DOI:
10.1039/C9SC01649J
(Edge Article)
Chem. Sci., 2019,
10, 7887-7897
Observation of osmotically driven, highly controllable and reconfigurable oil/water phase separation†
Received
3rd April 2019
, Accepted 21st June 2019
First published on 21st June 2019
Abstract
Liquid–liquid phase separation has been proven to be a valuable method for producing structured materials and creating chemical systems. Although several strategies have been developed to date, osmotically driven oil/water phase separation has never been achieved owing to the limited solubility of inorganic salts in conventional organic solvents and thus the insufficient osmotic driving force to counterbalance the Laplace pressure associated with the interfacial tension. Herein, we report the discovery that a mixture of 1-alkyl-3-vinylimidazolium bis(trifluoromethanesulfonyl)imide and LiTf2N can generate sufficient and widely tunable osmotic pressure in oil to realize water transport from the surrounding aqueous phase into the oil phase, triggering spontaneous phase separation. This osmotically driven phase separation could be modulated with unprecedented flexibility, offering unlimited possibilities to facilely access diverse thermodynamically metastable structures using one system. Importantly, this oil system can serve as a general phase separation carrier platform for realizing phase separation of various substances.
Introduction
Osmotic pressure is a thermodynamic driving force that can be exploited to control the transit of solvent molecules across a semipermeable membrane separating two liquid solutions.1 In biological creatures, osmotic pressure plays a central role in many processes such as fluid exchange in cells, elaborate osmo-sensing and osmo-signalling mechanisms as well as water transport in plants.2–4 It has also found tremendous applications in various fields including power production,5,6 water purification,7,8 and fabrication of functional materials.9–12 In particular, in the water/oil/water multiple emulsion systems widely used in food, pharmaceuticals, cosmetics, and agricultural products osmotic pressure provides a facile means of regulating their internal texture structure, rheology behaviour and delivery properties of encapsulated active components.13–16 In this respect, unbalanced osmotic pressure induced solvent (water) flow takes place between inner and outer aqueous phases of the multiple emulsions, and the water molecules only migrate across the oil layer to give rise to a volume change (swelling and shrinking) of aqueous phases without any phase transition.17–19 As conventional organic oils have limited solubility of the osmolyte (e.g. inorganic salts) and thus the generated osmotic pressure cannot counterbalance the Laplace pressure induced by water uptake,20,21 to the best of our knowledge, osmotically driven phase separation in common homogeneous oil phases has never been observed.
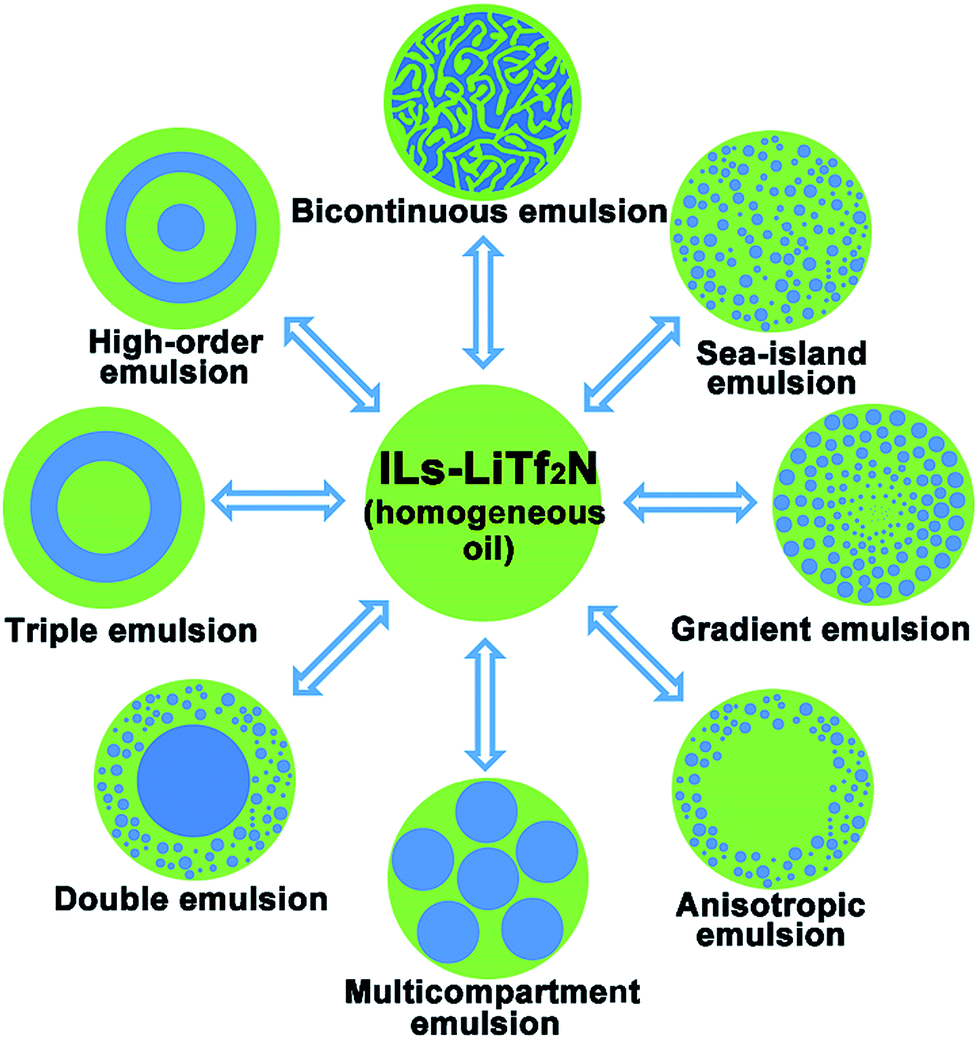
Ionic liquids (ILs) are a class of molten salts consisting of organic cations and anions, and are often viewed as an “intermediate” between organic molecular liquids and molten salts.22–24 Stemming from their dual ionic and organic nature, ionic liquids (ILs) exhibit a series of unique physicochemical properties. One of the distinctive properties of ILs is their excellent solvency for a broad range of substances.25,26 Compared with normal organic solvents hydrophobic ILs can even display peculiar solubility for different inorganic salts to form homogeneous oil solutions.27–30 In this work, we report the discovery that one of such special oil solutions, namely the mixture (Cnvim[Tf2N]–LiTf2N) of 1-alkyl-3-vinylimidazolium bis(trifluoromethanesulfonyl)imide (Cnvim[Tf2N]) and LiTf2N, can generate sufficient and widely tunable osmotic pressure in oil to realize water transport from the surrounding aqueous phase into the oil phase. Completely different from osmotically driven water flow in common emulsion systems reported so far, in which mass transport only occurs between two aqueous phases, the osmotically driven water transport described here from the aqueous phase into the oil phase can trigger spontaneous oil/water phase separation. It was found that the thermodynamic driving force, kinetic barrier and direction of this osmotically driven phase separation (OPS) could be facilely modulated with unprecedented flexibility by varying the amount of LiTf2N dissolved in Cnvim[Tf2N], changing the molecular structure of Cnvim[Tf2N] and altering the salt concentration in the aqueous phase. Surprisingly, by adding salt into the aqueous phase, the already phase-separated heterogeneous state could even be reverted to restore the homogeneous oil phase by redrawing the water molecules that are taken up, indicating that the OPS is reconfigurable. Thus, compared with those of the liquid–liquid phase separation methods reported so far, these attractive attributes enable precise design and control over the speed and kinetic pathways of the phase separation process, which offers unlimited possibilities to facilely access and capture diverse thermodynamically metastable structures using one phase separation system. Complex emulsions with a broad-spectrum of internal structures, including bicontinuous, sea-island, gradient, anisotropic, multicompartmental, and high-order multilayer structures could be easily achieved in a simple single-step OPS using one system, as shown in Fig. 1. Importantly, we found that without a significant change of its phase separation behaviour the extraordinary solubility of ionic liquids towards a broad range of substances allows the Cnvim[Tf2N]–LiTf2N system to serve as a general phase separation carrier platform for realizing the phase separation of various substances, especially those inaccessible using conventional methods, providing enormous opportunities to realize the fabrication of various nano- or micro-structured materials with widely tailorable internal structures through a simple one-step phase separation process.
