
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A thiochromenone antibiotic derived from the Pseudomonas quinolone signal selectively targets the Gram-negative pathogen Moraxella catarrhalis†
Dávid
Szamosvári‡
a,
Tamara
Schuhmacher‡
b,
Christof R.
Hauck
 *b and
Thomas
Böttcher
*a
*b and
Thomas
Böttcher
*a
aDepartment of Chemistry, Konstanz Research School Chemical Biology, Zukunftskolleg, University of Konstanz, 78457 Konstanz, Germany. E-mail: Thomas.Boettcher@uni-konstanz.de
bDepartment of Biology, University of Konstanz, 78457 Konstanz, Germany. E-mail: Christof.Hauck@uni-konstanz.de
First published on 23rd May 2019
Abstract
The Pseudomonas quinolone signal (PQS) is an important quorum sensing signal of the pathogen Pseudomonas aeruginosa. We discovered an additional activity of PQS as a narrow spectrum antibiotic. Exploiting the privileged structure of PQS by the synthesis of heteroatom-substituted analogues led to a class of 2-alkyl-3-hydroxythiochromen-4-ones with highly potent antibiotic activity against the nasopharyngeal pathogen Moraxella catarrhalis. Synthetic optimization resulted in minimum inhibitory concentrations in the nanomolar range even for clinical isolates of M. catarrhalis. Surprisingly, the growth of other human pathogens and commensals, including closely related Moraxella species, was not inhibited, indicating exceptional species selectivity. Mechanistic studies revealed that the antibiotic was bactericidal and likely inhibits a target in the primary energy metabolism causing rapid depletion of the cellular ATP pool.
Antibiotic-resistant pathogens limit the treatment of infectious diseases and are a major threat for human health. Thus, the development of novel antibiotics and expansion of the target spectrum are of utmost urgency.1 However, the discovery of new classes of potent antibiotics is rare, especially against Gram-negative bacteria, which are intrinsically harder to kill and often use multiple redundant resistance mechanisms.2 We have recently investigated the class of 2-alkyl-4-quinolone N-oxides of Pseudomonas aeruginosa and found an unsaturated compound as a major antibiotic weapon against Staphylococcus aureus.3,4 Other quinolones like the Pseudomonas quinolone signal (PQS) and its biosynthetic precursor 2-heptyl-4(1H)-quinolone (HHQ) are quorum sensing signals that coordinate the expression of virulence factors in P. aeruginosa.5 However, additional roles of these compounds have also been reported including growth inhibitory effects of HHQ against S. aureus and Vibrio species,6,7 as well as trapping of ferric iron8,9 and the formation of outer membrane vesicles by PQS.10,11 Activity-based protein profiling with photoreactive versions of HHQ and PQS revealed numerous putative targets and protein interactions in P. aeruginosa which indicate additional roles beyond classical signaling.12 In addition, PQS has been implicated in impairing the human immune response.13 We reasoned that PQS might have further unexplored effects in interspecies interactions and thus investigated potential antibiotic effects of PQS on various Gram-negative pathogens, which may compete with P. aeruginosa in the human body.
Here we report a highly selective antibacterial effect of the quorum sensing signal PQS of Pseudomonas aeruginosa against the human pathogen Moraxella catarrhalis, which we further exploited by organic synthesis. We hereby developed a new class of antibiotics based on a thiochromenone scaffold that exhibited submicromolar activity and unprecedented species selectivity by targeting the primary metabolism of M. catarrhalis.
We synthesized PQS (1) by the cyclization of 2-oxononyl 2′-aminobenzoate14 and tested it on the growth of 10 different species (Fig. S1†). Enterobacteria such as Klebsiella pneumoniae, Enterococcus faecalis, and Escherichia coli appeared to be unaffected by PQS concentrations of up to 50 μM. In contrast, the growth of Moraxella catarrhalis and Neisseria meningitidis was significantly impaired at concentrations as low as 5 μM (Fig. 1).
