
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ real-time tracing of hierarchical targeting nanostructures in drug resistant tumors using diffuse fluorescence tomography†
Qianqian
Guo‡
a,
Yangyun
Wang‡
b,
Limin
Zhang‡
c,
Peng
Zhang
 d,
Yunjian
Yu
a,
Yanqi
Zhang
c,
Chaoxing
Li
a,
Shaoyi
Jiang
*d and
Xinge
Zhang
*a
d,
Yunjian
Yu
a,
Yanqi
Zhang
c,
Chaoxing
Li
a,
Shaoyi
Jiang
*d and
Xinge
Zhang
*a
aKey Laboratory of Functional Polymer Materials of Ministry of Education, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: zhangxinge@nankai.edu.cn
bState Key Laboratory of Radiation Medicine and Protection, School for Radiological & Interdisciplinary Sciences (RAD-X), Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou 215123, China
cCollege of Precision Instrument and Optoelectronics Engineering, Tianjin University, Tianjin 300072, China
dDepartment of Chemical Engineering, University of Washington, Seattle, WA 98195, USA. E-mail: sjiang@uw.edu
First published on 2nd July 2019
Abstract
Nanoparticles that respond to specific endogenous or exogenous stimuli in tumor tissues are actively being developed to address multidrug resistance owing to multiple advantages, including a prolonged circulation time, enhanced permeability and retention effect, and superior cellular uptake. Although some exciting results have been obtained, existing nanoparticles have limited routes to overcome the drug resistance of tumor cells; this limitation results in a failure to ablate resistant tumors via intravenous administration. To resolve this dilemma, we developed a smart theranostic nanoplatform with programmable particle size, activatable target ligands and in vivo multimodal imaging. This nanoplatform, which includes stealth zwitterionic coating, was shown to be quickly trapped in tumor tissue from the blood circulation within 5 min. Subsequently, the targeting moieties were activated in response to the acidic tumor microenvironment by triggering the zwitterionic shell detachment, driving the peeled nanoparticles to penetrate into tumor cells. These smart nanoparticles completely inhibited drug-resistant tumor growth and did not cause any damage to normal organ tissues in live animals. The designed nanoplatforms simultaneously acted as a nanoprobe for fluorescence imaging. Moreover, we also used noninvasive pharmacokinetic diffuse fluorescence tomography (DFT) to dynamically monitor and in situ real-time trace the nanoplatforms' behavior throughout the entire tumor in live animals. The nanoplatforms enabled rapid drug accumulation and deep penetration throughout the entire tumor. The rate of drug accumulation after the administration of nanoplatforms was five-fold higher compared with that after the administration of the free drug, which resulted in increased drug delivery efficiency and improved antitumor efficacy. Collectively, this hierarchical vehicle design provides promising insights for the development of theragnosis for multidrug resistant tumors.
Multidrug resistance (MDR) has become a major obstacle to the success of cancer chemotherapy. The major mechanism of MDR involves the P-glycoprotein (P-gp), which is overexpressed in cancer cells and is able to pump various chemotherapeutics out of the cells, resulting in a reduced intracellular drug concentration with a limited therapeutic effect.1 Tumor-targeting drug delivery nanoparticles have been actively developed to overcome MDR,2 owing to their preferential accumulation at tumor sites due to enhanced permeability and retention (EPR) and active targeting effects.3 In contrast to small drug molecules, these nanoparticles are internalized into cells via endocytosis, and thus they are not affected by the overexpression of the P-gp efflux pump on the MDR cell surface.2d However, the remarkable abnormality and complexity of the tumor microenvironment often pose barriers to the supply, penetration and distribution of nanoparticles.4 To address these predicaments, taking into consideration the intra-tumor tissue accumulation, retention and penetration of drugs, an emerging delivery strategy was developed wherein vehicles respond to the specific endogenous or exogenous stimuli within tumor tissues.5 Although exciting results have been obtained previously, existing nanoparticles have limited routes to inhibit the resistance of tumor cells, and this limitation results in the failure to ablate resistant tumors via intravenous administration. The majority of these self-assembled nanostructures encounter problems with drug-loading stability, which is influenced by the in vivo environment, in which blood components can act as competing drug acceptors,6 and interactions between polymeric nanoparticles and blood components can lead to drug leakage in the circulation.7 Therefore, it is vital to develop a nanoscale delivery system for improving delivery efficiency and, eventually, therapeutic efficacy.
In addition to the efficient delivery of anticancer therapeutics to tumor cells, the capacity for in vivo imaging to trace drug distribution and accumulation in situ in real-time is also a desired property of theragnostic systems. Although the conventional labeling strategy can track the collective and metabolic behaviors of a nanocarrier in planar hierarchy, it is not currently possible to visualize the real-time trail throughout the entire tumor. Only limited visualization of the surface of the tumor is possible due to the limited spatial resolution and long imaging time. It is indispensable to dynamically trace nanocarriers throughout the entire tumor to understand their behavior at the targeted tissue. In this study, to complement the traditional radioisotopic tracing methods and realize noninvasive in situ nanocarrier monitoring in vivo, pharmacokinetic diffuse fluorescence tomography (DFT) was exploited for the first time to dynamically monitor and trace the nanocarrier behavior throughout the entire tumor in live animals.
Based on the aforementioned considerations, we proposed a smart hierarchical targeting theranostic nanoplatform utilizing dynamic covalent chemistry with a programmable particle size and activatable surface ligands to decrease drug leakage into the circulation, enhance the permeability of cargos and boost penetration into tumors. Boronate esters, which are formed by the reaction of phenylboronic acid (PBA) with diols, are stable under physiological conditions but they easily dissociate in the acidic tumor microenvironment.8 This chemistry was adopted not only to form a sheddable zwitterionic shell to stabilize the nanovehicles in the blood circulation, but also to act as an acid-responding switch to decorticate the stealth corona in the tumor tissue. When the switch is “on”, the zwitterionic shell is pared to release a much smaller core to facilitate tissue penetration. Simultaneously, the targeting ligands on the peeled nanoparticle surface are also activated to enhance cellular internalization. The intratumoral pharmacokinetics of the designed nanocarriers were explored using a real-time, noninvasive and radiation-free imaging technology at the spatial level. Simultaneously, BODIPY was also used to trace the biodistribution of nanocarriers in vivo via fluorescent imaging to supplement this DFT technology. A multidrug resistant tumor model was used to further evaluate the mechanism and treatment efficiency of the designed smart theragnostic system.
A nanoplatform for use as an anticancer drug vehicle was fabricated by the self-assembly of sophisticated copolymers, namely boron-dipyrromethene dye (BODIPY)-conjugated poly(carboxybetaine acrylate)-b-poly(2-(acrylamido) glucopyranose) (pCBAA-b-pAGA) and poly(2-lactobionamidoethyl methacrylate-b-poly(3-acrylamidophenylboronic acid)) (pLAMA-b-pAAPBA). Two block fluorescent copolymers were prepared by RAFT polymerization (Scheme S1†) and were subsequently characterized (Fig. S1–8 and Table S1†). Then, the two copolymers were self-assembled into hierarchical nanostructures via the hydrophilic–hydrophobic interaction and the phenylboronic acid–polysaccharide interaction. Particle size greatly influences the circulation time, tumor accumulation and penetration of nanocarriers.2e,9 It is generally accepted that nanoparticles with a diameter of more than 400 nm exhibit a superior circulating half-life and less extravasation from the leaky vasculature into the tumor interstitium, while smaller nanoparticles in the range of 10–100 nm can achieve higher tumor penetration owing to the reduced steric and diffusion hindrance.10 Consequently, an ideal drug delivery system should exhibit size-switching behavior from large to small in response to endogenous or exogenous stimuli within tumor tissues. Such a strategy has been demonstrated to be effective for achieving enhanced drug retention, penetration, cellular internalization and nuclear uptake within tumors.11 Generally, both asialoglycoprotein receptor (ASGP-R) and sialic acid (SA) are overexpressed on the membrane of cancer cells, which facilitates the active targeting via the specific LAMA-ASGP-R and PBA-SA interactions. Based on this principle, we designed a programmable size-switching nanoplatform in a fairly simple manner that employs a large size within the circulatory system to decrease drug leakage in circulation to efficiently enhance tumor accumulation, and then switches to a smaller size in the tumor tissue to promote drug penetration and cellular uptake (Scheme 1). The “switch” is realized by the incorporation of dynamic boronate ester bonds. At a pH of 7.4, pCBAA-b-pAGA and pLAMA-b-pAAPBA were shown to self-assemble into nanoparticles, abbreviated as pCA/pLB nanoparticles. These nanoparticles consist of a superhydrophilic zwitterionic pCBAA/pLAMA shell, a cross-linked boronate ester layer and a hydrophobic pAAPBA core. As previously mentioned, each block was designed to endow the nanocarrier with a specific desired function. The zwitterionic pCBAA stabilizes the nanostructures and increases their half-life in the circulatory system, the ligands pLAMA and pAAPBA facilitate tumor-targeted cell internalization, and the pAGA cross-linked with pAAPBA acts as the acid-responding switch in the acidic tumor microenvironment.
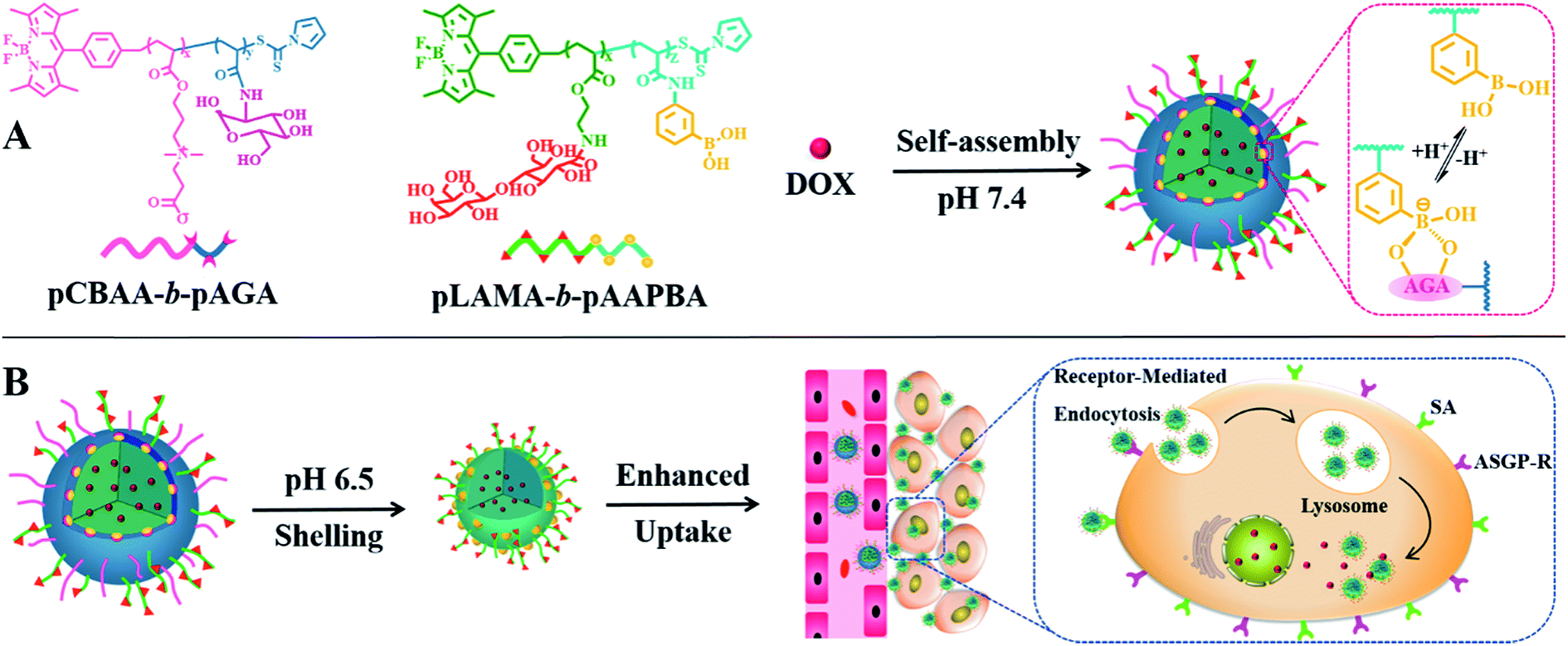 | ||
| Scheme 1 (A) Self-assembly of pCBAA-b-pAGA and pLAMA-b-pAAPBA copolymers into nanoparticles in phosphate buffered saline (PBS) at pH 7.4; (B) shell detachment and drug release in extracellular/intracellular environments. | ||
The fluorescence emission and UV-vis absorption spectra of BODIPY dye-labeled pCA/pLB nanoparticles were recorded (Fig. S9†), and the excitation/emission spectra presented a well-defined bandwidth with a visible absorption maximum at 506 nm and emission maximum at 523 nm. To evaluate the effect of the copolymer composition on the particle properties, three nanoparticles with different PBA contents (named pCA/pLB Np1, pCA/pLB Np2 and pCA/pLB Np3) were prepared (Table S2†). Dynamic light scattering (DLS) was used to demonstrate that the majority of the particles had a size in the range of 110–170 nm with a narrow polydispersity index (Table S2 and Fig. S10†). The zeta potential was slightly negative (Table S2†), which may contribute to enhanced biocompatibility and the avoidance of undesirable clearance by the reticuloendothelial system.12 The particle size exhibited no significant variation in serum within 48 h of incubation, indicating that the nanostructures are stable in complex blood medium (Fig. S11†). To enable a larger core size to increase the drug loading capacity, pCA/pLB Np3 was chosen for the following study.
To demonstrate their size-switching behavior (Fig. 1A), pCA/pLB Np3 was treated with pH 7.4 and 6.5 buffers before being subjected to measurements. The particle size, as measured by DLS at pH 7.4, was mainly approximately 165 nm (Fig. 1B). The shape was spherical, and the polydispersity was narrow (Fig. 1D). When treated with pH 6.5 buffer, the nanoparticle size decreased to ∼48 nm (Fig. 1C), confirmed by both DLS and TEM (Fig. 1C and E). The 1H NMR spectra (Fig. S12†) showed less pCBAA content in the nanoparticles that were treated in a weakly acidic medium compared with those treated in a medium at pH 7.4, demonstrating that the boronate ester dissociation mediated the zwitterionic shell detachment.
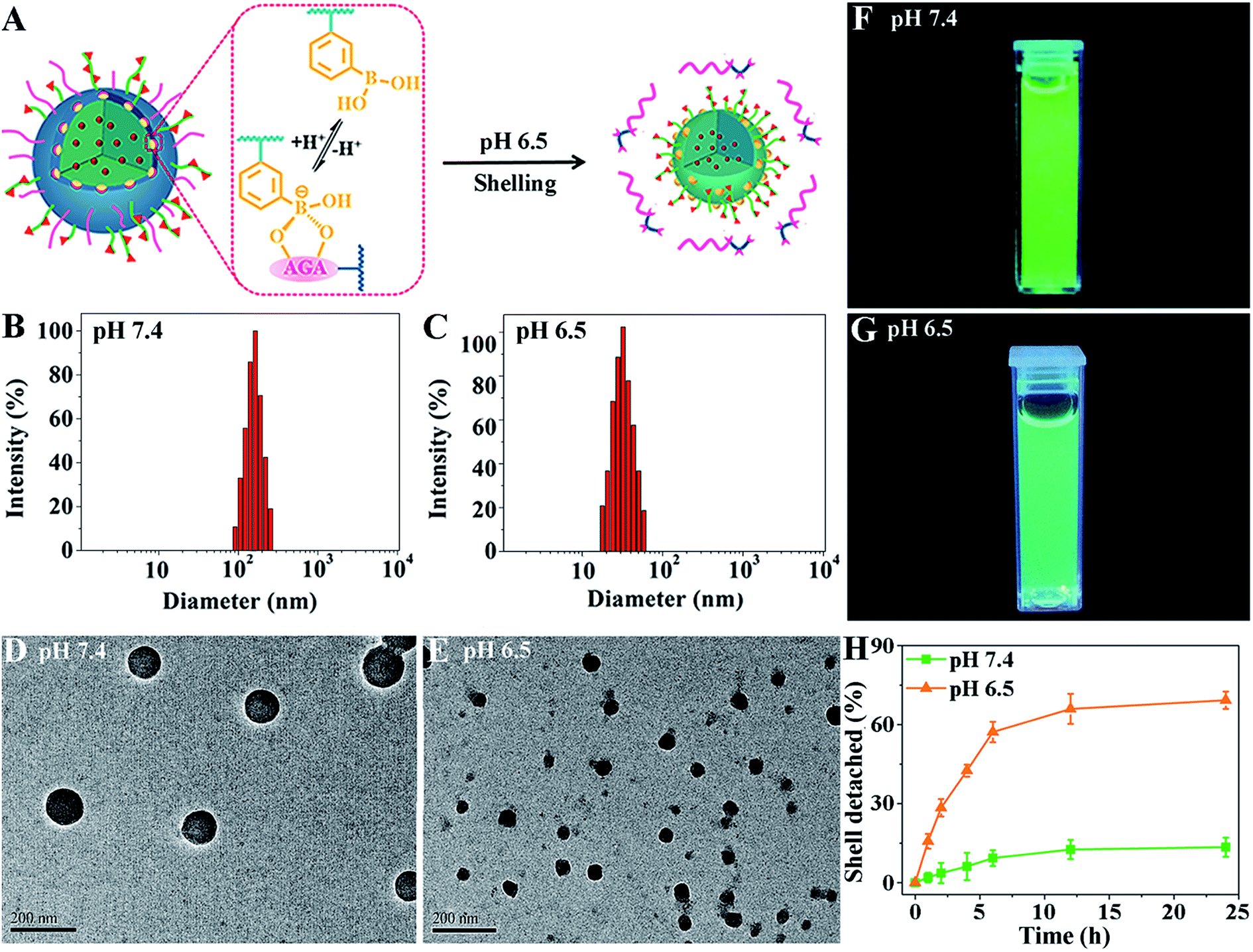 | ||
| Fig. 1 (A) The size-switching behavior in response to the pH 6.5 medium. The size distribution measured by DLS (B and C), TEM micrograph (D and E) and fluorescence graph (F and G) of pCA/pLB Np. (H) Quantitative analysis of the dissociation of pCA/pLB Np-NF in different media. | ||
To further validate the switching mechanism, pLAMA30-b-pAAPBA20 without fluorescence was synthesized (Scheme S2†). Then, a new nanoparticle was obtained by the self-assembly of the fluorescent pCBAA-b-pAGA with non-fluorescent pLAMA-b-pAAPBA; this new particle was denoted as pCA/pLB Np-NF. After incubation under different pH conditions (pH 7.4 or 6.5), the fluorescence intensity of the nanoparticles was measured. As shown in Fig. 1F and G, the much weaker fluorescence at pH 6.5 indicates that a considerable amount of pCBAA-b-pAGA was detached from the nanostructures. This detachment was also quantified; as shown in Fig. 1H, nearly 69% of pCBAA-b-pAGA had dissociated within 24 h in the pH 6.5 medium.
The dynamic boronate ester in this nanostructure facilitates the rapid release of drugs in cancer cells because the boronate ester is stable under physiological conditions but easily dissociates in the acidic tumor microenvironment.13 This property of boronate ester prevents drug leakage during circulation but ensures the rapid cellular internalization of vehicles in tumors. To explore drug loading and release behavior of this system, doxorubicin (DOX) was used as the model drug. The encapsulation efficiency (EE) and loading capacity (LC) of DOX are shown in Table S3.† The LC of pCA/pLB Np3 was higher (11.3%) than that of pCA/pLB Np1 (8.9%) and pCA/pLB Np2 (9.4%), indicating that the chain length of hydrophobic pAAPBA plays an important role in drug loading. Since insufficient intracellular drug release remains the rate-limiting step for reaching a drug concentration level within the therapeutic window, and given that the pH values of endosomes and lysosomes in tumor cells are 5.0–5.5, while those in the blood are ∼7.4, we investigated the in vitro DOX-release behavior from drug-loaded pCA/pLB Np3 in different media (pH 5.4 and 7.4, respectively). As shown in Fig. 2A, we determined that the cumulative DOX release at pH 7.4 was only 11% over 60 h, implying that DOX was steadily sequestrated in the nanoparticles. In the pH 5.4 medium, the DOX release profile presented a burst release of ∼21% after 3 h and 73% after 96 h, possibly due to the dissociation of boronate ester bonds. In general, dynamic boronic ester bonds could be cleaved not only under the acidic conditions but also under the condition where other glucose molecules are neighbors. Since the normal blood glucose level is 0.7–1.1 mg mL−1 in the human body, which may present a hidden danger to the nanocarriers by disrupting the boronate ester bonds, DOX release was further analyzed in a pH 7.4 medium that contained glucose (1 mg mL−1). The DOX release was only 18% over 60 h, indicating that the normal blood glucose levels would not trigger DOX leakage during circulation.
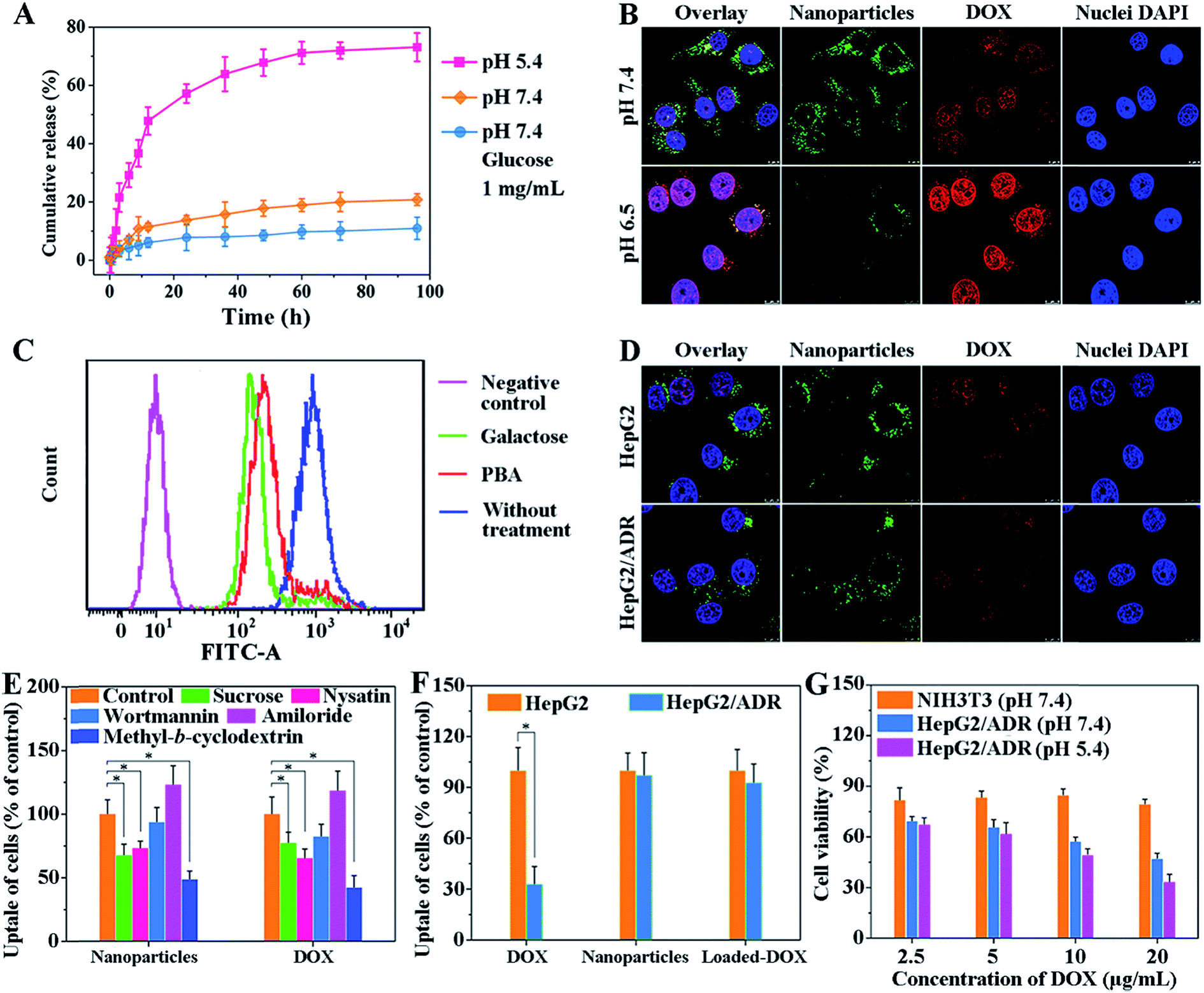 | ||
| Fig. 2 (A) In vitro DOX release from pCA/pLB Np3 in different media. (B) Confocal images of HepG2/ADR cells after incubation with DOX-loaded pCA/pLB Np3-NF at 10 μg mL−1 of DOX at pH 7.4 or 6.5. (C) Flow cytometry results of HepG2/ADR cells incubated with galactose and PBA-NH2, respectively. (D) Cellular uptake of DOX-loaded pCA/pLB Np3 in the presence of inhibitors detected by CLSM. (E) Cellular uptake of DOX-loaded pCA/pLB Np3 in the presence of inhibitors determined by flow cytometry. (F) Naked DOX uptake in HepG2 and HepG2/ADR cells determined by flow cytometry after the cells were incubated with naked DOX for 3 h. (G) Cell viability of NIH3T3 and HepG2/ADR cells after exposure to different concentrations of drug-loaded pCA/pLB Np3. | ||
Zwitterionic polymers prolong the in vivo circulation of nanoparticles by inhibiting nonspecific protein adsorption and cellular-uptake.14 However, this property may become an obstacle when nanoparticle cellular internalization is needed inside the tumor tissue. The incorporation of targeted ligands, such as galactose and PBA, onto the nanoparticle surface can enhance cellular internalization.15 In our nanocarrier design, the weakly acidic tumor microenvironment triggers the detachment of the zwitterionic shell and simultaneously activates the surface ligand PBA to enhance cellular internalization. We compared the nanoparticle cellular uptake in different media with different pH values using multidrug resistant HepG2/ADR tumor cells. As shown in Fig. 2B, much weaker green but stronger red fluorescent signals were observed at pH 6.5 compared with those at pH 7.4. The green signal came from the BODIPY groups, representing the polymer molecules, while the red fluorescence was emitted by DOX. Weak acids triggered the dissociation of dynamic boronate esters and peeled off the pCBAA-b-pAGA polymer shell, resulting in a much weaker green fluorescence. The detachment of the zwitterionic shell greatly enhanced cellular uptake and drug release, and the released drug was primarily detected in the nuclear region. This finding is attributed to the fact that the PBA and galactose targeting moieties were activated in response to the acidic medium triggering the zwitterionic shell detachment, driving the peeled nanoparticles to penetrate into the tumor cells. It has been validated that a typical PBA-rich nanoplatform can be transported into the nuclei for effective action.15d DOX is a DNA toxin that induces cell death by targeting nuclear DNA to cause DNA damage and inhibiting topoisomerase II to block DNA replication inside the nucleus.16 The significantly elevated drug nuclear concentration in the nucleus indicates the successful intracellular release and potentially enhanced therapeutic efficacy.
The emergence and development of multidrug resistance have limited the success of cancer chemotherapy, and addressing the cellular uptake mechanism of nanocarriers is critical for solving this problem. A competitive inhibition assay was performed to study the cellular internalization pathway of the peeled nanoparticles. As shown in Fig. 2C, the addition of galactose or PBA resulted in decreased nanoparticle cellular uptake, indicating that the nanoparticle surface ligands (galactose and PBA moieties) effectively facilitated internalization via receptor-mediated endocytosis. Fig. 2D shows that there was no obvious difference between the fluorescent signals of the nanoparticles and DOX, indicating that the drug-loaded nanoparticles were taken up by certain pathways but not via the P-glycoprotein. Selective inhibitors and nanocarriers were cocultured with cells to determine the intracellular drug content using flow cytometry to explore the major pathway of cellular internalization. Fig. 2E shows that the inhibition of clathrin and raft/caveolae-mediated endocytosis with sucrose, nystatin and methyl-β-cyclodextrin, respectively remarkably reduced the amount of intracellular DOX and nanoparticles. However, the inhibition of macropinocytosis with macrowortmannin and amiloride did not have a strong influence on cellular uptake, indicating that the clathrin and raft/caveolae-mediated endocytosis is the major pathway of cellular internalization for the obtained nanoparticles.
The cellular uptake behavior was further compared between normal HepG2 cells and drug resistant HepG2/ADR cells. We first used flow cytometry to verify that the DOX-induced HepG2/ADR cells were resistant to DOX treatment. Fig. 2F shows that significantly less DOX accumulated in HepG2/ADR cells owing to the overexpression of the multidrug resistance efflux pump.2e When DOX-loaded pCA/pLB Np3 was incubated with HepG2 or HepG2/ADR cells, no significant difference was observed between the two groups, indicating that the nanocarrier is capable of overcoming the multidrug drug resistance barrier (Fig. 2D and F).
For drug carriers, good cytocompatibility is essential for clinical applications. The in vitro cytotoxicity of blank pCA/pLB Np3 and the anticancer efficacy of DOX-loaded pCA/pLB Np3 in NIH3T3 and HepG2/ADR cells were analyzed by MTT assay. As shown in Fig. S13,† no obvious cytotoxicity was observed after NIH3T3 and HepG2/ADR cells were treated with blank pCA/pLB Np3. However, drug-loaded pCA/pLB Np3 displayed stronger cytotoxicity to HepG2/ADR cells, likely due to the ligand-mediated enhanced cellular uptake. The cell-killing efficacy of DOX-loaded pCA/pLB Np3 in media at different pH levels was evaluated by incubating drug-loaded pCA/pLB Np3 with HepG2/ADR cells for 24 h. Compared with pH 7.4, cell growth was strongly inhibited at pH 5.4 after the treatment with DOX-loaded pCA/pLB Np3 (Fig. 2G), due to the enhanced internalization and drug release of the peeled nanoparticles.
The zwitterionic pCBAA coating on the surface of the nanoparticles is expected to resist opsonization, inhibit nonspecific cell uptake, and prolong the circulation time in vivo.17 As shown in Fig. S14,† pCA/pLB Np3 exhibited a much longer circulation profile (t1/2 = ∼13.59 h) in mice compared with their counterparts, which were assembled solely from the pLAMA30-b-pAAPBA20 copolymer (t1/2 = ∼5.57 h). The significantly extended vascular residence time of pCA/pLB Np3 is expected to greatly facilitate drug accumulation in the tumor and reduce adverse effects.
To evaluate the antitumor efficacy of the obtained nanoparticles, two groups of HepG2/ADR tumor-bearing Balb/c mice were intravenously administered the same (doxorubicin equivalent) dose of free DOX and DOX-loaded nanoparticles, and the control group was administered PBS only. As shown in Fig. 3A, the tumors grew rapidly without treatment, and a 4.16-fold increase in tumor volume was observed in a period of two weeks. Free DOX showed a moderate inhibitory effect, with only a 3.27-fold increase in tumor volume during the same period. In contrast, the nanoparticle-treated group displayed complete tumor inhibition following the administrations. The rapid blood clearance and insufficient tumor accumulation of free DOX limited its therapeutic efficacy, while pCA/pLB Np3 restrained tumor growth more effectively by protecting DOX from premature clearance as well as enhancing tumor accumulation and retention. No obvious body weight loss was observed, suggesting that the drug dosage was safe for mice (Fig. 3B). Administered DOX is generally metabolized via the slow hepatobiliary route, and long-term drug retention in the reticuloendothelial system increases the risk of organ toxicity.18 To evaluate drug-induced organ damage, a histological analysis was performed on the major organs (heart, liver, spleen, lungs and kidneys). As shown in Fig. 3C, multifocal hepatic necrosis and apparent inflammatory cell infiltration were observed in the livers of the mice treated with free DOX, while no apparent cell or tissue injuries were detected in the PBS or DOX-loaded nanoparticle groups.
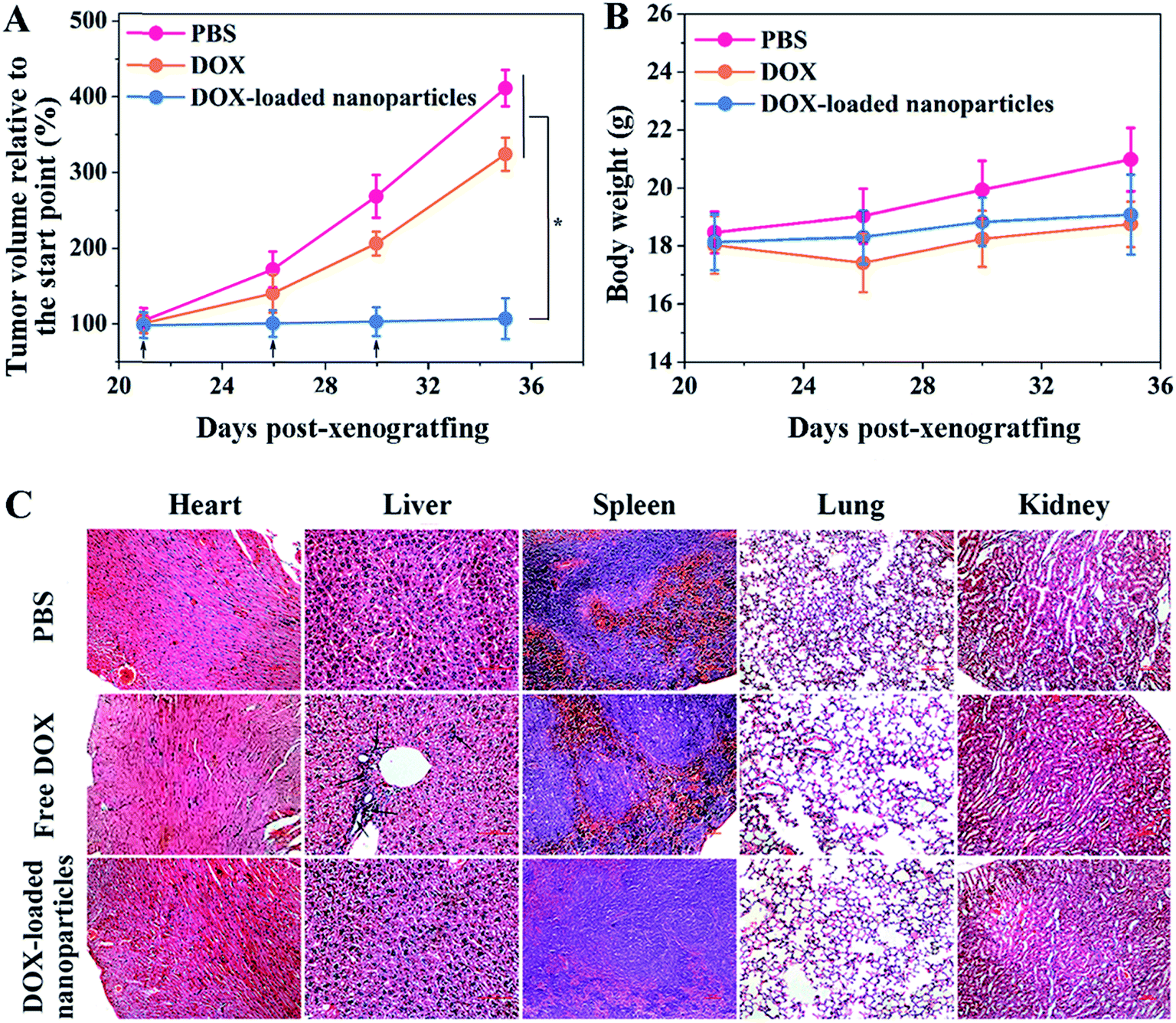 | ||
| Fig. 3 (A) Inhibition of tumor growth in a murine HepG2/ADR xenograft model after treatment with different formulations (p < 0.05). The mice were treated through repeated tail vein injection at 1 mg per kg of DOX on days 21, 26 and 30 (marked by arrows). (B) Body weight changes of different groups of mice after treatment with PBS, free DOX and DOX-loaded nanoparticles. (C) Histological analysis of H&E-stained liver, spleen and kidney tissue sections from mice treated with PBS, free DOX and DOX-loaded nanoparticles. The level of statistical significance was set at p < 0.05. | ||
Currently, the integration of diagnosis and therapy has become an inevitable tendency in multifunctional nanoparticle-based drug delivery systems to achieve targeted drug transport, real time tracing and therapy. Herein, a HepG2/ADR tumor-bearing mouse model was exploited to study the in vivo fate of the designed nanocarrier, using its integrated extraordinary imaging capacity. As shown in Fig. S15A,† pCA/pLB Np3 exhibited remarkable fluorescence signals in the tumor postinjection, indicating its rapid tumor accumulation. A similar phenomenon was detected in the tumor tissues harvested from tumor acid-sensitive polymeric vectors 2 h postinjection.5b Importantly, rapid tumor accumulation and a long retention time are useful for cancer therapy, though many previous nanoparticles have been shown to accumulate in other tissues. The decisive step for the rapid tumor accumulation of this nanocarrier is its robust tumor penetration owing to the smaller size resulting from the pH-triggered detachment of its zwitterionic shell at tumor sites. It has been validated that the penetration of nanoparticles into the tumor relies heavily on particle size, with the consensus that smaller particles have enhanced tissue penetration.5h The tumor fluorescence signals were quantified and shown to reach 1.8 × 107 ps−1 cm−2 sr−1 even at 24 h post-injection. Since the short excitation/emission wavelength (506/523 nm) of BODIPY limits deep photon tissue penetration, the fluorescence in BODIPY was not very intense. Twenty-four hours after the intravenous administration of the nanocarriers, the mice were sacrificed to isolate the major organs and tumors for ex vivo imaging. Fig. S15B† shows the biodistribution and quantified BODIPY fluorescence signal intensity. A significant fraction of the injected nanoparticles was still observed at the tumor site, while far less accumulation was detected in the heart, suggesting that these nanoparticles may effectively mitigate DOX cardiotoxicity.
To thoroughly understand the rapid accumulation and long retention time of the nanoparticles within the tumor, it is indispensable to monitor the nanocarrier pharmacokinetics inside the targeted tumor tissue in situ in real-time with a high spatial resolution. In this work, a combination of DFT technology (Fig. S16†) and indocyanine green (ICG) as a tracer agent was used to track the fate of the nanocarriers and semi-quantitatively monitor the content of the released drug by combining pharmacokinetic principles with mathematical models. A sequence of fluorescence yield images was reconstructed, and then pharmacokinetic images were estimated from the fluorescence concentration–time curve. The relationship between fluorescence yield γ(t) and concentration C(t) at time t could be linearly described using eqn (1).
γ(t) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 ξC 10 ξC | (1) |
The concentration–time curve was obtained by a biexponential-curve-fitting method based on the normally used two-compartment model.
| C(t) = −Aexp(−αt) + Bexp(−βt) | (2) |
As shown in Fig. 4A–D, S16 and S17,† the location of fluorescence in the tumor was in accordance with that in the slice images of the mice, indicating the nanoparticle exposure throughout the entire tumor and not on the tumor surface alone. It is indispensable to use DFT technology to verify that the nanoparticles can penetrate into the tumor and accumulate throughout the entire tumor, rather than only imaging nanoparticles on the surface of the tumor through live imaging technology similar to the results shown in Fig. 4A. The nanocarriers enabled rapid drug accumulation in the tumor within 5 min, which was consistent with the BODIPY fluorescence observations. Subsequently, a drug concentration–time curve and the pharmacokinetic parameters were obtained by the least squares fitting method, as shown in Fig. 4E and F. The parameters A and B of the ICG-loaded nanoparticle group were significantly increased compared with those of the free ICG group, indicating the enhanced drug accumulation in the tumor cells. Furthermore, the larger α and smaller β indicate more efficient drug accumulation and longer retention in the tumor, respectively. Each mouse was given a single dose of 20 μg of ICG, and the pCA/pLB Np3 group exhibited a five-fold higher (6.03 μg) tumor accumulation than the free ICG group (1.13 μg) within 5 min after administration. The free ICG, however, was then quickly cleared from the tumor, whereas the nanoparticle delivered ICG exhibited much higher tumor retention at 24 h. The intratumor AUC of free and nanoparticle delivered ICG from 5 min to 24 h was extrapolated to 47.78 and 139.05 a.u. min−1 (p < 0.05), respectively. The lack of nanomedicine accumulation throughout the entire tumor may be one of the primary reasons for the compromised therapeutic efficacy, which is caused by both the tumor pathological characteristics and the inappropriate physicochemical properties of nanomedicines. In this study, our nanoparticle system enables its basic physicochemical properties to adaptively change in response to the endogenous stimuli of the tumor microenvironment to achieve improved therapeutic efficacy by successively increasing the blood circulation time, improving tumor accumulation, facilitating cell internalization and accelerating intracellular drug release. We used the acidic extracellular pH as the stimulus to release small particles and activate the targeting moieties at the tumor sites, driving the peeled nanoparticles to penetrate into tumor cells. It should be noted that the post shrinkage size is the pivotal determinant of the therapeutic efficacy of nanoparticles.11a To the best of our knowledge, this is the first report of real-time tracing of nanovehicles inside tumors at the spatial level.
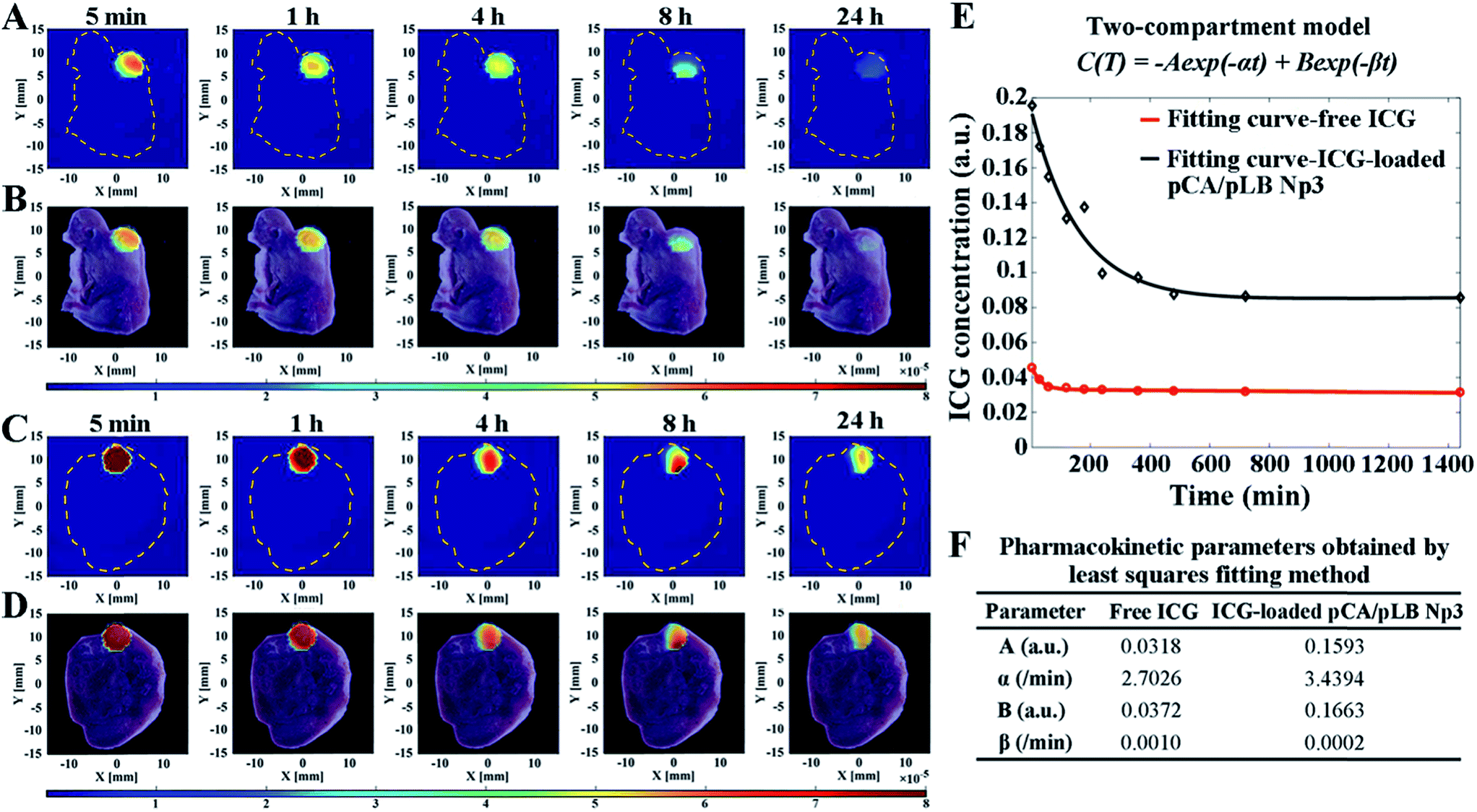 | ||
| Fig. 4 In vivo fluorescence imaging of the Balb/c nude mice bearing HepG2/ADR tumor injected with (A and B) free ICG and (C and D) ICG-loaded pCA/pLB Np3 over 24 h using a DFT system. (A and C) DFT images, and (B and D) the overlay of DFT and slice images. (E) Fitting curves of ICG concentrations vs. time for the tumor via tail intravenous injection with free ICG and ICG-loaded PCA/PLB Np3. (F) Double-exponential and pharmacokinetic parameters obtained by the least squares fitting method. | ||
In conclusion, we have designed a hierarchical nanoassembly for efficient drug delivery to overcome multidrug resistant tumors and to facilitate real-time imaging. This strategy utilized dynamic covalent chemistry to surmount a series of drug delivery obstacles. A zwitterionic polymer shell is retained in the systemic circulation for a long time to achieve rapid accumulation in the tumor and to mitigate side effects. When the designed nanoparticle reaches the tumor tissue, the acidic microenvironment triggers the detachment of the zwitterionic shell and activates the surface-binding ligands. The particle size decreases from 165 to 48 nm, which greatly facilitates tissue penetration and cellular uptake. Collectively, these merits endow the nanocarriers with a superior anti-tumor therapeutic efficacy. In addition, the intrinsic fluorescence from the BODIPY groups makes these nanocarriers a potential theragnostic platform. Finally, the intratumoral pharmacokinetic properties of these nanocarriers were evaluated in situ in real-time using noninvasive and radiation-free 3D imaging technology. Collectively, our findings advance the understanding of the behavior of nanovehicles in vivo, and provide guidance to improve drug delivery strategies.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21774062, 51673102, 51503139, 61475115 and 81671728). Male Balb/c nude mice (4-week-old) were purchased from Beijing HFK Bioscience Co., Ltd (Beijing, China), and all the protocols for animal experiments were carried out according to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. The procedures were approved by the Animal Care and Use Committee of Nankai University.References
- (a) H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994 CrossRef CAS; (b) X. Guo, X. Wei, Y. Jing and S. Zhou, Adv. Mater., 2015, 27, 6450 CrossRef CAS PubMed.
- (a) K. Cho, X. Wang, S. Nie, Z. G. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310 CrossRef CAS; (b) L. Qiu, T. Chen, I. Ocsoy, E. Yasun, C. Wu, G. Zhu, M. You, D. Han, J. Jiang, R. Yu and W. Tan, Nano Lett., 2015, 15, 457 CrossRef CAS; (c) Y. Tian, X. Jiang, X. Chen, Z. Shao and W. Yang, Adv. Mater., 2014, 26, 7393 CrossRef CAS; (d) F. Wang, Y. C. Wang, S. Dou, M. H. Xiong, T. M. Sun and J. Wang, ACS Nano, 2011, 5, 3679 CrossRef CAS; (e) X. Guo, X. Wei, Y. Jing and S. Zhou, Adv. Mater., 2015, 27, 6450 CrossRef CAS.
- (a) C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, Nat. Rev. Drug Discovery, 2015, 14, 239 CrossRef CAS; (b) T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320 CAS; (c) S. Mitragotri, D. G. Anderson, X. Chen, E. K. Chow, D. Ho, A. V. Kabanov, J. M. Karp, K. Kataoka, C. A. Mirkin, S. H. Petrosko, J. Shi, M. M. Stevens, S. Sun, S. Teoh, S. S. Venkatraman, Y. Xia, S. Wang, Z. Gu and C. Xu, ACS Nano, 2015, 9, 6644 CrossRef CAS; (d) V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813 CrossRef CAS.
- (a) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37 CrossRef; (b) T. Mizuhara, K. Saha, D. F. Moyano, C. S. Kim, B. Yan, Y. K. Kim and V. M. Rotello, Angew. Chem., Int. Ed., 2015, 54, 6567 CrossRef CAS.
- (a) C. F. Xu, H. B. Zhang, C. Y. Sun, Y. Liu, S. Shen, X. Z. Yang, Y.-H. Zhu and J. Wang, Biomaterials, 2016, 88, 48 CrossRef CAS; (b) C. Y. Sun, S. Shen, C. F. Xu, H. J. Li, Y. Liu, Z. T. Cao, X. Z. Yang, J. X. Xia and J. Wang, J. Am. Chem. Soc., 2015, 137, 15217 CrossRef CAS; (c) C. Y. Sun, Y. Liu, J. Z. Du, Z. T. Cao, C. F. Xu and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 1010 CrossRef CAS; (d) Z. Poon, D. Chang, X. Zhao and P. T. Hammond, ACS Nano, 2011, 5, 4284 CrossRef CAS; (e) X. Z. Yang, J. Z. Du, S. Dou, C. Q. Mao, H. Y. Long and J. Wang, ACS Nano, 2012, 6, 771 CrossRef CAS; (f) Z. Liu, M. Xiong, J. Gong, Y. Zhang, N. Bai, Y. Luo, L. Li, Y. Wei, Y. Liu and X. Tan, Nat. Commun., 2014, 5, 4280 CrossRef CAS; (g) Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289 RSCL. Zhu, P. Kate and V. P. Torchilin, ACS Nano, 2012, 6, 3491 CrossRef CAS; (h) L. Zhu, T. Wang, F. Perche, A. Taigind and V. P. Torchilin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17047 CrossRef CAS PubMed.
- (a) S. Kim, Y. Shi, J. Y. Kim, K. Park and J. X. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49 CrossRef CAS PubMed; (b) A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543 CrossRef CAS; (c) C. J. Cheng, G. T. Tietjen, J. K. Saucier-Sawyer and W. M. Saltzman, Nat. Rev. Drug Discovery, 2015, 14, 239 CrossRef CAS; (d) Y. Zhao, F. Fay, S. Hak, J. Manuel Perez-Aguilar, B. L. Sanchez-Gaytan, B. Goode, R. Duivenvoorden, C. de Lange Davies, A. Bjorkoy, H. Weinstein, Z. A. Fayad, C. Perez-Medina and W. J. Mulder, Nat. Commun., 2016, 7, 11221 CrossRef CAS; (e) K. Letchford and H. M. Burt, Mol. Pharm., 2012, 9, 248 CrossRef CAS.
- H. Chen, S. Kim, W. He, H. Wang, P. S. Low, K. Park and J. X. Cheng, Langmuir, 2008, 24, 5213 CrossRef CAS.
- (a) J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850 CrossRef CAS; (b) Y. Li, W. Xiao, K. Xiao, L. Berti, J. Luo, H. P. Tseng, G. Fung and K. S. Lam, Angew. Chem., Int. Ed., 2012, 51, 2864 CrossRef CAS.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344 CrossRef CAS.
- (a) J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195 CrossRef CAS; (b) K. Huang, H. Ma, J. Liu, S. Huo, A. Kumar, T. Wei, X. Zhang, S. Jin, Y. Gan, P. C. Wang, S. He, X. Zhang and X. J. Liang, ACS Nano, 2012, 6, 4483 CrossRef CAS.
- (a) H. J. Li, J. Z. Du, X. J. Du, C. F. Xu, C. Y. Sun, H. X. Wang, Z. T. Cao, X. Z. Yang, Y. H. Zhu, S. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164 CrossRef CAS; (b) H. J. Li, J. Z. Du, J. Liu, X. J. Du, S. Shen, Y. H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753 CrossRef CAS; (c) Y. Wang, K. Zhou, G. Huang, C. Hensley, X. Huang, X. Ma, T. Zhao, B. D. Sumer, R. J. DeBerardinis and J. Gao, Nat. Mater., 2014, 13, 204 CrossRef CAS.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32, 3435 CrossRef CAS.
- (a) Y. E. Aguirre-Chagala, J. L. Santos, Y. Huang and M. Herrera-Alonso, ACS Macro Lett, 2014, 3, 1249 CrossRef CASJ. Kim, Y. M. Lee, H. Kim, D. Park, J. Kim and W. J. Kim, Biomaterials, 2016, 75, 102 CrossRef CAS; (b) Y. Li, W. Xiao, K. Xiao, L. Berti, J. Luo, H. P. Tseng, G. Fung and K. S. Lam, Angew. Chem., Int. Ed., 2012, 51, 2864 CrossRef CAS; (c) V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86 CrossRef CAS.
- (a) L. Zhang, H. Xue, Z. Cao, A. Keefe, J. Wang and S. Jiang, Biomaterials, 2011, 32, 4604 CrossRef CAS; (b) P. Zhang, F. Sun, C. Tsao, S. Liu, P. Jain, A. Sinclair, H. C. Hung, T. Bai, K. Wu and S. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12046 CrossRef CAS.
- (a) A. Matsumoto, H. Cabral, N. Sato, K. Kataoka and Y. Miyahara, Angew. Chem., Int. Ed., 2010, 49, 5494 CrossRef CAS; (b) H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng, S. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603 CrossRef CAS PubMed; (c) S. Deshayes, H. Cabral, T. Ishii, Y. Miura, S. Kobayashi, T. Yamashita, A. Matsumoto, Y. Miyahara, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2013, 135, 15501 CrossRef CAS; (d) R. Tang, M. Wang, M. Ray, Y. Jiang, Z. Jiang, Q. Xu and V. M. Rotello, J. Am. Chem. Soc., 2017, 139, 8547 CrossRef CAS PubMed.
- H. Mizutani, S. Tada-Oikawa, Y. Hiraku, M. Kojima and S. Kawanishi, Life Sci., 2005, 76, 1439 CrossRef CAS.
- (a) Y. Y. Yuan, C. Q. Mao, X. J. Du, J. Z. Du, F. Wang and J. Wang, Adv. Mater., 2012, 24, 5476 CrossRef CAS; (b) H. Kang, J. Gravier, K. Bao, H. Wada, J. H. Lee, Y. Baek, G. El Fakhri, S. Gioux, B. P. Rubin, J. L. Coll and H. S. Choi, Adv. Mater., 2016, 28, 8162 CrossRef CAS.
- (a) R. Kumar, I. Roy, T. Y. Ohulchanskky, L. A. Vathy, E. J. Bergey, M. Sajjad and P. N. Prasad, ACS Nano, 2014, 4, 699 CrossRef; (b) E. Lilienberg, B. C. Ebeling, R. Nyman, M. Hedeland, U. Bondesson, N. Axén and H. Lennernäs, Mol. Pharm., 2014, 11, 131 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Polymer synthesis, self-assembly, and characterization; drug loading and in vitro drug release; treatment of tumour cells with drug and drug-loaded nanoparticles; cytocompatibility assay; in vitro cellular internalization; in vivo administration of nanoparticles; in vivo imaging. See DOI: 10.1039/c9sc01841g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |