, 9, 196–206 CrossRef CAS PubMed.
- D. Xu and J. D. Esko, Annu. Rev. Biochem., 2014, 83, 129–157 CrossRef CAS PubMed.
- A. Brito, Y. M. Abul-Haija, D. S. Da Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem. Sci., 2019, 10, 2385–2390 RSC.
- A. J. Garton and S. J. Yeaman, Eur. J. Biochem., 1990, 191, 245–250 CrossRef CAS PubMed.
- L. Chen, X. Zhao, Y. Lin, Z. Su and Q. Wang, Polym. Chem., 2014, 5, 6754–6760 RSC.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- P. Duan, L. Qin, X. Zhu and M. Liu, Chem.–Eur. J., 2011, 17, 6389–6395 CrossRef CAS PubMed.
- P. Duan, H. Cao, L. Zhang and M. Liu, Soft Matter, 2014, 10, 5428–5448 RSC.
- T. Wang, Y. Li and M. Liu, Soft Matter, 2009, 5, 1066–1073 RSC.
- P. Duan, X. Zhu and M. Liu, Chem. Commun., 2011, 47, 5569–5571 RSC.
- S. K. Rastogi, H. E. Anderson, J. Lamas, S. Barret, T. Cantu, S. Zauscher, W. J. Brittain and T. Betancourt, ACS Appl. Mater. Interfaces, 2018, 10, 30071–30080 CrossRef CAS PubMed.
- A. M. Rosales, K. M. Mabry, E. M. Nehls and K. S. Anseth, Biomacromolecules, 2015, 16, 798–806 CrossRef CAS PubMed.
- S. L. Chin, Q. Lu, E. L. Dane, L. Dominguez, C. J. McKnight, J. E. Straub and M. W. Grinstaff, J. Am. Chem. Soc., 2016, 138, 6532–6540 CrossRef CAS.
- R. U. Kadam, M. Bergmann, M. Hurley, D. Garg, M. Cacciarini, M. A. Swiderska, C. Nativi, M. Sattler, A. R. Smyth, P. Williams, M. Cámara, A. Stocker, T. Darbre and J. Reymond, Angew. Chem., Int. Ed., 2011, 50, 10631–10635 CrossRef CAS.
- H. C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- T. F. Mah, B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart and G. A. O'Toole, Nature, 2003, 426, 306–310 CrossRef CAS.
- L. Wu, Y. Zhang, Z. Li, G. Yang, Z. Kochovski, G. Chen and M. Jiang, J. Am. Chem. Soc., 2017, 139, 14684–14692 CrossRef CAS PubMed.
- M. W. Jones, L. Otten, S. J. Richards, R. Lowery, D. J. Phillips, D. M. Haddleton and M. I. Gibson, Chem. Sci., 2014, 5, 1611–1616 RSC.
- A. d. J. Cortés-Sánchez, H. Hernández-Sánchez and M. E. Jaramillo-Flores, Microbiol. Res., 2013, 168, 22–32 CrossRef PubMed.
- J. Li, K. Zhang, L. Ruan, S. F. Chin, N. Wickramasinghe, H. Liu, V. Ravikumar, J. Ren, H. Duan, L. Yang and M. B. Chan-Park, Nano Lett., 2018, 18, 4180–4187 CrossRef CAS PubMed.
- R. Joseph, A. Naugolny, M. Feldman, I. M. Herzog, M. Fridman and Y. Cohen, J. Am. Chem. Soc., 2016, 138, 754–757 CrossRef CAS PubMed.
- W. A. Velema, J. P. Van Der Berg, M. J. Hansen, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924–928 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular synthesis, structural characterization, MD simulations and biofilm assays. See DOI: 10.1039/c9sc00193j |
| This journal is © The Royal Society of Chemistry 2019 |