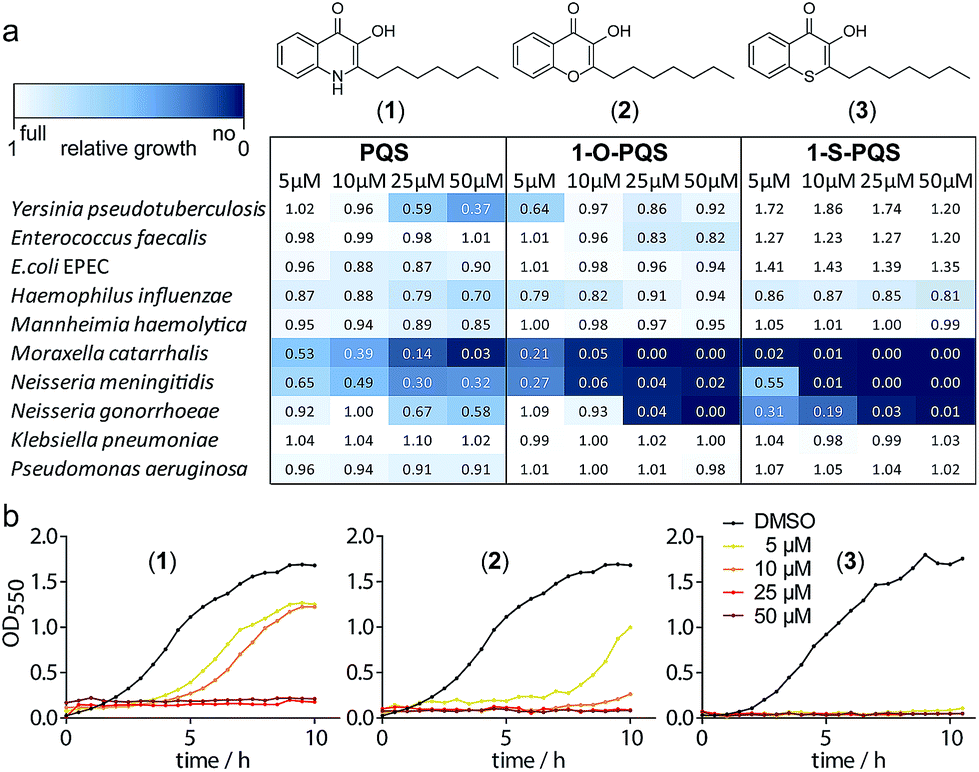 | ||
| Fig. 1 Antibacterial activity of the natural product PQS (1) and the synthetic derivatives (2) and (3) against Gram-negative bacteria. (a) Integrals of growth curves color-coded from dark blue (no growth) to white (full growth). (b) Example growth curves for the three compounds with M. catarrhalis ATCC 25238. | ||
In cultures of P. aeruginosa, PQS is produced in multi-micromolar quantities,15 suggesting that inhibiting the growth of certain competing species may be one additional, signaling-unrelated role of PQS. Next, we aimed to exploit this surprisingly selective activity of native PQS against certain Gram-negative bacteria by evolving a scaffold of this privileged structure using synthetic chemistry. In the first step, we generated heteroatom-substituted PQS derivatives with a 3-hydroxychromen-4-one and a 3-hydroxythiochromen-4-one scaffold. Chromen-4-one (2a) was synthesized by esterification of 2-hydroxyacetophenone with octanoyl chloride followed by Baker–Venkataraman rearrangement.16 1-O-PQS (2) was obtained after epoxidation of chromen-4-one (2a) with tert-butyl hydroperoxide and subsequent arene oxide rearrangement.17 The related 1-S-PQS (3) was synthesized from thiophenol and trans-2-decenoic acid in a superacid-catalyzed alkylation followed by a cyclic acylation reaction to give thiochroman-4-one (3b),18 which was subsequently oxidized by nitrosation with isoamyl nitrite and ensuing oximation and oxime hydrolysis (Scheme 1).19
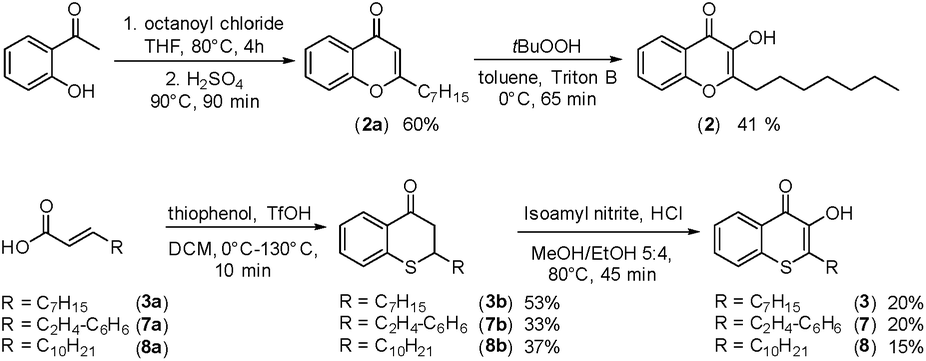 | ||
| Scheme 1 Synthesis of chromenone (2) and thiochromenones (3, 7, and 8). | ||
