
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective photoredox dehalogenative protonation†
Meimei
Hou‡
a,
Lu
Lin‡
a,
Xiangpei
Chai‡
a,
Xiaowei
Zhao
a,
Baokun
Qiao
*a and
Zhiyong
Jiang
 *ab
*ab
aHenan University, Jinming Campus, Kaifeng, Henan 475004, China. E-mail: jiangzhiyong@htu.edu.cn; chmjzy@henu.edu.cn
bHenan Key Laboratory of Organic Functional Molecules and Drug Innovation, Key Laboratory of Green Chemical Media and Reactions, Ministry of Education, Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
First published on 7th June 2019
Abstract
We report an enantioselective photoredox dehalogenative protonation as a new type of asymmetric protonation. As a paradigm, with a cooperative catalytic system consisting of a chiral H-bonding catalyst and a dicyanopyrazine-derived chromophore (DPZ) photosensitizer that is irradiated with visible light, a range of cyclic and acyclic ketones with labile chiral secondary C–F, C–Cl and C–Br bonds at the α-position were obtained in high yields with good to excellent enantioselectivities (up to >99% ee) by using a secondary amine as the terminal reductant. Given the ready accessibility of halides, the success of this work should provide inspiration for constructing diverse chiral α-tertiary carbonyls and their variants.
Introduction
Enantioselective protonation is a fundamental method for synthesizing enantioenriched α-tertiary carbonyl compounds.1 In the past decade, a number of efficient ground-state catalytic platforms have been established, allowing the precise delivery of a proton to prochiral carbanion intermediates in a highly enantioselective manner.2–8 Nonetheless, the approach of constructing secondary α-carbon–halogen bonds for ketones still remains underdeveloped, as few examples with insufficient enantioselectivities have been described (Scheme 1a).3b,4a,6a The stereoselective formation of C–F bonds is of critical importance in pharmaceutical chemistry given the ability of fluorine atoms to act as the isostere of hydrogen atoms in decreasing the rate of metabolic degradation without influencing the pharmacological effects.9 Meanwhile, chiral C–Cl and C–Br bonds are applicable for introducing various important molecular architectures via simple and stereospecific transformations and are thus extensively adopted in the asymmetric synthesis of chiral complex molecules with significant biological activities.9a,b,10 In this context, the development of a new and generic catalytic asymmetric protonation approach to form valuable chiral tertiary α-haloketones9,10 represents a highly desirable task that would be a complementary and more versatile method than direct catalytic enantioselective halogenation. Of note, a few direct asymmetric α-chlorination and α-fluorination reactions of aliphatic ketones via enamine catalysis have been established (Scheme 1b);11 however, the catalytic systems are not compatible with aromatic variants, likely because of the slow enamine formation and the approximately equimolar enamine rotational isomers. An approach through indirect enantioselective chlorination of β-ketocarboxylic acids12 also failed to generate chiral α-secondary chloro-ketones in high enantioselectivities (Scheme 1c). | ||
| Scheme 1 Previous studies on the enantioselective construction of secondary α-C–X bonds for ketones. | ||
Due to the strong electron-withdrawing properties of ketones and the high electronegativity of halogens, the stereocenter of secondary α-haloketones should be particularly labile, especially for those aromatic variants.6a,11b As such, a mild and neutral protonation strategy to avoid racemization of the products in the reaction process is inarguably crucial. Recently, we reported the asymmetric photoreduction of 1,2-diketones to chiral α-hydroxy ketones enabled by a synergistic photosensitizer and H-bonding catalysis with weakly basic tertiary amines as the terminal reductant (Scheme 2a).13 The excellent enantioselectivity clearly demonstrated the applicability of such an open-shell single-electron-transfer (SET) approach14 for the formation of the labile tertiary carbon stereocenter of α-hydroxyl ketones via an asymmetric protonation procedure. Accordingly, we wondered whether the ability to combine this dual-catalysis15 reduction system with competent oxidative feedstocks, from which halogenated carbon anions α to ketones could be produced via two SET reductions, might address this challenging task.
 | ||
| Scheme 2 Previous studies. (a) Photoredox catalytic asymmetric reduction of 1,2-diketones via enantioselective protonation to build the labile α-secondary alcohols of ketones. (b) Photoredox catalytic reductive dehalogenation of α,α-dibromoketones in a racemic manner. | ||
Alkyl halides (X) are an important class of readily accessible starting substrates in organic synthesis.16 Since Tanaka and co-workers introduced the visible light-driven reductive dehalogenation of benzyl bromide to benzene in 1984,17 the groups of Kellogg,18a Fukuzumi,18b Stephenson,18c–e Zeitler,18f and Hisaeda18g and so on18h–o have accomplished the transformation of diverse alkyl halides to alkanes via photoredox catalysis with tertiary amines as the reductant. This conceptual strategy was developed by the Reiser group19 by establishing an alternative reductive system, i.e., 1,5-dimethoxynaphthalene (DMN) with ascorbic acid, wherein the selective debrominative protonation of α,α-dibromoketones to the racemic monobromo ketones was described (Scheme 2b). Furthermore, Hyster and co-workers recently reported an enzyme catalytic debromination-enantioselective hydrogen atom transfer (HAT) to access enantioenriched α-tertiary esters from α-bromoesters.20 Encouraged by these elegant studies, we hypothesized that the enantioselective platform of α,α-dihaloketones would be an expedient and modular strategy for accessing the desired chiral organohalides. Several significant challenges should remain in this promising task, namely the weaker basicity of ketones relative to ketyls which would impair the stereocontrol of H-bonding catalysis, and both the competitive racemic background reaction and the possible racemization of the stereocenter which should diminish the enantioselectivity. More importantly, even employing a powerful enzyme catalysis and capturing a hydrogen atom from the bulky flavin still could not provide a good enantiofacial result for the formation of a C–F bond.20a Therefore, the direct delivery of the smallest proton to the prochiral intermediates through the distinctly weaker asymmetric H-bonding induction to provide stereocontrolling environments would be more formidable.
Results and discussion
Optimization of reaction conditions
We began our study with 2-chloro-2-fluoro-tetralone 1a as the model substrate (Table 1). A range of chiral H-bonding catalysts, photoredox catalysts, amine reductants, and inorganic bases as the acid-binding agents to remove the generated HCl were examined in conjunction with diverse reaction parameters (see Tables S1 and S2 in the ESI†). As a result, the desired product 2a was obtained in 92% yield with 96% ee when the reaction was performed in 1,2-dichloroethane at 5 °C for 36 h in the presence of dicyanopyrazine-derived chromophore (DPZ, Et(S*/S˙−) = +0.91 V vs. SCE, Ered1/2 = −1.45 V vs. SCE in CH2Cl2, ET = 46.4 kcal mol−1),13,16b,21 5 mol% L-tert-leucine-based squaramide-tertiary amine C1,22 0.6 equiv. Amine-1 as the secondary amine, 1.0 equiv. H2O and 2.5 equiv. Na2CO3 (entry 1). The structural features of the chiral catalyst, consisting of the aryl substituent of squaramide (C2), the structural skeleton of the tertiary amine (C3) and the type of H-bonding donor (C4 and C5), were modified to affect the enantiofacial selectivity (entries 2–5). When Amine-2 was the reductant, the yield of 2a decreased to 76% with a similar enantioselectivity (entry 6). Other plausible photoredox catalysts, including Ru(bpy)3Cl2·6H2O ( vs. SCE, EII/I1/2 = −1.33 V vs. SCE in CH3CN, ET = 46.5 kcal mol−1), Rose Bengal (Et(S*/S˙−) = +0.99 V vs. SCE, Ered1/2 = −0.68 V vs. SCE in CH3CN, ET = 40.9 kcal mol−1) and Ir(ppy)2(dtbbpy)PF6 (
vs. SCE, EII/I1/2 = −1.33 V vs. SCE in CH3CN, ET = 46.5 kcal mol−1), Rose Bengal (Et(S*/S˙−) = +0.99 V vs. SCE, Ered1/2 = −0.68 V vs. SCE in CH3CN, ET = 40.9 kcal mol−1) and Ir(ppy)2(dtbbpy)PF6 ( vs. SCE, EIII/II1/2 = −1.51 V vs. SCE in CH3CN, ET = 49.2 kcal mol−1), were tested (entries 7–9), but better comprehensive results were not achieved. The moderate yield obtained with Rose Bengal stemmed from an unclear reaction interruption, which resulted in unsatisfactory chemical conversion. The transformation in the absence of catalyst C1 offered rac-2a in 48% yield with >95% chemical conversion after 36 h, indicating that a racemic background reaction occurred (entry 10). The lower yield was due to deterioration of the chemical selectivity, as a few unknown side products were observed. In the absence of H2O, the yield of 2a decreased to 74% with a similar enantioselectivity (entry 11), suggesting its important effect in increasing the solubility of inorganic salts. DPZ as the photoredox catalyst was found to accelerate the transformation (entry 12), although the type of EDA23 is involved in the process (see ESI† for details). Control experiments also confirmed that reductants, inorganic bases and visible light were essential for the reaction (entries 13–15).
vs. SCE, EIII/II1/2 = −1.51 V vs. SCE in CH3CN, ET = 49.2 kcal mol−1), were tested (entries 7–9), but better comprehensive results were not achieved. The moderate yield obtained with Rose Bengal stemmed from an unclear reaction interruption, which resulted in unsatisfactory chemical conversion. The transformation in the absence of catalyst C1 offered rac-2a in 48% yield with >95% chemical conversion after 36 h, indicating that a racemic background reaction occurred (entry 10). The lower yield was due to deterioration of the chemical selectivity, as a few unknown side products were observed. In the absence of H2O, the yield of 2a decreased to 74% with a similar enantioselectivity (entry 11), suggesting its important effect in increasing the solubility of inorganic salts. DPZ as the photoredox catalyst was found to accelerate the transformation (entry 12), although the type of EDA23 is involved in the process (see ESI† for details). Control experiments also confirmed that reductants, inorganic bases and visible light were essential for the reaction (entries 13–15).
|
|
|||
|---|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) | eec (%) |
| a The reaction was performed on a 0.05 mmol scale. Entries 1–9, the chemical conversion of 1a was >95% determined by crude 1H NMR. b Yields were determined from the isolated compound following chromatographic purification. c Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. d The chemical conversion of 1a was >95%, as determined by crude 1H NMR. e The chemical conversion of 1a was 45%. | |||
| 1 | None | 92 | 96 |
| 2 | C2 instead of C1 | 87 | 81 |
| 3 | C3 instead of C1 | 64d | 23 |
| 4 | C4 instead of C1 | 89 | 77 |
| 5 | C5 instead of C1 | 87 | 65 |
| 6 | Amine-2 instead of Amine-1 | 76 | 94 |
| 7 | Ru(bpy)3Cl2·6H2O instead of DPZ | 87 | 95 |
| 8 | Rose Bengal instead of DPZ | 67 | 96 |
| 9 | Ir(ppy)2(dtbbpy)PF6 instead of DPZ | 82 | 91 |
| 10 | No C1 | 48d | N.A. |
| 11 | No H2O | 74 | 95 |
| 12 | No DPZ | 38e | 94 |
| 13 | No Amine-1 | 0 | N.A. |
| 14 | No Na2CO3 | 0 | N.A. |
| 15 | No light | 0 | N.A. |
With the optimized conditions in hand, we next evaluated the substrate scope (Table 2). A range of 2-chloro-2-fluoro-tetralones with diverse electron-withdrawing and electron-donating substituents on the aryl ring were first examined. The reactions were finished within 24–36 h, and a series of chiral α-fluorinated tetralones 2a–o were obtained in 68–96% yield and with 92 to >99% ee. The satisfactory results achieved with alkyne (2h), hydroxyls protected by tosyl (Ts, 2j), allyl (2k), mesyl (Ms, 2m) and tert-butyl-dimethylsilyl (TBS, 2n) groups and even the naked hydroxyl (2o) as the substituent underscore the high functional-group tolerance of this catalytic system. The reaction to furnish 2a was attempted on a 1.0 mmol scale, and a similar yield and enantioselectivity were observed with a slightly longer reaction time (48 h, see footnote b), indicating the promising synthetic utility of this method. Other significant cyclic ketones, including 4-chromanone (2p), thiochroman-4-one (2q), 1-indanones with distinct substituents on the aromatic ring (2r–2x) and 1-benzosuberone (2y), were subsequently tested, leading to a variety of corresponding enantioenriched α-fluoroketones in 55 to 90% yield and 84 to 93% ee. Linear and nonaromatic ketones were also evaluated. To achieve the best enantioselectivity, catalyst C25 was used for the preparation of 2za and 2zc–e (82–90% ee), and C26 was used for 2zb (90% ee) (see ESI† for the structures of tertiary-squaramide-based C25 and C26). The generality of this method was thus illustrated. We also carried out enantioselective reductive debromination of 2-bromo-2-fluoro-tetralone 3; 2a was obtained with a similar excellent enantioselectivity but in a lower yield than that achieved via the dechlorination of 1a. Note that these α-fluoroketones are stable under the reaction conditions, as indicated by the ee value being maintained and no defluorinated parent ketone being observed when the reaction was performed for a longer time. Moreover, the determination of 0% ee of 2a before the reaction completed excluded the possibility of kinetic resolution of 2a.
 | (1) |
| a Reaction performed on a 0.10 mmol scale. Yields were determined from the isolated material after chromatographic purification. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. b When the reaction was performed on a 1.0 mmol scale, t = 48 h, 86% yield, 97% ee. c Catalyst C25 was used instead of C1. d Catalyst C26 was used instead of C1. e The modified reaction parameters: Amine-2 instead of Amine-1, no H2O in THF at 40 °C for 60 h. |
|---|

|
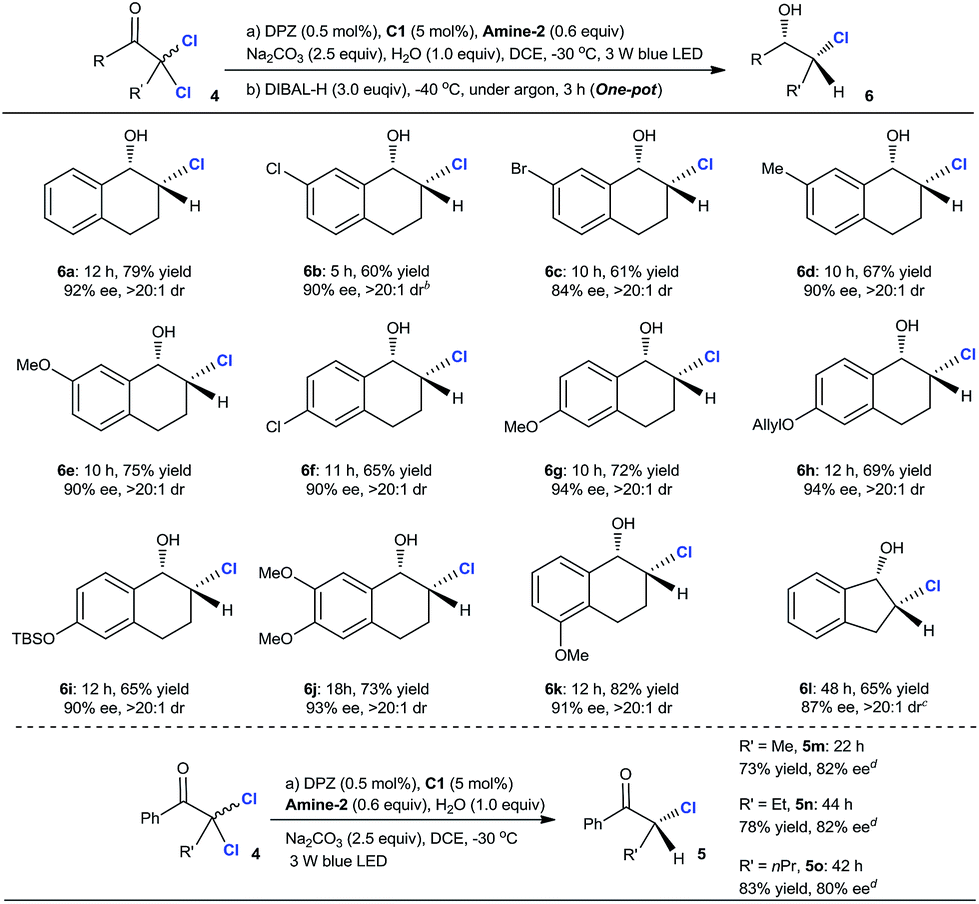
The aforementioned success encouraged us to further evaluate this protocol in constructing chiral tertiary α-chloroketones from α,α-dichloroketones. The preliminary studies with 2,2-dichloro-tetralone 4a as the starting substrate showed that the slightly modified reaction conditions, wherein the reductant was Amine-2 and the temperature was −30 °C, afforded chiral 2-chloro-tetralone 5a in 87% yield and with 92% ee within 12 h (eqn (1)).25 However, the ee value of this product was observed to deteriorate when flash column chromatography was performed, revealing that such a tertiary stereocenter is considerably labile.
Accordingly, we planned to explore a sequential strategy involving subsequent one-pot reduction of ketone to chlorohydrin, thus facilitating purification. After careful examination, we found that the desired product 6a was obtained in 79% yield over two steps with a maintained ee of 92% and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr when the reaction mixture from the first reductive dechlorination step was directly treated with 3.0 equiv. of diisobutylaluminum hydride (DIBAL-H) at −40 °C under an argon atmosphere for 3 h (Table 3). In the presence of the established tandem protocol, a series of 2,2-dichloro-tetralones with different substituents on the aryl ring were examined, leading to the important chlorohydrins246b–k in 60–82% yield with 84–94% ee and >20:1 dr. With respect to 2,2-dichloro-1-indanone, the reaction conditions were further modified for achieving higher enantioselectivity (see footnote c). As a result, product 6l was obtained in 65% yield, 87% ee and >20:1 dr. Inarguably, these results demonstrated that a secondary C–Cl bond was successfully introduced at the α-position of these cyclic ketones with high enantioselectivities, indicating the compatibility of this catalytic system even though these α-secondary C–Cl bonds of ketones are unstable. The attempt with linear ketones showed that C25 was a suitable H-bonding catalyst, leading to a series of α-chloroketones 5m–o in 73–83% yield with 80–82% ee. Because the products were stable during flash column chromatography, the further reduction of ketone to alcohol was unnecessary. Notably, some amount of doubly dechlorinated products was found before the reactions were finished. Hence, we tested the ee of 5a at different reaction stages and after prolonging the reaction beyond completion and found that the amount of tetralone continued to increase. The results demonstrated that all the ee values were consistent, indicating that kinetic resolution is not possible in the dechlorination of monochlorinated ketones.
:1 dr when the reaction mixture from the first reductive dechlorination step was directly treated with 3.0 equiv. of diisobutylaluminum hydride (DIBAL-H) at −40 °C under an argon atmosphere for 3 h (Table 3). In the presence of the established tandem protocol, a series of 2,2-dichloro-tetralones with different substituents on the aryl ring were examined, leading to the important chlorohydrins246b–k in 60–82% yield with 84–94% ee and >20:1 dr. With respect to 2,2-dichloro-1-indanone, the reaction conditions were further modified for achieving higher enantioselectivity (see footnote c). As a result, product 6l was obtained in 65% yield, 87% ee and >20:1 dr. Inarguably, these results demonstrated that a secondary C–Cl bond was successfully introduced at the α-position of these cyclic ketones with high enantioselectivities, indicating the compatibility of this catalytic system even though these α-secondary C–Cl bonds of ketones are unstable. The attempt with linear ketones showed that C25 was a suitable H-bonding catalyst, leading to a series of α-chloroketones 5m–o in 73–83% yield with 80–82% ee. Because the products were stable during flash column chromatography, the further reduction of ketone to alcohol was unnecessary. Notably, some amount of doubly dechlorinated products was found before the reactions were finished. Hence, we tested the ee of 5a at different reaction stages and after prolonging the reaction beyond completion and found that the amount of tetralone continued to increase. The results demonstrated that all the ee values were consistent, indicating that kinetic resolution is not possible in the dechlorination of monochlorinated ketones.
 | (2) |
| a Reaction performed on a 0.10 mmol scale. The reaction time stated below the product structure refers to the reductive dechlorination process. Yields were determined from the isolated material after chromatographic purification. Enantiomeric excesses were determined by HPLC analysis on a chiral stationary phase. The dr was determined by the crude 1H NMR analysis. b 2.0 mL DCE was used. c The modified reaction parameters: 20 mol% C1, 1.5 equiv. Na2CO3 in DCM at −60 °C. d Catalyst C25 was used instead of C1. |
|---|
|
Given the almost identical utility of chiral C–Cl and C–Br bonds in organic synthesis, 2,2-dibromo-tetralone 7 was selected as a representative to evaluate the viability of enantioselective photoredox debrominative protonation. The investigation revealed that the chiral α-bromo-tetralone stereocenter exhibited lability similar to that of the chloro variant. Accordingly, the cascade reaction was performed, and chiral bromohydrin 8 was successfully achieved in 61% yield, 90% ee and >20:1 dr (eqn (2)).
Mechanism studies
The mechanism of these reactions via a photoredox dehalogenation–protonation process is established.18,19 Our Stern–Volmer experiments demonstrated the occurrence of the same catalytic cycle in which the transformation was triggered by reductive quenching of the photoredox catalyst (see the ESI†). Notably, all chiral catalysts (C1, C25 and C26) contain a squaramide group, and this diketone moiety (e.g., Ered1/2 of C1 = −0.22, −0.79 V vs. SCE in CH3CN) should be reduced by DPZ˙− more readily than α,α-dihaloketones (e.g., Ered1/2 of 1a = −0.80, −1.23 V vs. SCE in CH3CN). In this context, the structure of C1 after the transformation of 1a to 2a was analyzed. As shown in Scheme 3a, C1 was recovered in 90% yield and no reduced product was observed. In the absence of 1a, a high recovery yield of C1 was still obtained (Scheme 3b). These results suggest a perfect tolerance of C1 under the reaction conditions, likely because of its evidently lower concentration than the substrates in the reaction system. For the formation of the α-secondary stereocenter, the use of structurally different Amine-1 and Amine-2 as the reductants offering similar enantioselectivities (entries 1 and 6, Table 1) could roughly prove a protonation process rather than HAT. The deuterium labelling studies could further demonstrate this protonation process (see ESI†). | ||
| Scheme 3 aDetermining the structure of C1 after the reaction completed. bDetermining the structure of C1 when without the starting substrate 1a. cThe reaction of 1a to 2a was performed under standard reaction conditions but with C28 instead of C1 as the chiral catalyst. | ||
To probe the role of C1's bifunctional groups, i.e., tertiary amine and squaramide, the transformation of 1a was performed under the standard reaction conditions, as shown in Table 1, but by using C1-derived quaternary ammonium salt C28 as the catalyst. Product 2a was obtained in 78% yield with 84% ee (Scheme 3c). The negligible change in enantioselectivity and the maintained absolute configuration suggest that the tertiary amine moiety of C1 might not participate in proton transfer but instead provides a steric factor only for establishing stereocontrol. From the similar enantioselective results in the construction of different C–X bonds with the same chiral catalyst C1 (see the results for 2a, 5a and 8 in Table 2, eqn (1) and (2), respectively), the hydrogen-bonding interaction between the squaramide of C1 and halogen of ketones might be excluded. Meanwhile, the remarkably different enantioselectivity with C1 and C3 catalysts (entries 1 and 3, Table 1) indicates a slight possibility of the strongly acidic squaramide participating in proton transfer interchange to present H+ to carbanion intermediates in the protonation step. Accordingly, a plausible transition state involving H-bonding interactions between the squaramide of C1 and ketone for facilitating stereocontrol and improving the chemoselectivity was proposed and is shown in the ESI.†
Conclusions
In summary, we have developed a novel asymmetric protonation strategy that is the visible-light-driven photoredox-catalyzed dehalogenation–enantioselective protonation. Through the establishment of a dual organocatalytic system with a secondary amine reductant, a range of important cyclic and acyclic ketones with labile chiral secondary C–F, C–Cl and C–Br bonds at the α-position were collectively obtained in high yields and with high ee values. Although the stereocenters of these products are liable to proceed racemization in the reaction process and a competitive racemic background reaction exists in the reaction system, the satisfactory results suggest the amazing feasibility and compatibility of this strategy. Given the convenient preparation of alkyl halides, we expect this method to be extensively utilized in the construction of diverse significant and, especially, complex chiral α-tertiary carbonyls and their variants, thus leveraging the broad applicability of enantioselective protonation in organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants from the NSFC (21672052) and Provincial Innovation Scientists and Technicians Troop Construction Projects Henan Provincial Science are gratefully acknowledged.Notes and references
- J. T. Mohr, A. Y. Hong and B. M. Stoltz, Nat. Chem., 2009, 1, 359 CrossRef CAS PubMed.
- For a selected example with amide enolates, see: E. Vedejs and A. W. Kruger, J. Org. Chem., 1998, 63, 2792 CrossRef CAS.
- For selected examples with silyl enol ethers, see: (a) K. Ishihara, S. Nakamura, M. Kaneeda and H. Yamamoto, J. Am. Chem. Soc., 1996, 118, 12854 CrossRef CAS; (b) T. Poisson, V. Gembus, V. Dalla, S. Oudeyer and V. Levacher, J. Org. Chem., 2010, 75, 7704 CrossRef CAS PubMed; (c) C. H. Cheon, O. Kanno and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 13248 CrossRef CAS PubMed; (d) S. Paladhi, Y. Liu, B. S. Kumar, M.-J. Jung, S. Y. Park, H. Yan and C. E. Song, Org. Lett., 2017, 19, 3279 CrossRef CAS PubMed.
- For selected examples with enol trifluoroacetates, see: (a) A. Claraz, J. Leroy, S. Oudeyer and V. Levacher, J. Org. Chem., 2011, 76, 6457 CrossRef CAS PubMed; (b) A. Yanagisawa, T. Sugita and K. Yoshida, Chem.–Eur. J., 2013, 19, 16200–16203 CrossRef CAS PubMed.
- For selected examples with ketenes, see: (a) B. L. Hodous, J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1999, 121, 2637 CrossRef CAS; (b) X. Dai, T. Nakai, J. A. C. Romero and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 4367 CrossRef CAS PubMed.
- For selected examples of catalytic enantioselective decarboxylative protonation, see: (a) M. A. Baur, A. Riahi, F. Hénin and J. Muzart, Tetrahedron: Asymmetry, 2003, 14, 2755 CrossRef CAS; (b) J. T. Mohr, T. Nishimata, D. C. Behenna and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 11348 CrossRef CAS PubMed; (c) C. Kingston and P. J. Guiry, J. Org. Chem., 2017, 82, 3806 CrossRef CAS PubMed.
- For selected examples of ketene disilyl/dithio acetals, see: (a) D. Uraguchi, N. Kinoshita and T. Ooi, J. Am. Chem. Soc., 2010, 132, 12240 CrossRef CAS PubMed; (b) J.-W. Lee and B. List, J. Am. Chem. Soc., 2012, 134, 18245 CrossRef CAS PubMed.
- For selected examples of catalytic enantioselective conjugate addition-protonation, see: (a) Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2006, 128, 3928 CrossRef CAS PubMed; (b) L. M. Repka, J. Ni and S. E. Reisman, J. Am. Chem. Soc., 2010, 132, 14418 CrossRef CAS PubMed; (c) B. Zhu, R. Lee, J. Li, X. Ye, S.-N. Hong, S. Qiu, M. L. Coote and Z. Jiang, Angew. Chem., Int. Ed., 2016, 55, 1299 CrossRef CAS PubMed.
- (a) G. Thomas, Medicinal Chemistry: An Introduction, Wiley, 2000 Search PubMed; (b) N. De Kimpe and R. Verhé, The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines, Wiley, 1990 Search PubMed; (c) V. Gouverneur and K. Müller, Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, Imperial College Press, London, 2012 CrossRef; (d) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708 CrossRef CAS.
- W. J. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396 CrossRef CAS PubMed.
- (a) M. Marigo, S. Bachmann, N. Halland, A. Braunton and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 5507 CrossRef CAS PubMed; (b) P. Kwiatkowski, T. D. Beeson, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 1738 CrossRef CAS PubMed.
- K. Shibatomi, K. Kitahara, N. Sasaki, Y. Kawasaki, I. Fujisawa and S. Iwasa, Nat. Commun., 2017, 8, 15600 CrossRef PubMed.
- L. Lin, X. Bai, X. Ye, X. Zhao, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2017, 56, 13842 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (c) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474 CrossRef CAS PubMed; (d) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735 CrossRef CAS PubMed; (c) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768 CrossRef CAS PubMed; (d) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed; (e) E. Meggers, Chem. Commun., 2015, 51, 3290 RSC; (f) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS.
- (a) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS PubMed; (b) J. Li, M. Kong, B. Qiao, R. Lee, X. Zhao and Z. Jiang, Nat. Commun., 2018, 9, 2445 CrossRef PubMed; (c) B. Qiao and Z. Jiang, ChemPhotoChem, 2018, 2, 703 CrossRef CAS.
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC.
- (a) S. H. Mashraqui and R. M. Kellogg, Tetrahedron Lett., 1985, 26, 1453 CrossRef CAS; (b) S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Phys. Chem., 1990, 94, 722 CrossRef CAS; (c) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed; (d) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (e) J. J. Devery III, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962 CrossRef; (f) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed; (g) K. Tahara and Y. Hisaeda, Green Chem., 2011, 13, 558 RSC; (h) G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342 CrossRef CAS PubMed; (i) C. D. McTiernan, S. P. Pitre, H. Ismaili and J. C. Scaiano, Adv. Synth. Catal., 2014, 356, 2819 CrossRef CAS; (j) P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084 CrossRef CAS PubMed; (k) T. McCallum, E. Slavko, M. Morin and L. Barriault, Eur. J. Org. Chem., 2015, 2015, 81 CrossRef CAS; (l) R. Matsubara, T. Shimada, Y. Kobori, T. Yabuta, T. Osakai and M. Hayashi, Chem.–Asian J., 2016, 11, 2006 CrossRef CAS PubMed; (m) C. Yang, F. Mehmood, T. L. Lam, S. L.-F. Chan, Y. Wu, C.-S. Yeung, X. Guan, K. Li, C. Y.-S. Chung, C.-Y. Zhou, T. Zou and C.-M. Che, Chem. Sci., 2016, 7, 3123 RSC; (n) P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083 RSC; (o) B. Michelet, C. Deldaele, S. Kajouj, C. Moucheron and G. Evano, Org. Lett., 2017, 19, 3576 CrossRef CAS.
- T. Maji, A. Karmakar and O. Reiser, J. Org. Chem., 2011, 76, 736 CrossRef CAS PubMed.
- (a) M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414 CrossRef CAS PubMed; (b) B. A. Sandoval, A. J. Meichan and T. K. Hyster, J. Am. Chem. Soc., 2017, 139, 11313 CrossRef CAS PubMed.
- (a) G. Wei, C. Zhang, F. Bureš, X. Ye, C.-H. Tan and Z. Jiang, ACS Catal., 2016, 6, 3708 CrossRef CAS; (b) Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083 CrossRef CAS PubMed; (c) Y. Liu, X. Liu, J. Li, X. Zhao, B. Qiao and Z. Jiang, Chem. Sci., 2018, 9, 8094 RSC; (d) X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao and Z. Jiang, Org. Lett., 2018, 20, 6298 CrossRef CAS PubMed; (e) K. Cao, S. M. Tan, R. Lee, S. Yang, H. Jia, X. Zhao, B. Qiao and Z. Jiang, J. Am. Chem. Soc., 2019, 141, 5437 CrossRef CAS.
- S. Chen, J. Pan, Y. Wang and Z. Zhou, Eur. J. Org. Chem., 2014, 7940 CrossRef CAS.
- (a) E. Arceo, I. D. Jurberg, A. Alvarez-Fernandez and P. Melchiorre, Nat. Chem., 2013, 5, 750 CrossRef CAS PubMed; (b) M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868 CrossRef CAS PubMed; (c) G. Filippini, M. Silvi and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 4447 CrossRef CAS PubMed; (d) D. Mazzarella, G. E. M. Crisenza and P. Melchiorre, J. Am. Chem. Soc., 2018, 140, 8439 CrossRef CAS PubMed; (e) X. Huang, J. Li, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454 CrossRef CAS PubMed.
- The ee was measured by directly using the crude product and employing the pure product purified by high-performance thin layer chromatography, in which UV detection was conducted at 254, 230 and 210 nm. Note that the stereocenter of 6a was stable after purification, even in isopropanol as the solvent.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1590512 and 1840318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02000d |
| ‡ The authors made equal contributions. |
| This journal is © The Royal Society of Chemistry 2019 |