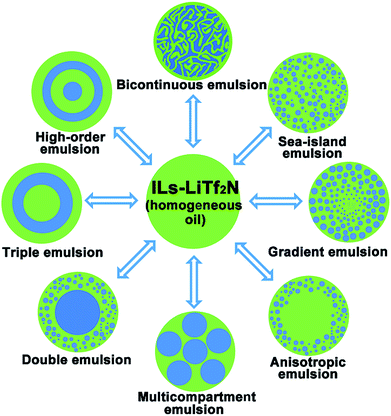 |
| | Fig. 1 One-step fabrication of diverse complex emulsion structures by using osmotically driven water–oil phase separation. | |
Results and discussion
LiTf2N solubility in Cnvim[Tf2N] ionic liquids
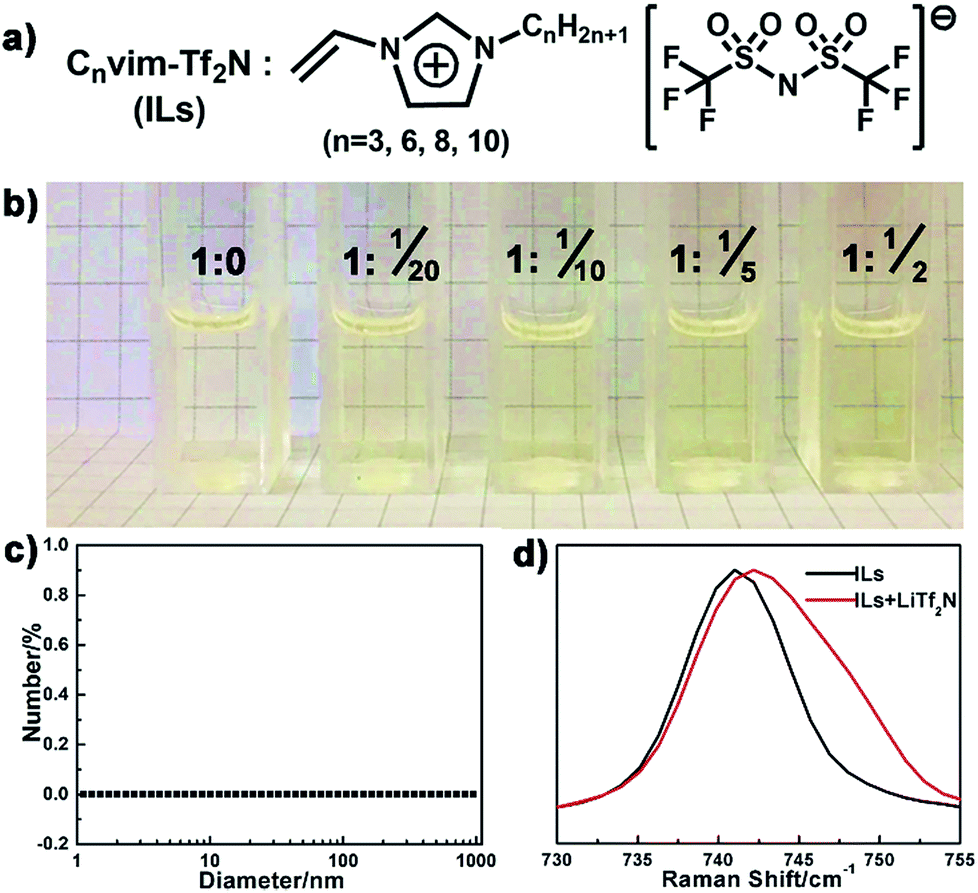
Compared to the conventional hydrophobic organic solvents, the unique dual organic and ionic nature of ionic liquids endows them with distinctive solubility of inorganic salts.27–30 The dissolved salts in the resultant IL oil solution should provide an osmotic driving force to induce phase separation upon exposure to the aqueous phase. In our case, a series of hydrophobic, low viscosity and polymerizable ionic liquids (Cnvim[Tf2N], n = 3, 6, 8, 10) were synthesized (Fig. S1,† the synthesis method and 1H NMR, 13C NMR and mass spectra are described in the ESI†) and chosen as oil solvents (Fig. 2a), and their solubility towards inorganic salts was first verified. As a demonstration, Fig. 2b clearly displays the solubility of LiTf2N in C3vim[Tf2N]. Pure C3vim[Tf2N] is a pale yellow liquid. When 0.05 molar LiTf2N was added in 1 molar C3vim[Tf2N], the salt was rapidly dissolved and a homogeneous solution is formed. With increasing amounts of LiTf2N, even 0.5 molar salt, it is found that in all cases the salts dissolved very well in 1 molar C3vim[Tf2N], affording homogeneous solutions with a transparent appearance (Fig. 2b). Dynamic light scattering (DLS) measurements show that no aggregation was detected in the resulting ionic liquid mixtures (Fig. 2c), definitely confirming that LiTf2N dissolved well in C3vim[Tf2N] at the molecule level. Besides C3vim[Tf2N], a similar dissolution capacity of LiTf2N is also found in the more hydrophobic Cnvim[Tf2N] (n = 6, 8, 10) cases. It should be noted that the solubility of LiTf2N in Cnvim[Tf2N] (1 molar Cnvim[Tf2N] dissolving 0.5 molar LiTf2N) exceeds the saturated solubility of NaCl in water (0.12 molar NaCl per mole of H2O). This extraordinary capacity of Cnvim[Tf2N] to dissolve LiTf2N can be attributed to the formation of a four-coordinated structure of the Li+ ion of LiTf2N with the O atoms of two bidentate Tf2N− ions of Cnvim[Tf2N].28Fig. 2d presents the comparison of the Raman absorption peaks of the Tf2N− anion in C3vim[Tf2N] before and after the addition of LiTf2N salt. The intense peak located at 740 cm−1 redshifts to 743 cm−1 in the presence of LiTf2N salt, indicative of the strong interaction between Li+ cations and Tf2N− anions of Cnvim[Tf2N], thus demonstrating the dissolution of LiTf2N in C3vim-Tf2N. In our work, we also tested the solubility of other types of hydrophobic ILs with their corresponding salts, including Cnvim[PF6] with LiPF6, Cnvim[BF4] with LiBF4 and Cnvim[C4F9SO3] with LiC4F9SO3. Due to the multiple interactions the Tf2N− based ILs (Cnvim[Tf2N]) exhibit the best solubility with their corresponding salts. Thus, the Cnvim[Tf2N]–LiTf2N solutions were used as special oil systems for studying their phase separation behaviour.
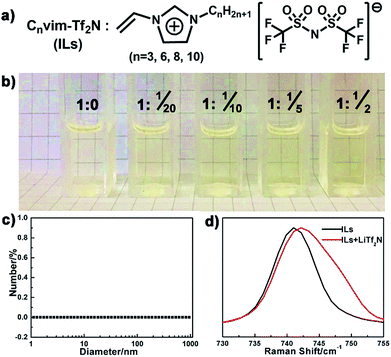 |
| | Fig. 2 (a) Chemical structure of the ILs used in this work. (b) Optical images of C3vim[Tf2N] and LiTf2N mixtures with different IL/salt ratios. (c) Dynamic light scattering (DLS) spectrum of the C3vim[Tf2N]–LiTf2N mixture at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/2. (d) Raman spectra of neat C3vim[Tf2N] and the C3vim[Tf2N]–LiTf2N (1:1/2) mixture. :1/2. (d) Raman spectra of neat C3vim[Tf2N] and the C3vim[Tf2N]–LiTf2N (1:1/2) mixture. | |
Osmotically driven phase separation (OPS) of the Cnvim[Tf2N]–LiTf2N system
Compared to conventional organic hydrophobic solvents, the remarkable amounts of LiTf2N dissolved in Cnvim[Tf2N] should provide a sufficient osmotic driving force to induce the phase separation of the Cnvim[Tf2N] oil phase when the ionic liquid solution is exposed to an aqueous phase. To efficiently monitor the phase separation behaviour of the Cnvim[Tf2N] oil phase, the osmotically driven phase separation (OPS) process was performed in a co-flow microfluidic device in our work. The co-flow geometry microfluidic chip used is shown in Fig. S2.† In the microfluidic system, Cnvim[Tf2N]–LiTf2N oil was used as the dispersed phase with a flow speed of 0.1–0.3 mL h−1 and a 10% low mW PVA aqueous solution was used as the continuous phase with a flow speed of 5–10 mL h−1. This microfluidic approach allows for facilely producing well-defined and uniform oil droplets in a continuous aqueous phase, and thus is beneficial for the capture and observation of kinetic pathways and speed of the OPS process as well as the internal structure evolution of a single droplet as the droplet moves downstream. Additionally, a large-quantity synthesis of uniform materials with well-defined morphologies, including spherical, fibrous and tubular materials, in a short time is another attractive feature of the microfluidic technique (shown in Movie 1†).31–33
Theoretically, when the osmotic driving force provided by the salts dissolved in Cnvim[Tf2N] oil exceeds the Laplace pressure associated with the oil/water interface, the water molecules from the continuous aqueous phase would have the capacity to spontaneously diffuse into Cnvim[Tf2N]–LiTf2N oil droplets, thus initiating the phase separation process. The Laplace pressure difference (ΔP) between an oil droplet of radius R and the external aqueous phase resulting from the oil/water interfacial tension (γ) is given by ΔP = 2γ/R. When the Cnvim[Tf2N]–LiTf2N mixture is simplified as a dilute ideal solution, its osmotic pressure Φ is 2ckT, where c is the concentration of the monovalent salt in the ideal solution. The interfacial tension between the Cnvim[Tf2N]–LiTf2N mixture and the aqueous solution was measured by using pendant drop tensiometry. For example, for a C3vim[Tf2N]–LiTf2N (molar ratio = 1:1/20) droplet of ca. 200 μm, the osmotic pressure Φ is already 1650 Pa, which far exceeds the Laplace pressure (ΔP ≈ 20 Pa). The huge difference will drive water transport from the external water phase into the IL oil phase, and the minimum aqueous droplet generated through OPS should be about Rmin = 2400 nm when Φ = ΔPmax. It should be noted that, although the LiTf2N salt shows good solubility in water, we neglected the possibility of LiTf2N salt diffusing from the oil droplets into the continuous aqueous phase because LiTf2N has multiple interactions with Cnvim[Tf2N].
The droplets of the Cnvim[Tf2N]–LiTf2N oil solution prepared in the microchannel are initially homogeneous and transparent. After being injected into the continuous aqueous phase, it is found that the droplets would spontaneously develop a cloudy appearance, and gradually change from clear to dark under transmission microscopy observation, as shown in Fig. S3.† These results clearly indicate that sufficient osmotic pressure drives the water permeation from the aqueous phase into the oil phase, leading to the liquid–liquid phase separation process inside the oil droplets. In the microchannel the phase separation evolution as well as the resultant emulsion structure is clearly observable. Importantly, we found that the remarkable solubility of LiTf2N in Cnvim[Tf2N] and the variation of the Cnvim[Tf2N] structure could provide great opportunities and flexibility to modulate the speed and kinetic pathways of this osmotically driven phase separation process. The highly kinetically controlled features of OPS would make it possible to access various phase-separated meta-structures, as illustrated qualitatively in the ternary phase diagram of the water–LiTf2N–Cnvim[Tf2N] system (Fig. S4†). In this work, we choose polymerizable Cnvim[Tf2N] because it can be polymerized by UV light to freeze the metastable state during the OPS process. The arrested metastable structure can be easily characterized and analysis by SEM.
Effect of LiTf2N concentration on the OPS process
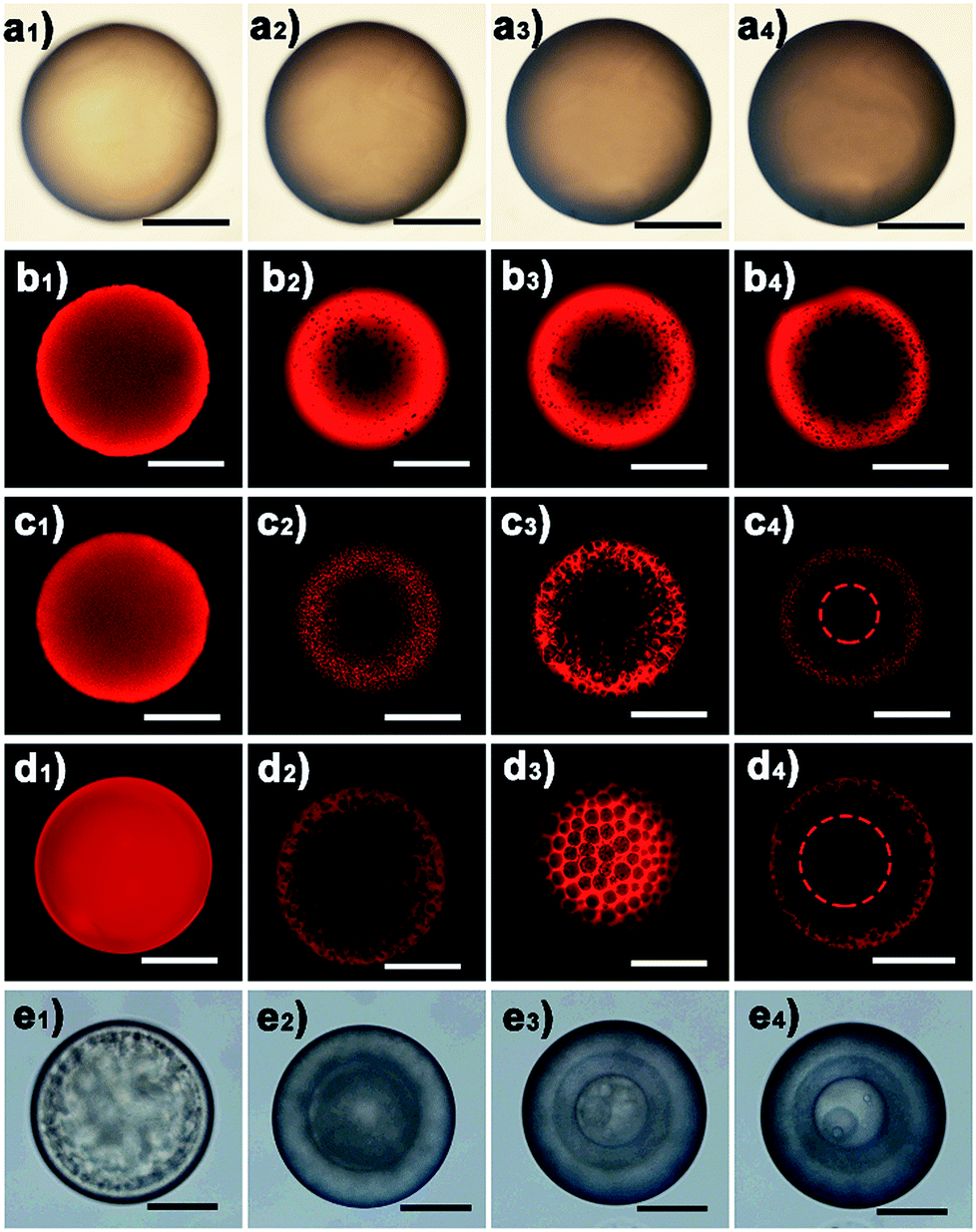
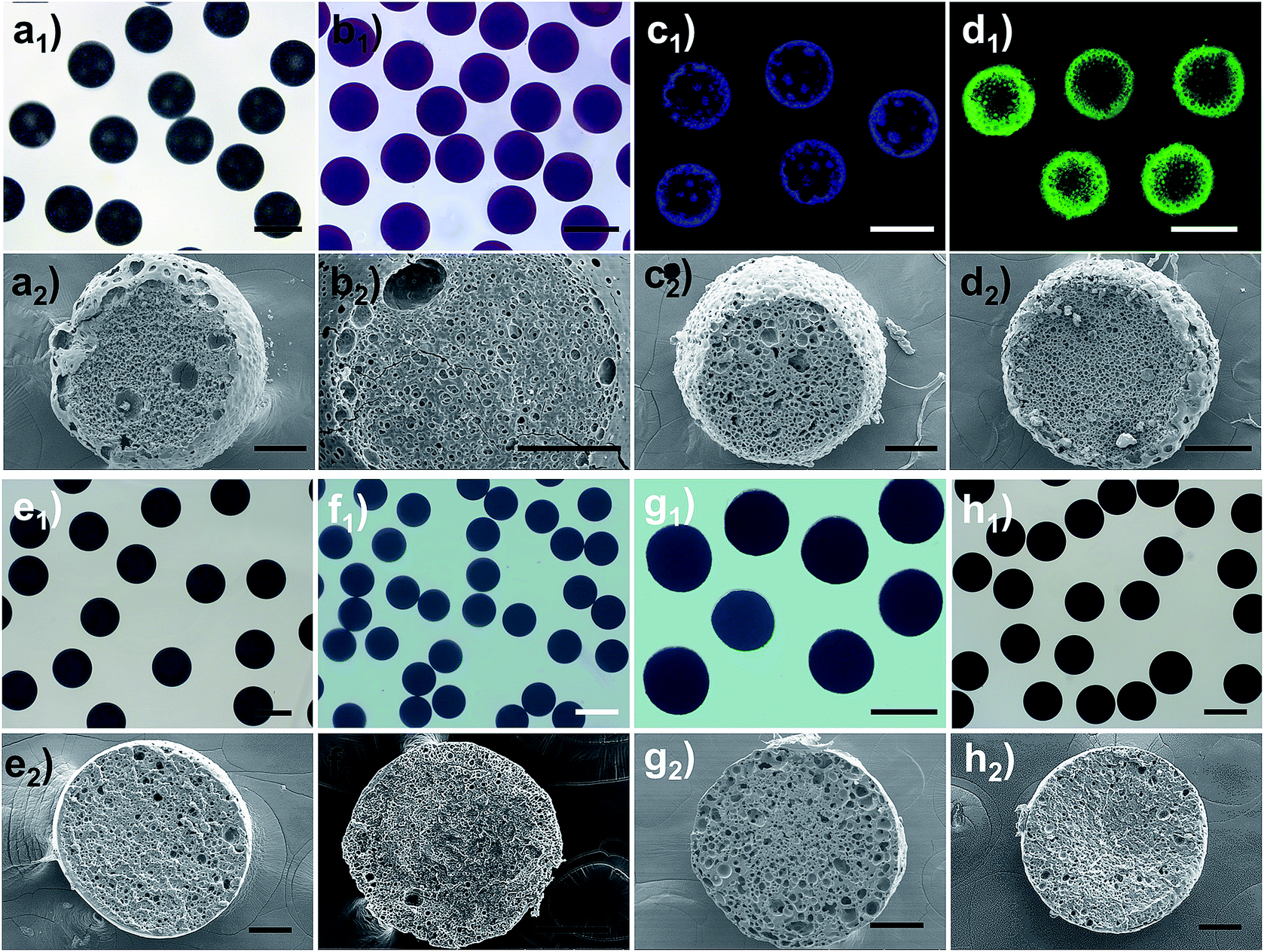
The water diffusion speed is dictated by the osmotic difference resulting from the dissolved LiTf2N salt. The kinetics of the observed phase separation in oil could be facilely controlled by varying the initial salt concentration. As a representative example, Fig. 3 shows the phase separation behaviour of a series of C3vim[Tf2N] oils with different amounts of dissolved LiTf2N salts. After prolonged exposure (ca. 60 min) to the aqueous phase, no appreciable phase separation was detected in the pure C3vim[Tf2N] droplet without the addition of LiTf2N (Fig. 3a). The boundary of the oil droplet turned dark with prolonged exposure (Fig. 3a1–a4); the reason for this phenomenon is probably the interfacial instability at the water–oil interface. Indeed, after the polymerization of the oil droplets under UV irradiation the resultant polymer particles were dense from the outside to inside (Fig. S5†), and no phase separation was observed. By contrast, when a small amount (1/40 molar) of LiTf2N salt was dissolved in 1 molar C3vim[Tf2N], the phase separation was initiated in a slow fashion. In this case, to better observe the phase separation process, the hydrophobic Nile red dye was added into the ionic liquid solution, which showed bright fluorescence in the oil phase and negligible fluorescence in the aqueous phase. As shown in Fig. 3b, the initial homogeneous phase (Fig. 3b1) became heterogeneous, and water molecules were driven into the oil phase to form discrete smaller aqueous droplets after 1 min (Fig. 3b2). As the oil droplet moved downstream in the channel, more water molecules continuously diffused into the oil phase, finally resulting in a sea-island emulsion structure (Fig. 3b3 and b4). In this case, due to the lower osmotic driving force the OPS took place in an ultraslow fashion, and 10 min was required to achieve OPS at equilibrium. With the increasing amounts of LiTf2N in C3vim[Tf2N], the enhanced driving force could expedite the OPS process. In the case of 1/10 or 1/5 molar LiTf2N in 1 molar C3vim[Tf2N], the OPS occurred immediately and the transparent oil droplet became cloudy as it was injected into the continuous aqueous phase. In addition to the speed of OPS, the kinetic pathway also varied with the amount of LiTf2N. As can be seen from Fig. 3c and d, the structural evolution of the oil droplet went from homogeneous (Fig. 3c1 and d1), bicontinuous (Fig. 3c2 and d2), and multicompartmental (Fig. 3c3 and d3) to a double emulsion structure (Fig. 3c4 and d4). The bicontinuous structure was further confirmed in the SEM image in Fig. S20.† Compared with the C3vim[Tf2N]–LiTf2N solution at a molar ratio of 1:1/5, the resulting double emulsion structure obtained by the 1:1/10 C3vim[Tf2N]–LiTf2N system shows a smaller inner core and a thicker porous shell (Fig. 3c4 and d4). When more LiTf2N salt was dissolved in C3vim[Tf2N], the phase separation behaviour of the C3vim[Tf2N]–LiTf2N system changed dramatically. Fig. 3e shows the OPS evolution process of the C3vim[Tf2N]–LiTf2N at a molar ratio of 1:1/2. Quite different from the induced structures described above, in this case the onion-like emulsion structure was formed through OPS. Such a violent change of the structure induced through OPS can be attributed to the rapid diffusion of a large amount of water into the C3vim[Tf2N]–LiTf2N oil droplet (Fig. 3e1). Spinodal decomposition was first induced, leading to a double emulsion structure with a C3vim[Tf2N] rich shell and a water rich core (Fig. 3e2). Inside the droplet the liquid–liquid phase separation then proceeds continuously through self-similar cycles (Fig. S4d†) of mass transfer, spinodal decomposition, and coalescence into a multiple layer structure (Fig. 3e3 and e4).34 The self-similar phase separation behaviour was quantitatively proved by the linear relationship of each shell diameter, as shown in Fig. S6.† It should be pointed out that the abovementioned phase separation behaviour is highly reproducible. As can be seen in Fig. S7,† large scale fabrication of the same phase separation droplets was achieved by using the microfluidic method. Clearly, these results demonstrate that the speed and kinetic pathways of OPS as well as the induced structure could be facilely modulated by simply varying LiTf2N in the C3vim[Tf2N]–LiTf2N system.
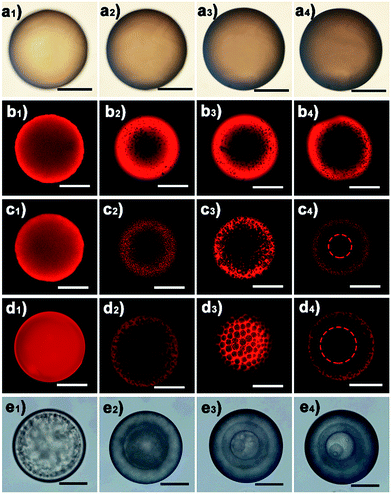 |
| | Fig. 3 Optical (a and e) and CLSM (b–d) images of the emulsion structures induced by the osmotically driven water–oil phase separation process using the C3vim[Tf2N]–LiTf2N mixture with a molar ratio of 1:0 (a), 1:1/40 (b), 1:1/10 (c), 1:1/5 (d), and 1:1/2 (e) as the oil phase. The scale bar is 150 μm. | |
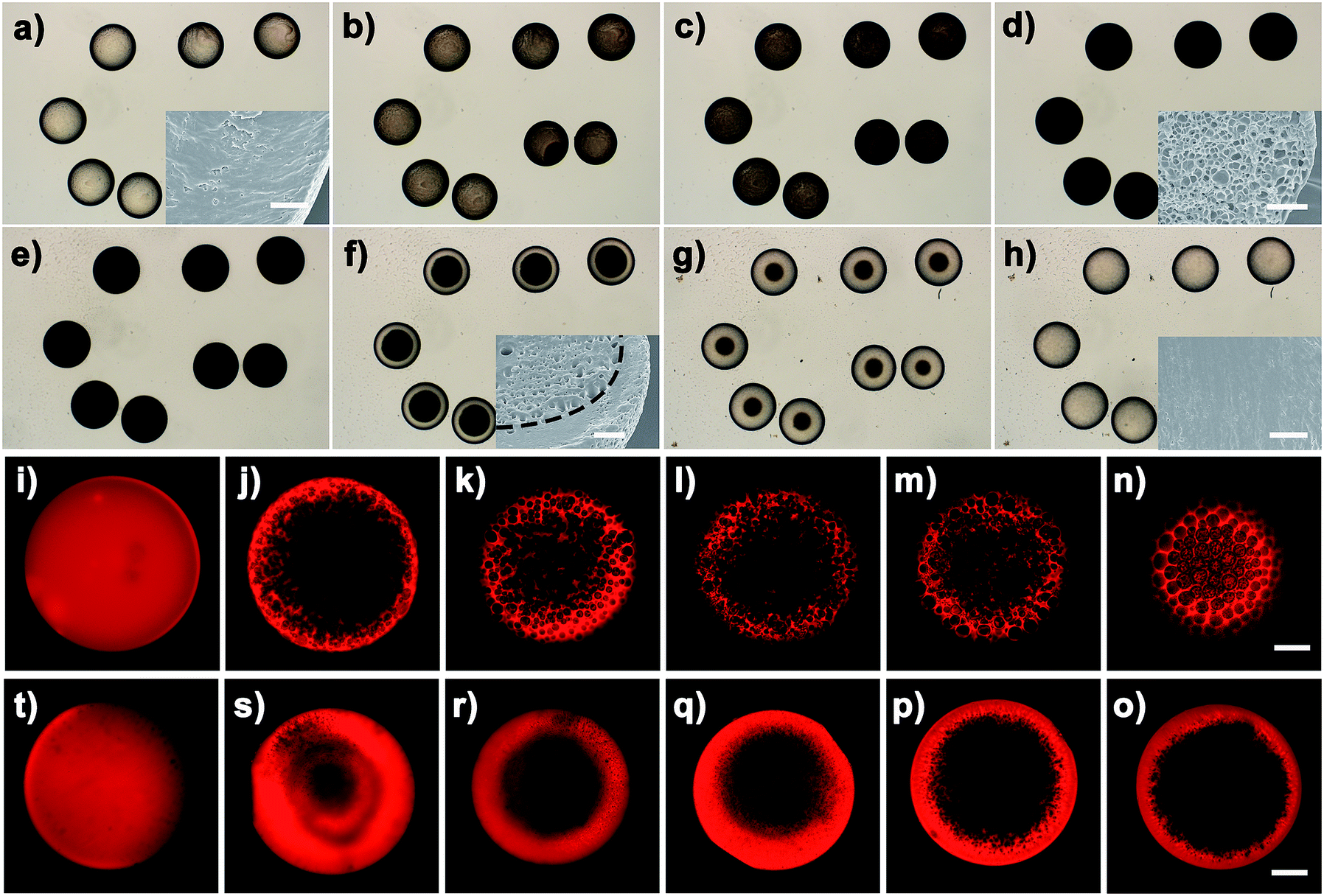
Effect of the alkyl chain on the OPS process
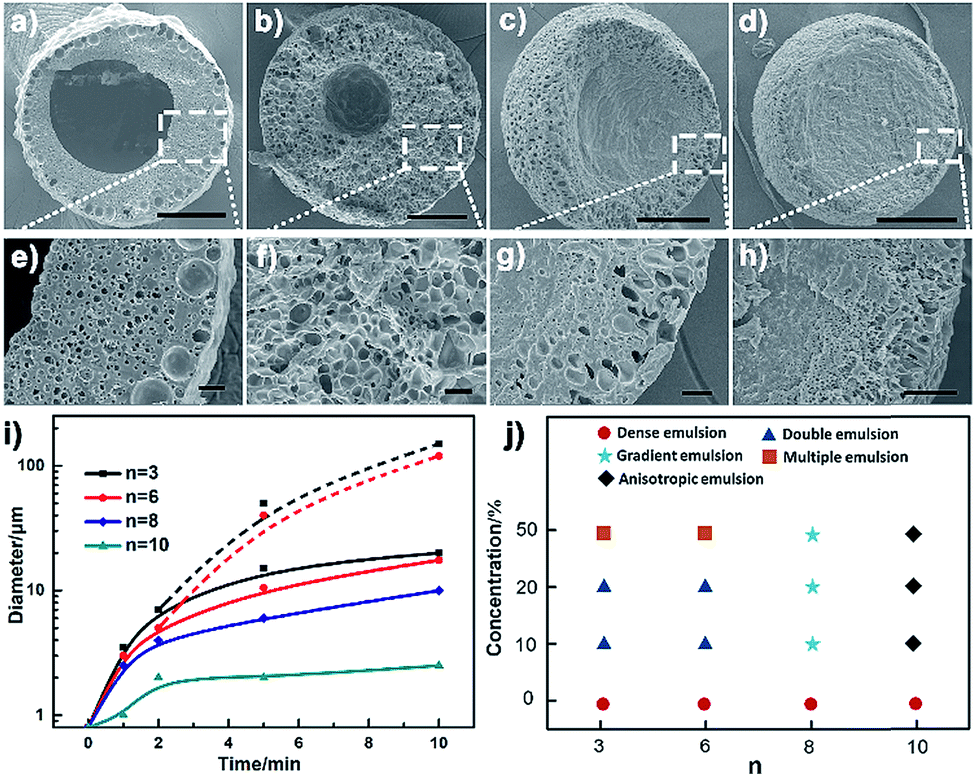
Besides the thermodynamic driving force, varying the alkyl length of the Cnvim[Tf2N] could adjust the hydrophobicity of ionic liquids, and thus provides another means to modulate the kinetics of the OPS through a change of the kinetic barrier. In the Cnvim[Tf2N]–LiTf2N system, increasing the hydrophobicity of ILs could decrease the speed of the osmotically driven water transport, leading to the change of kinetic pathways of the phase separation process (the hydrophobicity increased with the increase of the alkyl chain length, which is demonstrated by the solubility test in Fig. S18†). In the case of C3vim[Tf2N]–LiTf2N (molar ratio: 1:1/5), the ionic liquid shows a relative high affinity toward water molecules. The liquid–liquid OPS process occurred fast accompanied with the structural evolution from bicontinuous and multicompartmental to the double emulsion structure. When the C6vim[Tf2N]–LiTf2N system with the same concentration of LiTf2N salt was used, the structural evolution during OPS also went from bicontinuous and coalescence to sea-island, and Ostwald ripening to the double emulsion structure. However, the higher hydrophobicity of hexyl (n = 6) than of propyl (n = 3) gave rise to a clear reduction of the speed of the OPS process and the double emulsion structure with a smaller aqueous core and a thicker porous shell (Fig. 4a, b and e, f). Further increasing the alkyl length of ionic liquids would significantly enhance the kinetic barrier of water transport. As a result, the speed of the OPS process became remarkably slower. On the other hand, due to the change of the kinetic pathway, the structural evolution induced by OPS and the resultant equilibrium structure are quite different. For the more hydrophobic C8vim[Tf2N]–LiTf2N system no double emulsion structure could be formed during the OPS process, and instead only an anisotropic porous sphere with an isolated porous core and interconnected micrometer-sized porous shell was induced (Fig. 4c and g). When we further increase the alkyl chain length from octyl to decanyl, the OPS of C10vim[Tf2N]–LiTf2N under comparable conditions afforded the sphere with a dense core and thin porous shell, namely the “ring porous sphere”, at equilibrium (Fig. 4d and h). Fig. 4i summarizes the effect of the alkyl length of the ionic liquid on the structural evolution of the Cnvim[Tf2N]–LiTf2N system at the same molar ratio of 1 to 1/5. Fig. 4j shows the diagram of the structure induced by the OPS using ionic liquids with different alkyl chains under different driving forces (LiTf2N concentrations). It should be noted that with the increase of the alkyl chain length, the interfacial tension between Cnvim[Tf2N] and the 10% PVA aqueous solution was almost unchanged (Fig. S21†), which means it had no influence on the OPS process. In addition, when we used C8vim[Tf2N] or C10vim[Tf2N], increasing the salt concentration doesn't have a significant influence on the type of prepared emulsion. This is because with long alkyl chain lengths, the hydrophobicity of the ionic liquid generates a ‘kinetic barrier’, which could offset the driving force of osmotic pressure (Fig. S19†). These results clearly indicate that the variation of the alkyl length of ionic liquids could finely tune the kinetics of the OPS process and provide an efficient way to access diverse emulsion structures using a single-step procedure. To clearly prove that the OPS process is not a simple dissolution process, we prepared a double emulsion in which the Cnvim[Tf2N]–LiTf2N oil and water were separated by another immiscible oil (PDMS). In this case, the simple dissolution process would not take place because Cnvim[Tf2N]–LiTf2N oil and water were not in contact with each other. However, the water transport and phase separation still occurred. This is a solid demonstration that the OPS process is not a simple dissolution process.
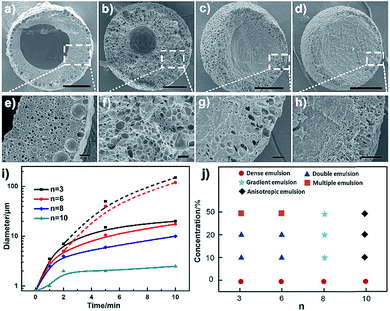 |
| | Fig. 4 SEM images of polymerized emulsions after 10 minutes of phase separation by using the Cnvim[Tf2N]–LiTf2N (1:1/5) mixture, n = 3 (a and e), n = 6 (b and f), n = 8 (c and g) and n = 10 (d and h). (i) Average pore diameter during the phase separation process by using the Cnvim[Tf2N]–LiTf2N (1:1/5) mixture (solid line: shell pore diameter, dashed line: core diameter) and (j) morphology diagram of polymerized complex emulsions induced by using ILs containing different LiTf2N amounts. Scale bars are 150 μm (a–d) and 20 μm (e–h). | |
Based on the above results, it is conceivable that adjusting the IL molecular structure together with varying the thermodynamic driving force (namely the LiTf2N concentration) would provide unprecedented freedom and flexibility in controlling or modulating the kinetics of OPS of the Cnvim[Tf2N]–LiTf2N system, and diverse thermodynamically metastable structures could be created and arrested using one system. As a demonstration, Fig. 5 shows several typical structures induced by adjusting the LiTf2N concentration and molecular structure, including the sea-island emulsion structure (Fig. 5a1 and a2), bicontinuous emulsion structure (Fig. 5b1 and b2), multicompartmental structure with different sizes (Fig. 5c1, c2 and d1, d2), dense core with thin porous shell structure (Fig. 5e1 and e2), gradient emulsion structure (Fig. 5f1 and f2), double emulsion structure (Fig. 5g1 and g2), and complex triple and multiple emulsion structure (Fig. 5h1 and h2). Besides the fact that the internal structure of the oil droplet could also be adjusted in a wide range, the shell thickness, the pore diameter and the pore distribution are also finely tunable (Fig. S8–S10†). It should be noted that a gradient emulsion (Fig. 5f1 and f2) could be prepared with the 5:1 C8vim[Tf2N]–LiTf2N mixture. During the OPS process, water was absorbed into the oil phase. Subsequent water migration from the outer to the inner region of the oil phase was quite slow because of the hydrophobicity of C8vim[Tf2N]. Consequently, the outer region of the oil phase contained more water than the inner region, which generated a water gradient from the outside to the inside.
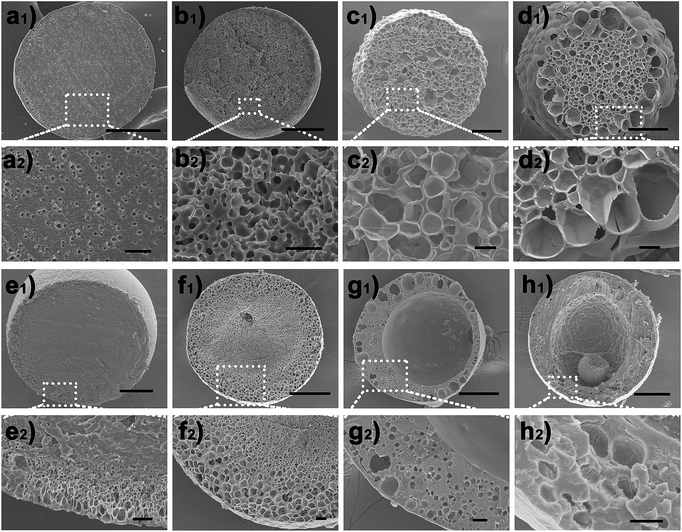 |
| | Fig. 5 (a–h) SEM images of diverse emulsion structures induced by judicious choosing of phase separation conditions. (a) Sea-island structure, (b) bicontinuous structure, (c and d) multicompartment structure, (e) anisotropic structure, (f) gradient structure, (g) double emulsion structure, and (h) multiple emulsion structure. Scale bars are 100 μm (a1–h1) and 10 μm (a2–h2). | |
Effect of salt concentration in the aqueous phase on the OPS process
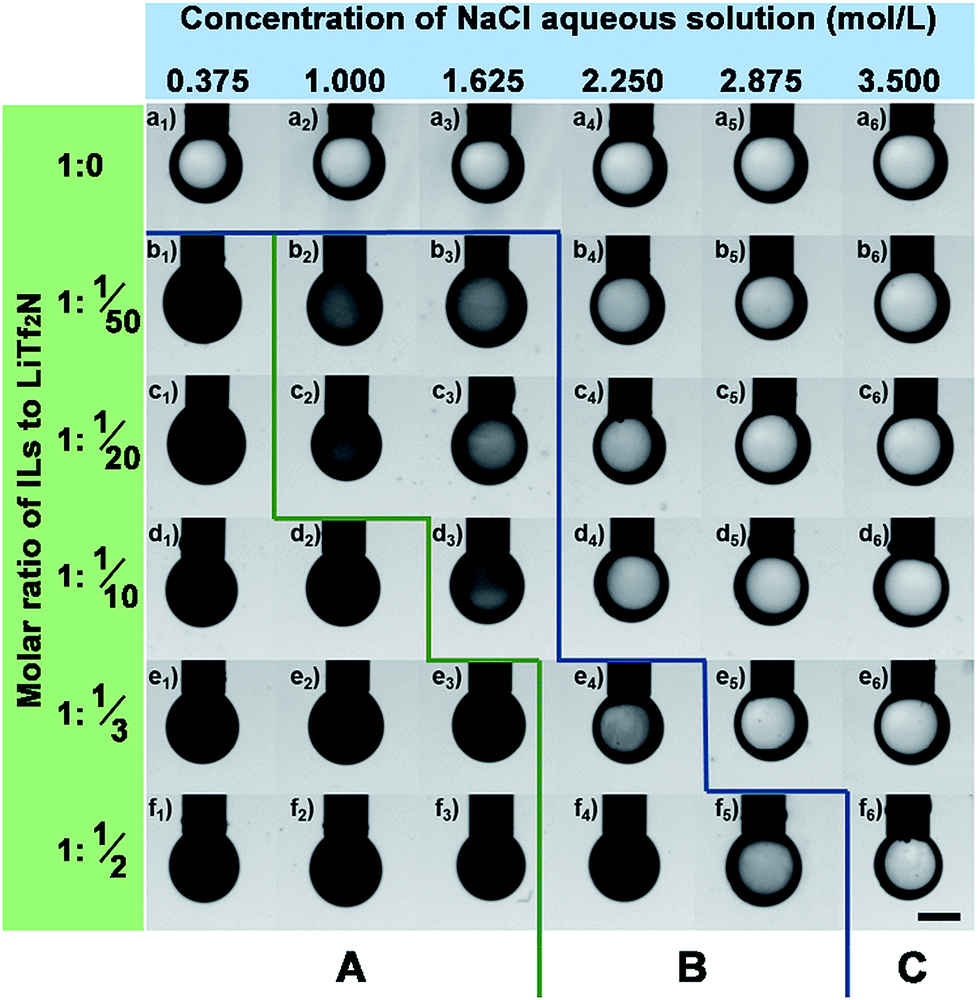
For a given Cnvim[Tf2N]–LiTf2N system the addition of salt in the aqueous phase could adjust the gradient of osmotic pressure between the oil phase and the aqueous phase, thus providing an alternative way of tailoring the thermodynamic driving force to further impact the kinetics involved in the OPS process. The transport of water molecules into or out of the IL oil phase during the phase separation process could be slowed down or stopped at any stage by simply adjusting the salt concentration in the aqueous phase. In our work, to demonstrate the ability to precisely control the kinetics through the alteration of the salt concentration in the aqueous phase, a series of parallel experiments was performed by exposing one oil droplet with a given C3vim[Tf2N]–LiTf2N composition to different concentrations of a NaCl aqueous phase (0.375 mol L−1 to 3.5 mol L−1), respectively. Fig. 6 presents the optical images of the pendent oil drops after their exposure to a NaCl aqueous solution for 10 min. Due to the insufficient osmotic pressure of pure C3vim[Tf2N], the OPS didn't take place as described above. As expected, the presence of the salt in the aqueous phase would further weaken the driving force for OPS, and the pendent droplets of pure C3vim[Tf2N] in all the cases remained transparent in appearance without the occurrence of the permeation of water molecules into the IL oil phase (Fig. 6a1–a6). However, in the case of C3vim[Tf2N] containing a small amount of LiTf2N (C3vim[Tf2N]:LiTf2N = 1:1/50), even in the presence of 0.375 mol L−1 NaCl the osmotic pressure difference between the IL oil and aqueous phase was still large enough to induce the OPS process quickly, and after 10 min the droplet became cloudy and dark as shown in the optical image (Fig. 6b1). But, with the increasing amount of the NaCl aqueous phase, the oil droplets only became gray and semi-transparent at a comparable time, indicating that the OPS process was appreciably slowed down (Fig. 6b). When a sufficient amount of NaCl was added (≥2.250 mol L−1 NaCl solution), the OPS process could be completely inhibited or blocked, and the droplets of C3vim[Tf2N]–LiTf2N remain transparent. As more LiTf2N salt was dissolved in C3vim[Tf2N], the enhanced osmotic pressure accelerated the transport of water molecules into the oil phase, and thus sped up the OPS process (see section A in Fig. 6). Nevertheless, for all C3vim[Tf2N]–LiTf2N cases it was found that the rigorous adjustment of the salt concentration in the aqueous phase could precisely regulate the speed of the OPS process (see section B in Fig. 6), stop it at any stage and even inhibit the OPS process (see section C in Fig. 6).
 |
| | Fig. 6 Optical images of the C3vim[Tf2N]–LiTf2N droplets after exposure to the NaCl aqueous solution for 10 minutes of phase separation. Molar ratio of C3vim[Tf2N]–LiTf2N is increased from 1:0 to 1:1/2. The concentration of the NaCl aqueous solution is increased from 0.375 to 3.5 mol L−1. The scale bar is 300 μm. | |
Reconfigurable OPS process
In this work, we also found that the direction of the mentioned OPS process could be effectively controlled by altering the salt (NaCl) concentration in the outer aqueous phase. Fig. 7a–d show the representative optical evolution of C8vim[Tf2N]–LiTf2N (molar ratio: 1:1/10) oil droplets exposed to a pure water phase. As described above, the osmotic pressure gradient between the IL oil phase and the aqueous phase drives the transport of water into oil to induce the OPS. With the permeation of water molecules the appearance of oil droplets changes gradually from initially transparent to cloudy semi-transparent, and finally to a dark emulsion state at equilibrium after several minutes. Interestingly, when a concentrated NaCl solution was subsequently added in the equilibrium system, the dehydration of the emulsion droplets was initiated and the water molecules absorbed inside the oil droplets could be retracted back into the aqueous phase and correspondingly the appearance of emulation droplets evolved from dark to transparent (Fig. 7e–h). Surprisingly, we found that the heterogeneous emulsion structure induced by OPS could be restored into the initial homogeneous oil phase. To better distinguish the structural evolution of oil droplets, a dye-doped C6vim[Tf2N]–LiTf2N (molar ratio: 1:1/5) oil system was employed for the OPS and its reverse process (dehydration). Clearly, similar results were observed (Fig. 7i–o). The heterogeneous structure induced by OPS could be returned to the initial homogeneous oil phase by changing the direction of the osmotic pressure imbalance. During the reverse process of phase separation (dehydration), an obvious boundary between the phase separation region and the non-phase separation region was clearly observed. Finally, the complex emulsion reverted to a homogeneous droplet. Indeed, DLS measurement confirmed that the restored phase is homogeneous (Fig. S11†), and no porous structure was detected after the polymerization of the restored oil droplets (see inset in Fig. 7h). This result implies that the process of the OPS in the Cnvim[Tf2N]–LiTf2N oil system could be modulated on demand by the aid of osmotic pressure imbalance. Over the past few decades, the osmotic pressure gradient has been widely exploited to control the direction of water transport across the oil layer between both aqueous phases in multiple emulsion systems. To the best of our knowledge, however, the described phenomenon that the heterogeneous multiple phase formed through phase separation was again restored to the initial homogeneous oil phase by changing the direction of the osmotic pressure imbalance has never been reported so far. This important finding together with the widely adjustable thermodynamic driving force and kinetic barrier indicates that the Cnvim[Tf2N]–LiTf2N mixture is a novel oil system, with which a highly kinetically controlled and reconfigurable OPS process could be realized.
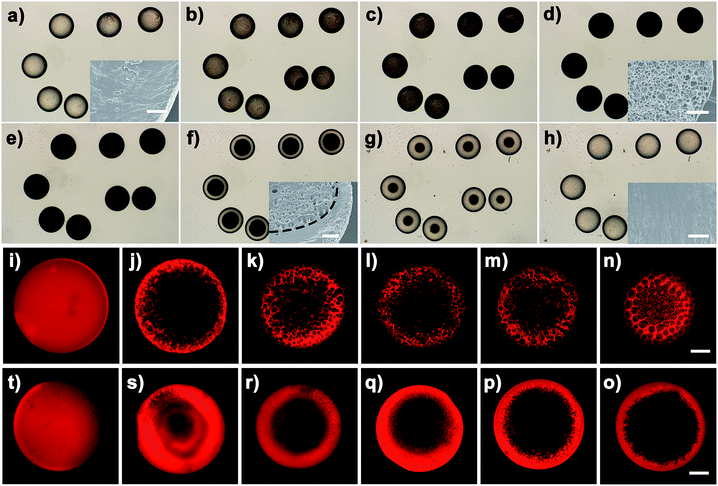 |
| | Fig. 7 (a–d) Optical images of the water absorption induced phase separation process by using C8vim-Tf2N/LiTf2N (1:1/10) microdroplets dispersed in water; (e–h) optical images of the reverted phase separation process from the phase-separated heterogeneous state to the homogeneous oil phase by redrawing the water molecules taken up after adding NaCl in the surrounding aqueous solution; the insets are SEM images of the complex emulsions. (i–t) CLSM monitoring of the reconfigurable OPS process by using C6vim[Tf2N]–LiTf2N (1:1/5) microdroplets doped with dye molecules. Scale bars are 400 μm (a–h), 15 μm (insets) and 100 μm (i–t). | |
Remarkable extendibility of the OPS using the Cnvim[Tf2N]–LiTf2N system
Due to the extraordinary multiple intermolecular interactions, including weak nonspecific forces (van der Waals, solvophobic, and dispersion forces) and strong specific forces (hydrogen bonding, dipole–dipole, and electron pair donor/acceptor interactions), ionic liquids exhibit a peculiar ability to dissolve or disperse a broad range of substances or nano-objects.35,36 This unique feature implies that the Cnvim[Tf2N]–LiTf2N system described here should serve as a general phase separation platform, and the phase separation process of numerous materials could be facilely achieved through dissolution or dispersion of the relevant materials in the Cnvim[Tf2N]–LiTf2N system. This strategy together with online photo-polymerization would afford various pore-structured functional materials, especially those that are inaccessible using conventional methods. As proof of our concept, several types of substances and nano-objects were dissolved or dispersed in Cnvim[Tf2N]–LiTf2N, and the OPS behavior of the Cnvim[Tf2N]–LiTf2N system remained even after the addition of other substances or nano-objects, and various emulsion mixtures were obtained. Indeed, it is found that the OPS structures were formed. In our case, eight classes of functional species, including ethoxylated trimethylolpropane triacrylate (cross-linker), an azobenzene-type monomer (photo-responsive compound), a tetraphenylethylene luminogen, N-acryloxysuccinimide (bioorthogonal reactive compound), graphene, carbon tubes, gold and TiO2 nanoparticles were incorporated into the Cnvim[Tf2N]–LiTf2N system, respectively (the chemical structures of the used molecules are shown in Fig. S12†). Fig. 8 shows the optical and SEM images after OPS followed by photopolymerization. Clearly, diverse functional porous materials were conveniently fabricated through the OPS strategy. And the successful incorporation of the functional species into the resulting porous materials was verified using different analytical methods, as shown in Fig. S13–S16.† In particular, for pristine carbon and other inorganic nanomaterials, which are difficult or impossible to be processed using conventional methods, our strategy offers an efficient way to produce pore-structured materials while still maintaining their intrinsic properties. Additionally, after the OPS process and polymerization, a series of functional porous materials could also be derived from the counter anion exchange of the original porous poly(ionic liquid)s (the Experimental method is in the ESI†). In this way, porous materials with oxidative (AuCl4−), reductive (vitamin C), and bio-active (dopamine) functions were created, as shown in Fig. S17.†
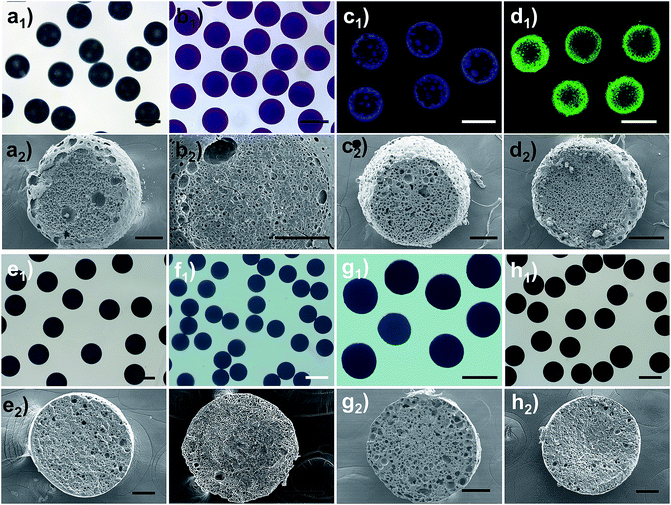 |
| | Fig. 8 Great extensibility of our proposed osmotically driven phase separation. Optical and SEM images of the formed pore-structured spheres by using C3vim[Tf2N]–LiTf2N as a general phase separation carrier platform for realizing the phase separation of various substances: (a1–2) ETPTA, (b1–2) azobenzene-type monomer, (c1–2) AIE-type monomer, (d1–2) bioorthogonal monomer (click reaction with GOX-FITC enzyme), (e1–2) TiO2 particles, (f1–2) Au nanoparticles, (g1–2) carbon nanotubes, and (h1–2) graphene. Scale bars are 400 μm (a1–h1) and 100 μm (a2–h2). | |
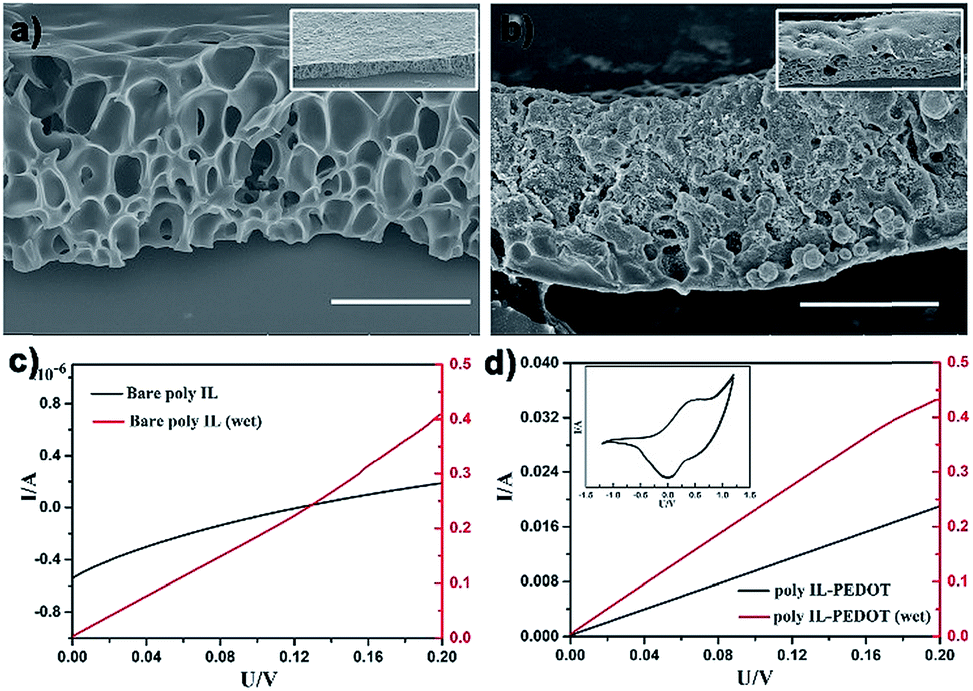
Apart from the spherical porous material, the OPS of the Cnvim[Tf2N]–LiTf2N system could be extended for large-scale fabrication of porous films or membranes, which have wide applications in separation, catalysis, electronics and so on. As a demonstration, C8vim[Tf2N]–LiTf2N (1:1/10) was employed for producing functional porous films. The C8vim[Tf2N]–LiTf2N oil film was first deposited on a glass slide by the spin-coating method. After immersion of the coated substrate in the water phase for OPS the resultant porous film was solidified by photopolymerization. This simple three-step procedure afforded large-area poly(IL) films with a well-defined interconnected pore structure, as shown in Fig. 9a. Poly(IL)s bearing Tf2N− are believed to have good ion transport efficiency.37 As can be seen in Fig. 9c, the prepared poly(IL) film interacting with LiTf2N salt shows ion transport properties, and these properties increased dramatically when the film composite was treated with aqueous vapor. In our work, the capillary force of the prepared porous film was exploited to introduce a 3,4-ethoxylenedioxythiophene (EDOT) monomer into the interconnected pore channels, and the subsequent polymerization using chloroauric acid as the oxidative agent led to an ion-conductive PIL film with integrated electric conductivity. The SEM image in Fig. 9b shows the structure of a composite film. The prepared composite film possesses a bi-channel structure for both ion and electron transport. As shown in Fig. 9d, in a drying state, the composite film shows electron conductivity of the PEDOT channel after being treated with I2 vapour. When the film was treated with water vapour, enhanced conductivity was detected, which is attributed to the additional ion conductivity of the PIL–LiTf2N channel. In principle, all recipes demonstrated above regarding droplets could also be used to construct functional porous materials in the film format. All these results indicate that the application scope of the OPS using the Cnvim[Tf2N]–LiTf2N system could be greatly extended, presenting tremendous potential for facilely developing functional porous materials through a simple OPS process.
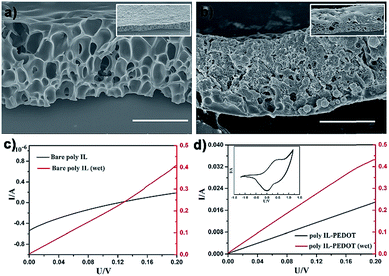 |
| | Fig. 9 SEM images of a bicontinuous poly(ionic liquid) membrane (a) and poly(ionic liquid)–PEDOT composite (b). Electrochemical curves of bare bicontinuous poly(ionic liquid) in the dry and wet state (c). Electrochemical curves for poly(ionic liquid)–PEDOT (doping with I2 vapour) in the dry and wet state (d); the inset image is the CV sweep of the poly(ionic liquid)–PEDOT membrane. The scale bar is 20 μm. | |
Discussion
Over the past few decades, exploiting mass transfer to induce water/oil phase separation for the creation of multiple emulsions has been intensively investigated. Most commonly, homogeneous ternary mixtures composed of two immiscible liquids and a cosolvent, miscible with both liquids (e.g., a short chain alcohol, THF, or acetonitrile), have been employed in most studies.34,38–41 Once the mixture systems are brought into contact with the aqueous phase, the rapid diffusion of cosolvent into the aqueous phase can induce local supersaturation near the interface, thus giving rise to water/oil liquid phase separation and spontaneous formation of an emulsion structure. Such spontaneous liquid–liquid phase separation, referred to as self-emulsification or spontaneous emulsification,41 has also been observed in many oil systems with oil-soluble surfactants or amphiphilic block copolymers. But in these cases the phase separation process is relatively slow, and only simple emulsion structures like multicore morphologies are generally induced. The mechanism of this phenomenon is still not completely understood, and interfacial turbulence, negative interfacial tension, and diffusion and stranding were prominently proposed for the explanation of these spontaneous processes.42 Recently, T. P. Russell and R. C. Hayward have reported that the presence of salt impurity aggregates within the chloroform solutions of amphiphilic block copolymers could provide an osmotic driving force, triggering a spontaneous emulsification process after the organic phase was brought into contact with water.43 Under the osmotic driving force water molecules were drawn out of the surrounding water phase through chloroform, each salt aggregate within the organic phase was inflated into a single emulsion droplet, and finally double emulsions with a controlled multicore architecture were formed by adjusting the initial characteristics of the salt aggregates. Nevertheless, as the solubility of inorganic salts in the common organic solvents is quite low, and thus the generated osmotic pressure is not sufficient to counterbalance the Laplace pressure of the tiny water droplets formed within the organic oil phase, to the best of our knowledge the osmotically driven water/oil liquid–liquid phase separation has not been achieved in a homogeneous oil phase so far.
As mentioned in the Introduction section, the unique dual ionic and organic nature of ionic liquids provide them with a special affinity to inorganic salts and thus exceptional solubility of inorganic salts, in particular in the case of Cnvim[Tf2N]–LiTf2N. Due to the multiple interactions between the Cnvim[Tf2N] ionic liquid and LiTf2N salt, the solubility ratio of LiTf2N in hydrophobic Cnvim[Tf2N] oil reaches 0.5, which much exceeds the saturated solubility ratio of NaCl in water (0.12). We discovered that the salt species dissolved in the special ionic liquid oils could provide sufficient osmotic stress to draw water out of the water phase and into the oil phase, initiating water/oil liquid–liquid phase separation and spontaneously forming emulsion structures. In the case described by T. P. Russell and R. C. Hayward, the osmotically driven water/oil liquid–liquid phase takes place in a heterogeneous system through water transport between salt impurity aggregates and the bulky water phase, and thus the final structures of the osmotically formed emulsions are not sensitive to the kinetics, but instead only to the initial characteristics of the salt aggregates.43,44 Completely different from this situation, the liquid–liquid phase separation osmotically driven by the molecule-level dissolved salts in oil described here could induce the formation of emulsions with numerous and diverse morphologies dependent on the adopted kinetic pathway using one system. Indeed, the remarkably high solubility of LiTf2N in Cnvim[Tf2N] oil provides an unprecedented degree and room for modulating the osmotic pressure, namely the thermodynamic driving force for phase separation. The facile variation in the alkyl length of the Cnvim[Tf2N] molecule can easily tailor the kinetic barrier during phase separation. Additionally, the alteration of the salt concentration in the bulky water phase can control the direction of the phase separation process, and even the water taken-up in the formed heterogeneous emulsions can be drawn back into the bulky water phase to restore the initial homogeneous oil phase. These attributes indicate that based on the discovery of osmotically driven reversible water flow between aqueous and IL oil phases a new system could be developed towards highly kinetically controlled and even reconfigurable phase separation. As a result, the kinetic pathway of phase separation can be modulated in an unprecedented broad range, and arresting a variety of thermodynamically metastable structures induced is possible. More importantly, due to the extraordinary multiple types of molecular interactions involved in ILs, including van der Waals force, electrostatic force, hydrogen bonding, hydrophobic interaction, and π–π interaction, one of the distinctive properties of ILs is their excellent solvency for a broad range of substances.25,26 ILs can even display peculiar solubility or dispersibility of these compounds or objects (e.g. biomass, carbon materials, or inorganic nanoparticles), whose processability is difficult or impossible using common organic solvents. This advantage implies that the Cnvim[Tf2N]–LiTf2N system could be extended or utilized as a general carrier platform for phase separation, providing great opportunities for achieving the phase separation of a broad range of substances and correspondingly the creation of nanostructured functional materials.
Conclusions
In summary, by exploiting the unique solubility of inorganic salts in hydrophobic ionic liquids and thus the generated osmotic pressure, for the first time osmotically driven spontaneous water/oil phase separation (OPS) was achieved using the Cnvim[Tf2N]–LiTf2N oil solution. Compared to the liquid–liquid phase separations reported so far, this osmotically driven phase separation process is characterized by the following distinct features. Firstly, owing to the extraordinary solubility of inorganic salts in ionic liquids as well as the easily tailorable molecular structure of Cnvim[Tf2N] the thermodynamic (osmotic) driving force and kinetic barrier of water transport during the OPS process could be facilely modulated with unprecedented flexibility and range. Secondly, by simply adding salt into the aqueous phase, the OPS process is further finely tunable and even the phase separation direction could be adjustable. Surprisingly, it is found that the already phase-separated heterogeneous state could even be reverted to restore the homogeneous oil phase by redrawing the water molecules taken up, indicating that the OPS is reconfigurable. Based on these attractive features, precise design and control of the speed and kinetic pathways of the phase separation process is possible, thus offering unlimited opportunities to facilely access and capture diverse thermodynamically metastable structures using one phase separation system. Importantly, the unique structure of ionic liquids allows the extraordinary solubility or dispersity of a broad range of substances in ionic liquids. Nevertheless, we found that after introduction of the various species the phase separation behavior of the Cnvim[Tf2N]–LiTf2N oil system still remained without significant change, indicating that the Cnvim[Tf2N]–LiTf2N system can serve as a general phase separation carrier platform for realizing the phase separation of various substances, especially those inaccessible using conventional methods. In this work, although only imidazolium-based ILs are explored for OPS, in principle our concept of osmotically driven oil/water phase separation could be extended to other ionic liquids, including pyridinium- and phosphonium-based ionic liquids. Therefore, with the simplicity, controllability, versatility, and potential scalability our findings would open up new avenues for efficiently producing oil/water phase separations, which could hold enormous potential for creating novel structures and chemical systems or in the development of new actuators or adaptive materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from the NSF China (No. 21773135, 21473098, 21121004 and 21421064), MOST (2017YFA0204501 and 2013CB834502), and the Deutsche Forschungsgemeinschaft DFG (TRR61).
Notes and references
-
P. Atkins and J. de Paula, Atkins' Physical Chemistry, Oxford University Press, United Kingdom, 2014 Search PubMed.
- V. W. Sidel and A. K. Solomon, J. Gen. Physiol., 1957, 41, 243–257 CrossRef CAS.
- E. L. Fiscus, Plant Physiol., 1975, 55, 917–922 CrossRef CAS.
- R. Munns, Aust. J. Plant Physiol., 1988, 15, 717–726 Search PubMed.
- S. E. Skilhagen, J. E. Dugstad and R. J. Aaberg, Desalination, 2008, 220, 476–482 CrossRef CAS.
- Y. C. Kim and M. Elimelech, J. Membr. Sci., 2013, 429, 330–337 CrossRef CAS.
- Y. Kim, M. Elimelech, H. K. Shon and S. Hong, J. Membr. Sci., 2014, 460, 206–212 CrossRef CAS.
- C. X. Guo, D. Zhao, Q. Zhao, P. Wang and X. Lu, Chem. Commun., 2014, 50, 7318–7732 RSC.
- J. G. Park, S. H. Kim, S. Magkiriadou, T. M. Choi, Y. S. Kim and V. N. Manoharan, Angew. Chem., Int. Ed., 2014, 53, 2899–2903 CrossRef CAS.
- A. Peyret, E. Ibarboure, A. Tron, L. Beaute, R. Rust, O. Sandre, N. D. Mcclenaghan and S. Lecommandoux, Angew. Chem., Int. Ed., 2017, 56, 1566–1570 CrossRef CAS.
- J. J. F. Sleeboom, P. Voudouris, M. T. J. J. M. Punter, F. J. Aangenendt, D. Florea, P. V. D. Schoot and H. M. Wyss, Phys. Rev. Lett., 2017, 119, 98001 CrossRef PubMed.
-
(a) W. Zhang, A. Abbaspourrad, D. Chen, E. Campbell, H. Zhao, Y. Li, Q. Li and D. A. Weitz, Adv. Funct. Mater., 2017, 27, 1700975 CrossRef;
(b) S.-H. Kim, J.-G. Park, T. M. Chio, V. N. Manoharan and D. A. Weitz, Nat. Commun., 2014, 5, 3068, DOI:10.1038/ncomms4068.
- N. Garti, LWT--Food Sci. Technol., 1997, 30, 222–235 CrossRef CAS.
- J. A. Omotosho, Int. J. Pharm., 1990, 62, 81–84 CrossRef CAS.
- D. J. Mcclements and K. Demetriades, Crit. Rev. Food Sci. Nutr., 1998, 38, 511–536 CrossRef CAS.
- S. Iqbal, M. K. Baloch, G. Hameed and D. J. McClements, Food Res. Int., 2013, 54, 1613–1620 CrossRef CAS.
- S. Matsumoto, T. Inoue, M. Kohda and K. Ikura, J. Colloid Interface Sci., 1980, 77, 555–563 CrossRef CAS.
- H. L. Rosano, F. G. Gandolfo and J. D. P. Hidrot, Colloids Surf., A, 1998, 138, 109–121 CrossRef CAS.
- L. Wen and K. D. Papadopoulos, J. Colloid Interface Sci., 2001, 235, 398–404 CrossRef CAS.
- R. Mezzenga, B. M. Folme and E. Hughes, Langmuir, 2004, 20, 3574–3582 CrossRef CAS.
- J. Zhu and R. C. Hayward, J. Colloid Interface Sci., 2012, 365, 275–279 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS.
- R. Shi and Y. Wang, Sci. Rep., 2016, 6, 19644 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- A. B. Pereiro, J. M. M. Araujo, F. S. Oliveira, J. M. S. S. Esperanca, J. N. C. Lopes, I. M. Marrucho and L. P. N. Rebelo, J. Chem. Thermodyn., 2012, 55, 29–36 CrossRef CAS.
- V. L. Martins, B. G. Nicolau, S. M. Urahata, M. C. C. Ribeiro and R. M. Torresi, J. Phys. Chem. B, 2013, 117, 8782–8792 CrossRef CAS.
- M. Y. Lui, L. Crowhurst, J. P. Hallett, P. A. Hunt, H. Niedermeyer and T. Welton, Chem. Sci., 2011, 2, 1491–1496 RSC.
- M. J. Monteiro, F. F. C. Bazito, L. J. A. Siqueira, M. C. C. Ribeiro and R. M. Torresi, J. Phys. Chem. B, 2008, 112, 2102–2109 CrossRef CAS PubMed.
- T. Nisisako and T. Torii, Lab Chip, 2008, 8, 287–293 RSC.
- M. F. Haase, K. J. Stebe and D. Lee, Adv. Mater., 2015, 27, 7065–7071 CrossRef CAS.
- Y. Cheng, F. Zheng, J. Lu, L. Shang, Z. Xie, Y. Zhao, Y. Chen and Z. Gu, Adv. Mater., 2014, 26, 5184–5190 CrossRef CAS.
- M. F. Haase and J. Brujic, Angew. Chem., Int. Ed., 2014, 53, 11793–11797 CrossRef CAS.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS.
- G. T. Wei, Z. Yang, C. Y. Lee, H. Y. Yang and C. R. C. Wang, J. Am. Chem. Soc., 2004, 126, 5036–5037 CrossRef CAS.
- W. Ogihara, S. Washiro, H. Nakajima and H. Ohno, Electrochim. Acta, 2006, 51, 2614–2619 CrossRef CAS.
- C. X. Zhao and A. P. J. Middelberg, Angew. Chem., Int. Ed., 2009, 48, 7208–7211 CrossRef CAS PubMed.
- C. H. Choi, D. A. Weitz and C. S. Lee, Adv. Mater., 2013, 25, 2536–2541 CrossRef CAS PubMed.
- C. Solans and I. Sole, Curr. Opin. Colloid Interface Sci., 2012, 17, 246–254 CrossRef CAS.
- C. Solans, D. Morales and M. Homs, Curr. Opin. Colloid Interface Sci., 2016, 22, 88–93 CrossRef CAS.
- J. C. Lopez-Montilla, P. E. Herrera-Morales, S. Pandey and D. O. Shah, J. Dispersion Sci. Technol., 2002, 23, 219–268 CrossRef CAS.
- J. Bae, T. P. Russell and R. C. Hayward, Angew. Chem., Int. Ed., 2014, 53, 8240–8245 CrossRef CAS.
- X. Wang, X. Feng, G. Ma, D. Zhang, Y. Chai, M. Ge and L. Yao, Adv. Mater., 2017, 1605932 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01649j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*