Alkylated 3-hydroxychromen-4-one (2) and 3-hydroxythiochromen-4-one (3) exhibited both significantly stronger growth inhibition compared to PQS and an identical species spectrum (Fig. S1†). 3-Hydroxythiochromen-4-one (3) was hereby the most active compound and completely inhibited the growth of M. catarrhalis at concentrations down to 5 μM (Fig. 1). We therefore synthesized a small focused library of substituted thiochromen-4-one derivatives. To test whether 3-OH was essential for the activity, we generated the HHQ derived thiochromen-4-one compound (4).20 Interestingly, the lack of the 3-OH group resulted in a severe drop in activity against M. catarrhalis so further refinement was restricted to the 3-hydroxythiochromen-4-one scaffold (Fig. 2a and b). Since Pseudomonas quinolone N-oxides are known respiratory chain inhibitors,3,21,22 we investigated the possibility that (3) is activated in situ by oxidation to a sulfoxide, which is isosteric with N-oxide. We thus synthesized thiochromen-4-one 1-oxide (5) and 1,1-dioxide (6) by treating (3) with mCPBA (Scheme 2). Both compounds with an oxidized sulfur exhibited strongly decreased activity against M. catarrhalis suggesting that (3) is not activated by 1-S-oxidation (Fig. 2b). We next explored the effects of variations on substitution and electronic changes to the thiochromen-4-one ring system. 2-Phenethyl (7) and 2-decyl (8) derivatives were synthesized using the same strategy as for (3). To obtain the thiopyranopyridin-4-ones (9 and 10) we used the described oxidation method on 2-heptyl-2,3-dihydro-4H-thiopyrano[2,3-b]pyridin-4-one (9c) and 2-heptyl-2,3-dihydro-4H-thiopyrano[3,2-c]pyridin-4-one (10c), which were synthesized according to Kobayashi et al. (Scheme 2).23 Derivatives 11–14 with substitutions at the aromatic region were not accessible by this synthetic approach. We therefore used copper catalyzed conjugate addition of heptylmagnesium bromide on the corresponding thiochromenones (11c–14c) as described by Luo et al.24 to obtain 2-heptylthiochroman-4-ones (11d–14d) in four steps. Riley oxidation with SeO2 gave 2-heptyl-3-hydroxythiochromen-4-ones (11–14) with less byproducts compared to oxidation with isoamyl nitrite (Scheme 2).
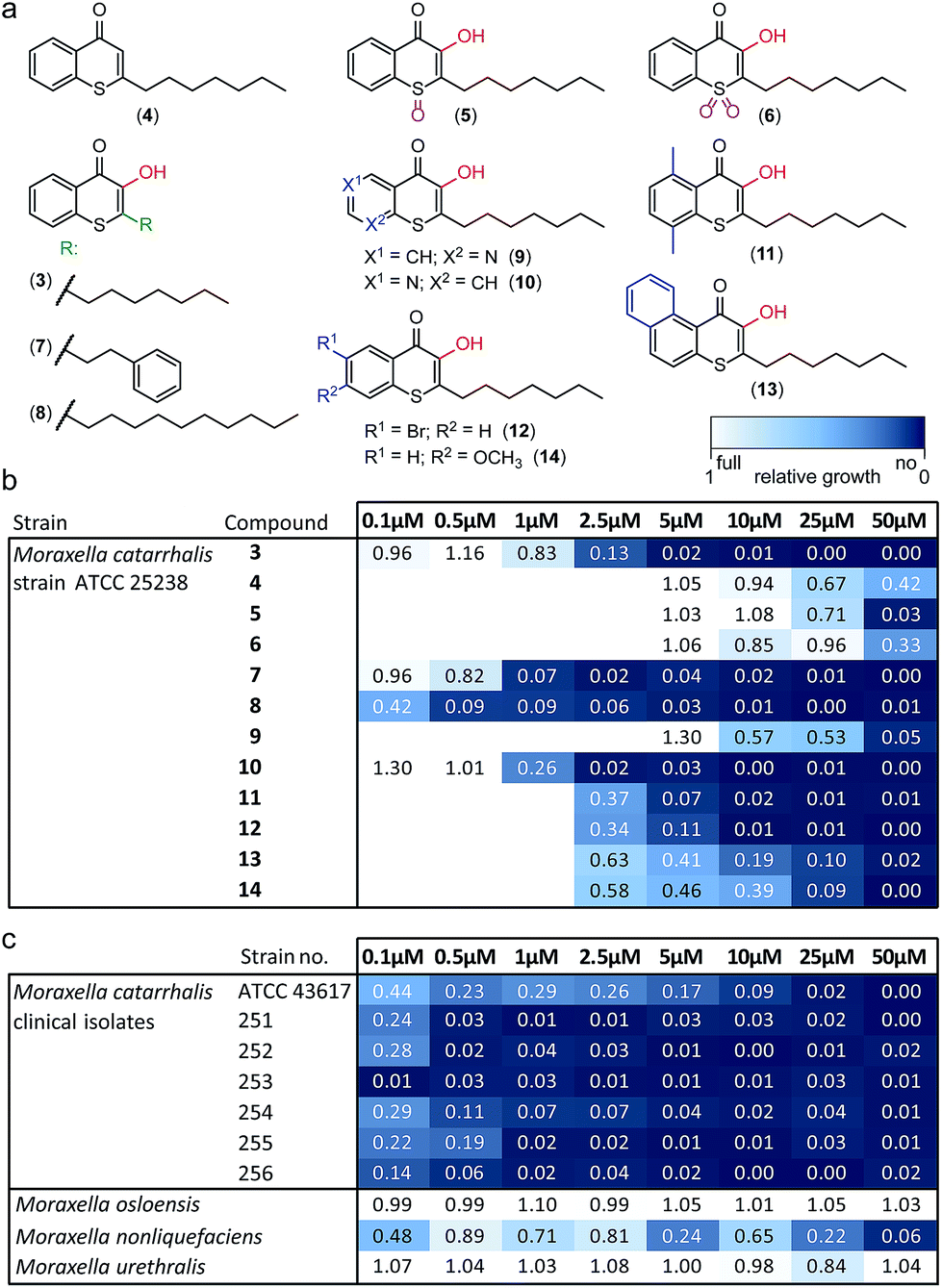 | ||
| Fig. 2 Derivatives of 1-S-PQS and their activity against Moraxella. (a) Focused synthetic library. (b) Antibacterial activity against M. catarrhalis ATCC 25238. (c) Activity of compound (8) against clinical strains and commensal Moraxella species. Integrals of growth curves color-coded from dark blue (no growth) to white (full growth). | ||
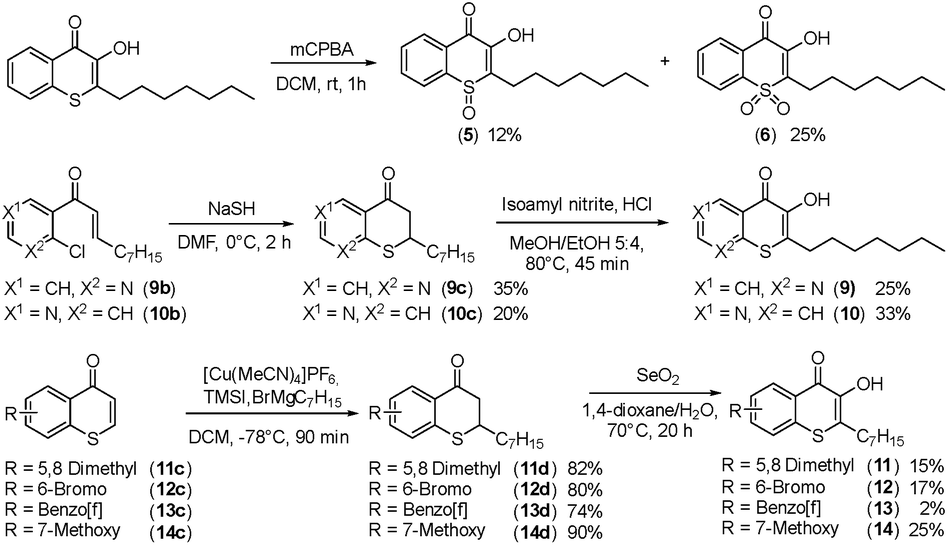 | ||
| Scheme 2 Synthesis of thiochromenones (5, 6 and 9–14). | ||
The phenethyl group of (7) increased the activity by more than two-fold compared to the parent compound (3). Strikingly, the decyl chain (8) further boosted the activity against M. catarrhalis by a factor of ten, while the thiopyranopyridin-4-one (10) slightly increased and (9) nearly completely abrogated the activity as compared to (3). Aromatic ring substitutions (11 and 12) were tolerated with a minor loss of activity, while (13) and (14) led to a four- to ten-fold drop in activity (Fig. 2b and S2†). None of the compounds displayed major activities against other nasopharyngeal pathogens such as Haemophilus influenzae or Streptococcus pyogenes (Fig. S3†). Since the most potent compound (8) in this study displayed unprecedented activity against the human pathogen M. catarrhalis in the nanomolar range with a MIC of only 0.5 μM (∼0.16 μg mL−1), we investigated if this compound also inhibited the growth of more problematic clinical isolates ranging from blood cultures to wound swabs and tracheal exudates. Indeed all clinical strains were inhibited with MIC values ranging from <0.1 μM to 1 μM (Fig. 2c and S4†). The antibiotic activity of (8) was hereby highly selective for M. catarrhalis and even the closely related commensal species Moraxella osloensis and Moraxella urethralis did not respond to the antibiotic at concentrations of up to 50 μM (Fig. 2c and S5†).
This unexpected selectivity indicates an extremely narrow-spectrum antibiotic, which is rare especially for Gram-negative pathogens. In order to test the cytotoxicity of our lead compound (8) against eukaryotic cells, we measured the metabolic activity and membrane integrity of A549 human lung carcinoma cells.
Adverse effects on metabolism were only observed at around 50 μM and loss of membrane integrity was found only after a prolonged incubation of 2–3 days at 50 μM, indicating that the sensitivity of eukaryotic cells is two orders of magnitude lower than the MIC against M. catarrhalis (Fig. S6a and b†). A similarly low sensitivity was observed for human kidney cells (Fig. S6c†). These properties make our lead compound an interesting and highly potent drug candidate for treating infections caused by M. catarrhalis including middle-ear infections in children as well as pneumonia, endocarditis, septicemia, and meningitis in adults.25Moraxella is also one of the few pathogens associated with exacerbations of chronic obstructive pulmonary disease, a pathologic lung condition of growing global significance.26 We thus aimed to further investigate the molecular mechanism by which the compound inhibits the growth of M. catarrhalis.
Since the 3-OH group was essential for the activity and the natural product PQS has a role in ferric iron trapping, we investigated the possible interference of (8) with Moraxella's iron uptake. While pre-incubation of (8) in an equimolar ratio with ferric chloride abrogated antibiotic activity, supplementing bacterial cultures separately with (8) and ferric iron restored growth inhibition (Fig. 3c). These results suggest that the catechol moiety of 3-hydroxythiochromene-4-one may be important for interactions with the cellular target and consequently a pre-formed complex with ferric iron would prevent target binding. The structurally related iron chelator deferiprone had no effect on the growth of M. catarrhalis even at 100 μM (Fig. 3c), confirming that iron chelation was not responsible for the antibiotic activity. Measuring membrane polarization using a fluorescent indicator dye showed no significant influence of compound (8) within two hours, while the proton ionophore CCCP or the cytochrome c reductase inhibitor antimycin A abrogated the membrane potential within 15 min (Fig. S7†). Also, no increase in reactive oxygen species (ROS) was detected (Fig. S8†). Scanning electron micrographs of M. catarrhalis cells treated with and without our antibiotic (8) did not show visible morphological differences compared to a known inhibitor of cell wall biosynthesis, suggesting that the membrane and cell wall of the bacteria were not targets of the compound (Fig. 3a). To investigate if DNA, RNA, or protein biosynthesis were targeted by the compound, we analyzed the incorporation of 3H radiolabeled precursors of the corresponding biopolymers. Control antibiotics known to interfere with DNA replication, transcription, or protein biosynthesis selectively prevented the incorporation of the radiolabeled precursors. In the samples treated for 40 min with (8), incorporation of the corresponding 3H labelled precursors was inhibited for each of the three biopolymers (Fig. 3b), indicating an effect on the primary metabolism of the cell, which ultimately affects biosynthesis of all macromolecules.
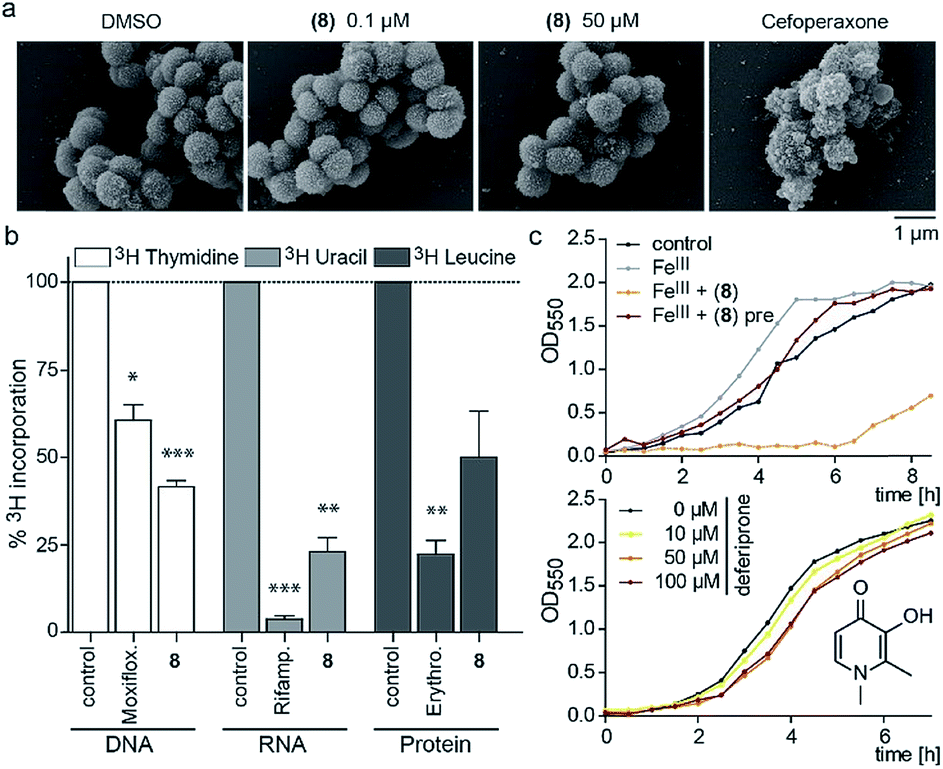 | ||
| Fig. 3 Morphological and physiological responses of M. catarrhalis ATCC 25238 to compound (8). (a) Scanning electron micrographs of cells after treatment with (8) in comparison to a DMSO control and the cell wall inhibitor cefoperaxone. (b) Radiolabelling experiments with 3H-thymidine (DNA), 3H-uridine (RNA), 3H-leucine (protein) and antibiotics moxifloxacin, rifampicin, and erythromycin as positive controls. (c) Upper panel: growth of M. catarrhalis with ferric iron (FeIII, 5 μM) and 5 μM compound (8); pre: pre-incubation. Lower panel: effect of the iron chelator deferiprone on the growth of M. catarrhalis. | ||
To investigate the survival rate of cells, we challenged the bacteria with (8) for 40 min and 3 h and then plated dilutions on antibiotic-free agar for counting of colony forming units (CFUs). 40 min of exposure already severely reduced the viability of the cells (Fig. 4a and S9†), which led us to conclude that (1) our compound is bactericidal and not bacteriostatic and (2) the antibiotic inhibits a pathway, which causes rapid cell death. Hereby the average colony size on the plates considerably decreased when pre-incubated with higher concentration of (8) (Fig. 4b). Small-colony morphology is typically associated with impaired electron transport and depletion of ATP.27 Indeed, the overall metabolic activity of M. catarrhalis as measured by the reduction of a tetrazolium salt was inhibited (Fig. 4c). We thus investigated if cellular ATP concentrations were affected by our antibiotic using a luciferase assay. After 20 min of exposure, ATP levels in M. catarrhalis had already dropped significantly depending on the concentration of (8). In contrast, ATP levels in M. osloensis, a closely related microbe not sensitive to (8), did not decrease (Fig. 4d). These results suggest that our thiochromenone antibiotic (8) inhibits a pathway in the energy metabolism of M. catarrhalis in a highly effective manner with remarkable species specificity. Continuous cultivation of M. catarrhalis ATCC 25238 at 0.1 μM of compound (8) for 10 days did not result in decreased susceptibility, suggesting that the compound was not prone to rapid resistance development (Fig. S10†).
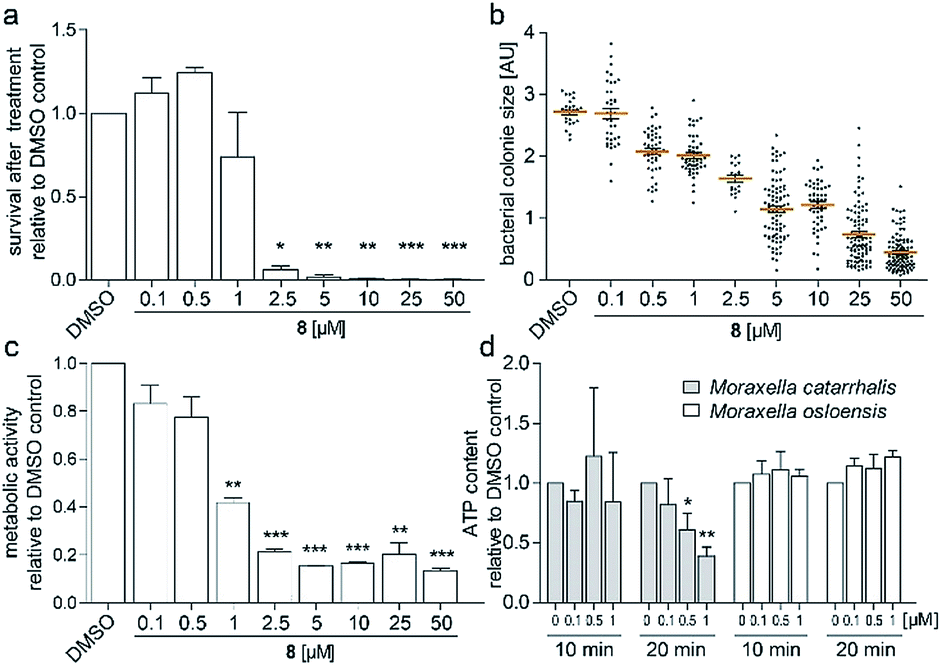 | ||
| Fig. 4 (a) Survival of M. catarrhalis ATCC 25238 after treatment for 40 min with (8) shown by the number of CFUs on agar relative to the DMSO control. (b) Relative sizes of the colonies quantified in (a). (c) Inhibition of the metabolic activity of M. catarrhalis after 3 h of incubation with (8). (d) Rapid concentration dependent depletion of ATP after 20 min of incubation with (8) for M. catarrhalis but not for M. osloensis. | ||
3-Hydroxythiochromen-4-ones in this study constitute a novel class of highly potent narrow spectrum antibiotics. Related quinolones like nalidixic acid or fluoroquinolones have comparable potency but lack this species selectivity.28
While these quinolone antibiotics are topoisomerase inhibitors, 3-hydroxythiochromen-4-ones did not selectively inhibit DNA replication and further mechanistic investigations point to primary energy metabolism as the target. This new class of antibiotics may pave the way for customized drugs against infectious diseases or serve as chemical precision tools for microbiome research.
In conclusion, we were able to exploit the selective antibacterial activity of the quorum sensing signal PQS against the Gram-negative pathogen Moraxella catarrhalis by developing a new class of highly potent thiochromenone antibiotics with unprecedented species selectivity. The antibiotic targets primary energy metabolism causing rapid depletion of the cellular ATP pool. Our results demonstrate that (1) quorum sensing signals like PQS can have roles beyond classical signaling and (2) even moderate activities of these natural products can be exploited by scaffold engineering and lead to highly potent antibiotics with unprecedented customized species selectivity against medically relevant Gram-negative pathogens.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Prof. Dr Andreas Marx and his group for their generous support. This work was funded by the Emmy Noether program (DFG), EU FP7 Marie Curie ZIF, SFB969 project C06, Fonds der Chemischen Industrie, DFG Ha2586/10-1, and a KoRS-CB PhD fellowship (DS). We thank Johannes Wirth and Mario Rapp for contributions in synthesis, Prof. Dr Reinhard Berner (TU Dresden) for providing Moraxella strains 251-256, Dr Norbert Kunze and Dr Sascha Beneke for help with scintillation, and Dr Michael Laumann and Dr Paavo Bergmann from the electron microscopy center as well as the FlowKon core facility for technical support.Notes and references
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29–40 CrossRef CAS PubMed.
- G. Taubes, Science, 2008, 321, 356–361 CrossRef CAS PubMed.
- D. Szamosvári and T. Böttcher, Angew. Chem., Int. Ed. Engl., 2017, 56, 7271–7275 CrossRef PubMed.
- D. Szamosvári and T. Böttcher, Synlett, 2018, 29, 542–547 CrossRef.
- H. Huse and M. Whiteley, Chem. Rev., 2011, 111, 152–159 CrossRef CAS PubMed.
- S. J. Wratten, M. S. Wolfe, R. J. Andersen and D. J. Faulkner, Antimicrob. Agents Chemother., 1977, 11, 411–414 CrossRef CAS PubMed.
- F. J. Reen, M. J. Mooij, L. J. Holcombe, C. M. McSweeney, G. P. McGlacken, J. P. Morrissey and F. O'Gara, FEMS Microbiol. Ecol., 2011, 77, 413–428 CrossRef CAS PubMed.
- F. Bredenbruch, R. Geffers, M. Nimtz, J. Buer and S. Haussler, Environ. Microbiol., 2006, 8, 1318–1329 CrossRef CAS PubMed.
- S. P. Diggle, S. Matthijs, V. J. Wright, M. P. Fletcher, S. R. Chhabra, I. L. Lamont, X. Kong, R. C. Hider, P. Cornelis, M. Camara and P. Williams, Chem. Biol., 2007, 14, 87–96 CrossRef CAS PubMed.
- L. M. Mashburn and M. Whiteley, Nature, 2005, 437, 422–425 CrossRef CAS PubMed.
- L. Mashburn-Warren, J. Howe, K. Brandenburg and M. Whiteley, J. Bacteriol., 2009, 191, 3411–3414 CrossRef CAS PubMed.
- R. Dandela, D. Mantin, B. F. Cravatt, J. Rayo and M. M. Meijler, Chem. Sci., 2018, 9, 2290–2294 RSC.
- J. Lin, J. Cheng, Y. Wang and X. Shen, Front. Cell. Infect. Microbiol., 2018, 8, 230 CrossRef PubMed.
- P. Hradil, J. Hlavac and K. Lemr, J. Heterocycl. Chem., 1999, 36, 141–144 CrossRef CAS.
- C. Florez, J. E. Raab, A. C. Cooke and J. W. Schertzer, mBio, 2017, 8(4), 1–13 CrossRef PubMed.
- S. B. Abdel Ghani, P. J. Mugisha, J. C. Wilcox, E. A. M. Gado, E. O. Medu, A. J. Lamb and R. C. D. Brown, Synth. Commun., 2013, 43, 1549–1556 CrossRef CAS.
- L. Duque, C. Zapata, B. Rojano, B. Schneider and F. Otalvaro, Org. Lett., 2013, 15, 3542–3545 CrossRef CAS PubMed.
- H. Vaghoo, G. K. Prakash, A. Narayanan, R. Choudhary, F. Paknia, T. Mathew and G. A. Olah, Org. Lett., 2015, 17, 6170–6173 CrossRef CAS PubMed.
- M. Ferrali, S. Bambagioni, A. Ceccanti, D. Donati, G. Giorgi, M. Fontani, F. Laschi, P. Zanello, M. Casolaro and A. Pietrangelo, J. Med. Chem., 2002, 45, 5776–5785 CrossRef CAS PubMed.
- D. Szamosvári, F. V. Reichle, M. Jureschi and T. Böttcher, Chem. Commun., 2016, 52, 13440–13443 RSC.
- J. W. Lightbown and F. L. Jackson, Biochem. J., 1956, 63, 130–137 CrossRef CAS PubMed.
- A. I. Zatsman, H. Zhang, W. A. Gunderson, W. A. Cramer and M. P. Hendrich, J. Am. Chem. Soc., 2006, 128, 14246–14247 CrossRef CAS PubMed.
- K. Kobayashi and A. Imaoka, Helv. Chim. Acta, 2013, 96, 624–632 CrossRef CAS.
- S. H. Luo, L. Meng, Q. X. Yang and J. Wang, Synlett, 2018, 29, 2071–2075 CrossRef CAS.
- C. M. Verduin, C. Hol, A. Fleer, H. van Dijk and A. van Belkum, Clin. Microbiol. Rev., 2002, 15, 125–144 CrossRef PubMed.
- S. Sethi and T. F. Murphy, N. Engl. J. Med., 2008, 359, 2355–2365 CrossRef CAS PubMed.
- R. A. Proctor, A. Kriegeskorte, B. C. Kahl, K. Becker, B. Loffler and G. Peters, Front. Cell. Infect. Microbiol., 2014, 4, 99 Search PubMed.
- S. Heeb, M. P. Fletcher, S. R. Chhabra, S. P. Diggle, P. Williams and M. Camara, FEMS Microbiol. Rev., 2011, 35, 247–274 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01090d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |