
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-catalysed vinyl 1,4-conjugate addition coupled with Sharpless asymmetric dihydroxylation in the synthesis of the CDE ring fragment of pectenotoxin-4†
Melodie S. W.
Richardson
a,
Christopher J.
Tame
b,
Darren L.
Poole
b and
Timothy J.
Donohoe
 *a
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: timothy.donohoe@chem.ox.ac.uk
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 24th May 2019
Abstract
Our synthesis of the CDE ring fragment of pectenotoxin-4 utilised two key steps to make the complex bicyclic ketal unit: (i) a rhodium-catalysed vinyl group 1,4-addition as the major C–C bond forming step; (ii) a stereoselective Sharpless Asymmetric Dihydroxylation (SAD) of the resulting 1,1-disubstituted homoallylic alcohol. Subsequent acid-catalysed cyclisation afforded the desired [5,6]-bicyclic ketal of the target molecule. This methodology was shown to be compatible with the desired E ring fragment 35 in order to construct the CDE fragment 37 of pectenotoxin-4.
Introduction
The pectenotoxins (PTXs) are a family of polyether macrolides containing a spiroketal (AB ring), three substituted tetrahydrofurans (C, E and F rings) and 19 or more stereocentres decorating the 40-carbon chain.1 These intriguing natural products were first isolated in 1985 by Yasumoto and coworkers,2 and have been shown to exhibit potent biological activity, including selective cytotoxicity against tumour cell lines.3The architectural complexity of these highly functionalised macrolactones have garnered significant interest within the synthetic chemistry community,4 however only two total syntheses of these molecules have been completed to date: PTX-4 by Evans in 2002 (ref. 5) and PTX-2 by Fujiwara in 2014.6

The Donohoe group has made several significant advances towards the total synthesis of PTX-4. We have successfully synthesised the C-1 to C-16 ABC fragment via a double osmium catalyzed oxidative cyclisation together with a hydride-shift-initiated spiroketalisation,7 as well as preparing the C-21 to C-40 EFG fragment via stereodivergent catalytic cobalt and osmium oxidative cyclisations (Scheme 1a).8
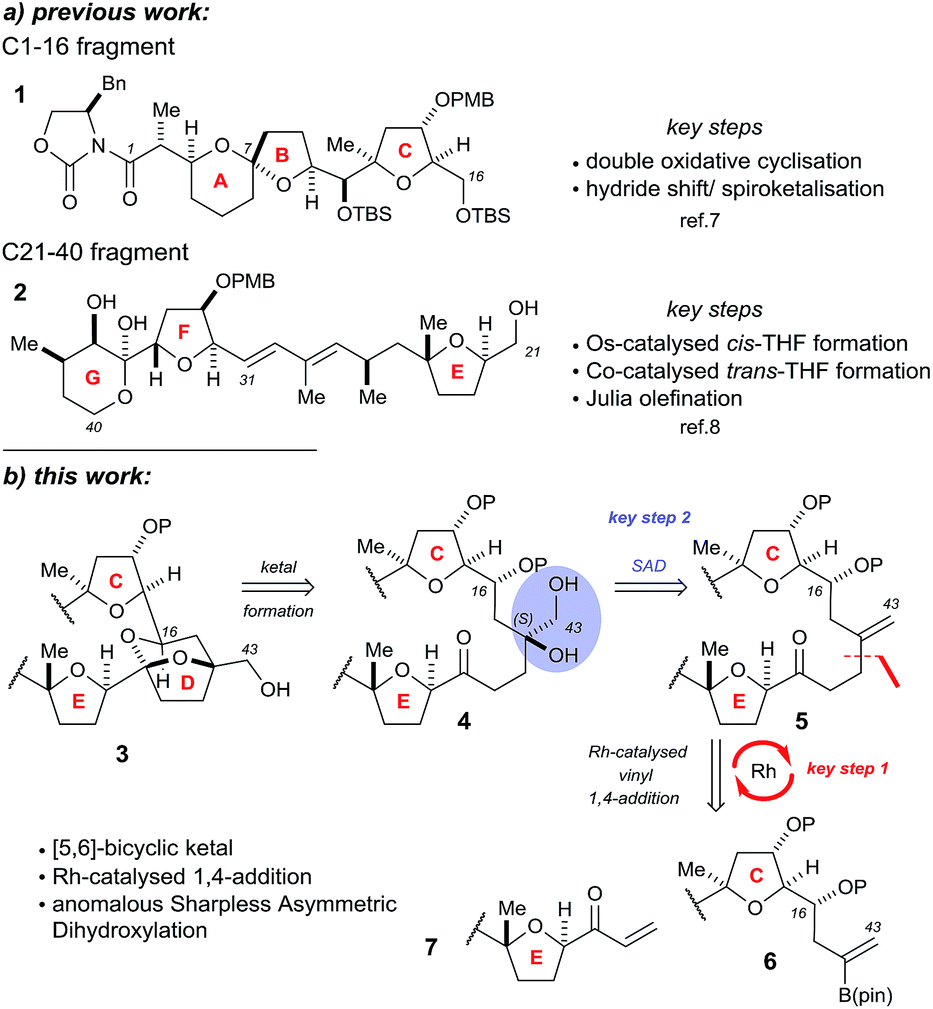 | ||
| Scheme 1 Previously synthesised fragments and key disconnections proposed in this work for the CDE fragments: (a) previous work; (b) this work. | ||
The major challenge remaining in our synthesis of PTX-4 is uniting these two complex fragments to synthesise the final [5,6]-bicyclic ketal, the D ring. A handful of approaches to this bicyclic structure for PTX-2 (ref. 4o, s, t, x, y, z and 6) and PTX-4 (ref. 4d, x and 5) have been published in the literature. Herein, we propose a novel route which proceeds via an unusual rhodium-catalysed vinyl 1,4-addition9 as the key C–C bond formation step to join the ABC and E ring fragments. It is important to note that this key reaction has the potential to allow complex molecular fragments to be joined under relatively mild conditions and without using a large excess of either, extremely valuable, component. A subsequent stereoselective dihydroxylation–ketalisation sequence should then afford the desired [5,6]-bicyclic ketal structure of the D ring of PTX-4 (Scheme 1b). Note here that the sensitive diene containing FG ring fragment will be constructed as it is attached to the E-ring by a Julia reaction, after cyclisation of the D ring, because the 1,3-diene fragment itself would be unlikely to survive the conditions needed for dihydroxylation and/or cyclisation.
Results and discussion
To begin, we chose to use model C ring boronate 15 as a substitute for the real ABC ring fragment required in the synthesis of PTX-4 (Scheme 2). Starting from commercially available enantiopure furanose 8, the hemiacetal was reduced to the corresponding THF 9, and the primary benzyl group removed in two steps, via acetate 10, to reveal 11.10 Oxidation of the primary alcohol to the aldehyde followed by a Hosomi–Sakurai reaction11 with bromoallylsilane 12 (ref. 12) afforded the (R)-homoallylic alcohol 13 as a single diastereoisomer in 56% yield over two steps. The stereochemistry arises from Felkin–Ahn-controlled addition of the bromoallylsilane 12 and was confirmed via Mosher's ester analysis.13 Direct conversion of the bromide to the desired boronate was unsuccessful; therefore protection of the secondary alcohol 13 with TESOTf was necessary. Miyaura borylation of TES-protected bromide 14 to the model C ring boronate 15 was then accomplished in 79% yield.14 | ||
| Scheme 2 Synthesis of the model C ring boronate 15. Reagents and conditions: (i) Et3SiH, BF3·OEt2, MeCN, 95%; (ii) Ac2O, TFA; (iii) NaOMe, MeOH, 89% over two steps; (iv) DMSO, SO3·py, Et3N, CH2Cl2, 0 °C; (v) 12, BF3·OEt2, CH2Cl2, −78 °C, 56% over two steps; (vi) TESOTf, imidazole, CH2Cl2, 85%; (vii) (B(pin))2, PdCl2(PPh3)2 (3 mol%), PPh3 (6 mol%), KOPh, PhMe, 50 °C, 79%. | ||
Similarly, we started with a less substituted THF ring in place of the desired E ring fragment in our initial studies (Scheme 3). Therefore, (R)-tetrahydrofurfuryl alcohol 16 was oxidised to the corresponding aldehyde 17, and vinyl Grignard reagent was added to afford volatile allyl alcohol 18 in 35% yield (1.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr at the hydroxyl centre) over two steps. Oxidation using DMP furnished the model E ring enone 19 in 89% yield.
:1 dr at the hydroxyl centre) over two steps. Oxidation using DMP furnished the model E ring enone 19 in 89% yield.
 | ||
| Scheme 3 Synthesis of the model E ring enone 19. Reagents and conditions: (i) DMSO, (COCl)2, Et3N, CH2Cl2, −78 °C; (ii) vinyl magnesium bromide, Et2O, −78 °C, 35% over two steps, 1.15:1 dr; (iii) DMP, CH2Cl2, 89%. | ||
Using rhodium-catalysed 1,4-addition conditions9 on model C ring boronate 15 with 19 was successful and afforded the desired adduct 20 in approximately 40% yield. However, the use of methanol as the solvent formed the methanol 1,4-addition adduct of compound 19 as a by-product, which often co-eluted with the desired products. Pleasingly, we found that replacing methanol with THF as the solvent prevented the formation of this by-product and improved the yield to 63% for the reaction between 15 and 19 (Scheme 4).
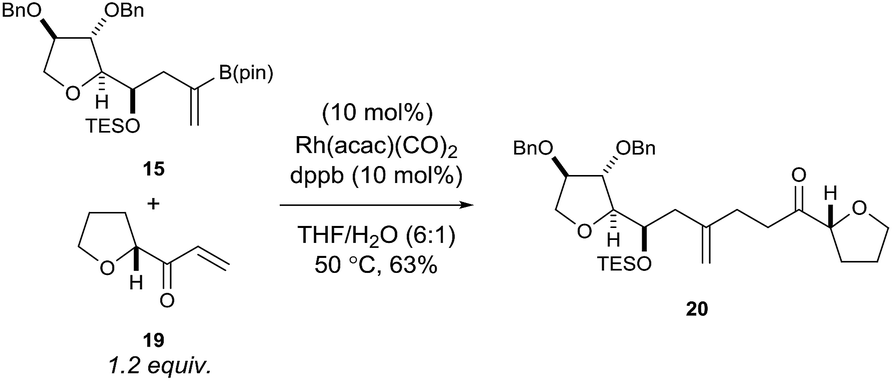 | ||
| Scheme 4 Rhodium-catalysed 1,4-addition reaction of model C ring boronate 15 with model E ring enone 19. | ||
In order to ensure that no epimerisation had taken place adjacent to the ketone carbonyl, we repeated the coupling between 15 and racemic 19 (compound S6 prepared separately, see ESI† for details). This reaction gave two diastereoisomeric compounds (in an approximately 1:1 ratio) and from the 13C NMR spectrum of this mixture we could rule out epimerisation in compound 20 formed from enantiopure 19.
In order to construct the bicyclic acetal D-ring system we next required a facially selective dihydroxylation of the alkene within 20 (to set the stereochemistry at C7, Scheme 5) followed by a ketalisation reaction. Although the stereochemical outcome of dihydroxylation of 1,1-disubstituted alkenes are difficult to predict,15 we chose to use the Sharpless Asymmetric Dihydroxylation (SAD) to control diol formation. It is worth noting that the original mnemonic proposed by Sharpless for the SAD reaction16 is often problematic when applied to 1,1-disubstituted alkenes, as first shown by Hale.15a In the case of substrate 20, even if we could achieve near “perfect” facial selectivity for the correct diol 21, acid-catalysed cyclisation could then result in three possible isomeric bicyclic ketal structures: the desired [5,6]-ketal 23, [6,7]-ketal 24 and [5,5]-ketal 25 (Scheme 5).
 | ||
| Scheme 5 Possible bicyclic ketal structures arising from diols 21 and 22. | ||
At first the dihydroxylation of 1,1-disubstituted alkene 20 was attempted using Upjohn conditions17 to reveal any substrate bias during oxidation, however purification and identification of the desired diol was difficult as a complex mixture of products were obtained, possibly due to TES group migration. It was proposed to cyclise the crude diol using mildly acidic conditions5b while also removing the TES group; however the use of this procedure still produced in a complex mixture.
Undeterred, we chose the Sharpless Asymmetric Dihydroxylation (SAD) conditions to obtain the desired diol stereochemistry. As it is difficult to predict which ligand is required, we used both (DHQ)2PHAL and (DHQD)2PHAL separately. According to the mnemonic,16 we predicted (DHQ)2PHAL would produce the desired diol. However, using (DHQ)2PHAL ligand in the dihydroxylation and acid-induced cyclisation sequence produced a mixture of products (Scheme 6). Nevertheless, upon derivatisation of the mixture with 4-bromobenzoic acid we identified [5,6]-bicyclic ketal 27, with a characteristic 13C NMR peak at 108.4 ppm.4d The connectivity, supported by COSY/HSQC/HMBC experiments, was shown to be the [5,6]-ketal over the [6,7] or [5,5] isomers. Moreover, the relative stereochemistry of dihydroxylation could also be assigned as shown, because within the [5,6]-ketal structure we did not observe an nOe enhancement across the ring system (i.e. between C-5 to either C-8 or C-9); this would be expected in the desired ketal structure. Looking at the full set of data we concluded that compound 27 contained the [5,6]-bicyclic ketal but with the opposite stereochemistry at C-7 (as set by the initial dihydroxylation).
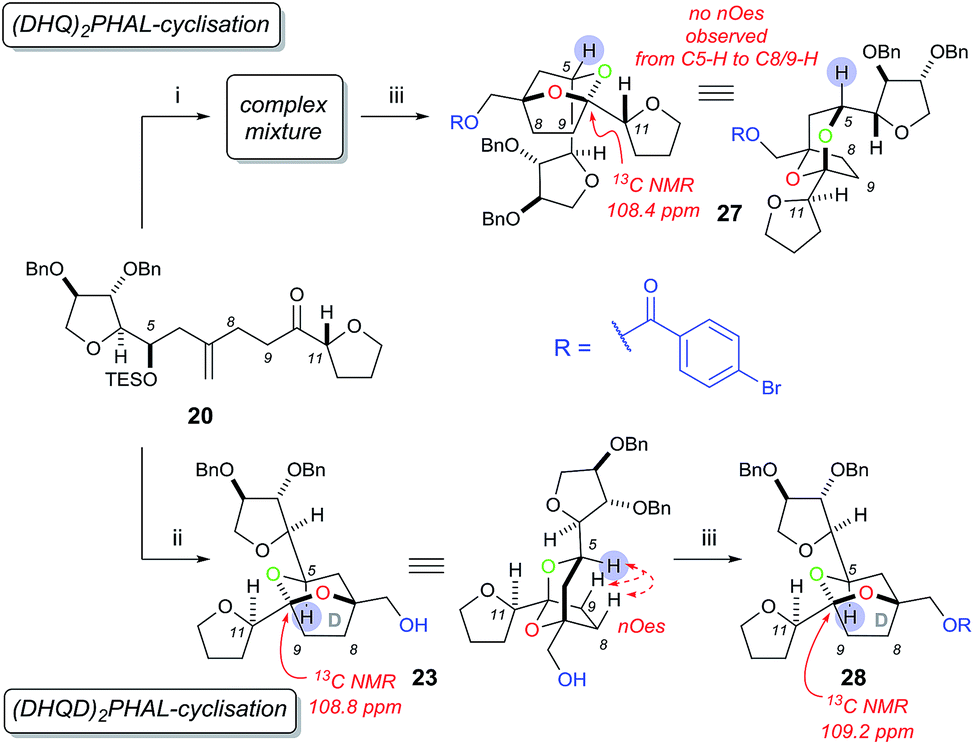 | ||
| Scheme 6 Sharpless asymmetric dihydroxylation and acid-catalysed cyclisation of 20. Reagents and conditions: (i) K2OsO2(OH)4, (DHQ)2PHAL, K3Fe(CN)6, K2CO3, MeSO2NH2, t-BuOH/H2O (1:1), 0 °C then PPTS, CH2Cl2/MeOH (1:1); (ii) K2OsO2(OH)4, (DHQD)2PHAL, K3Fe(CN)6, K2CO3, MeSO2NH2, t-BuOH/H2O (1:1), 0 °C then PPTS, CH2Cl2/MeOH (1:1), 91%; (iii) 4-bromobenzoic acid, DIC, DMAP, CH2Cl2, 5% over two steps for 27, 53% for 28. | ||
Interestingly, the use of (DHQD)2PHAL in the dihydroxylation–cyclisation sequence (oxidation being followed by treatment with acid) also provided one bicyclic ketal diastereoisomer 23 with a 13C NMR peak at 108.8 ppm (Scheme 6). Upon detailed NMR (COSY/HSQC/HMBC) analysis, 23 was again confirmed to have the [5,6]-bicyclic ketal connectivity. However, this time the molecule did exhibit key nOe enhancements across the bicyclic ring (C-5 to C-8 and C-9), showing it to be the desired [5,6]-bicyclic ketal 23 originating from the correct stereochemistry at C7. Further derivatisation of 23 with 4-bromobenzoic acid gave compound 28 which was different to the related ketal (27) formed from the (DHQ)2PHAL derived experiments.
We note that other bicyclic ketals ([6,7] and [5,5]) were not isolated in any reaction, however there have been reports that these types of structures may undergo facile degradation upon purification and may not be isolatable.4d Our studies show that in this system it is the (DHQD)2PHAL ligand that delivers the correct facial selectivity during dihydroxylation, and that acid-catalysed ketalisation then forms the desired [5,6]-ketal system as found in the natural product. The fact that (DHQD)2PHAL has formed the (S)-diol 21 during dihydroxylation is consistent with the reversed stereoselectivity that has been reported during the SAD reaction of 1,1-disubstituted alkenes.15a–e,i
To further test this methodology in the synthesis of pectenotoxin-4, we converted the desired E ring fragment 29 (ref. 8) into the desired enone 35, with the unsaturated ethyl ester side chain serving as a precursor for a Julia olefination coupling with the FG ring fragment. Therefore, the previously reported E fragment enantiopure 29 (ref. 8) was deprotected with TBAF, before Parikh–Doering oxidation to the aldehyde and vinyl Grignard reagent was added to afford allyl alcohol 30 (Scheme 7). The hydroxyl group was protected with TBSCl, before the Weinreb amide was reduced to the aldehyde with DIBAL-H and a Horner–Wadsworth–Emmons reaction with ylide 32 furnished (E)-unsaturated ethyl ester 33 (stereochemistry proven by nOe analysis). Finally, removal of the TBS group with TBAF followed by oxidation with DMP afforded the E ring enone 35.
 | ||
| Scheme 7 Synthesis of the E ring enone 35. Reagents and conditions: (i) TBAF, THF, 0 °C, 96%; (ii) SO3·py, Et3N, DMSO, CH2Cl2, 0 °C; (iii) vinyl magnesium bromide, Et2O, 0 °C, 69% over two steps; (iv) TBSCl, imidazole, DMAP, CH2Cl2, 88%; (v) DIBAL-H, THF, −78 °C; (vi) 32, benzene, Δ, 87% over two steps; (vii) TBAF, THF, 78%; (viii) DMP, NaHCO3, CH2Cl2, 90%. | ||
Initial rhodium-catalysed 1,4-addition reaction conditions between model C ring boronate 15 and E ring enone 35 were moderately successful, affording adduct 36 in 30% yield (Scheme 8). Repeating the reaction with a more active catalyst system, [Rh(cod)OH]2,18 improved the yield of 36 to 53%. Pleasingly, the dihydroxylation–cyclisation sequence (using (DHQD)2PHAL ligand) then afforded the desired CDE fragment 37 in 40% yield, as a single compound, with a characteristic 13C NMR peak at 108.0 ppm. Other ligands tested in the osmium catalyzed dihydroxylation of 36, such as (DHQD)2PYR and (DHQD)2AQN, did not improve the yield. It should be noted that the omission of methanesulfonamide and careful monitoring of the reaction progress was required to avoid over-oxidation of the ethyl ester substituted alkene. Moreover, the structure of 37 was confirmed with COSY/HSQC/HMBC NMR experiments to be the desired [5,6]-ketal and the stereochemistry was then assigned by the nOes observed across the bicyclic ring system (C-16 to C-19 and C-20) as was the case for compound 23. In this case, experiments performed to dihydroxylate and cyclise 36 using the opposite chiral ligand (i.e. (DHQ)2PHAL) only resulted in the formation of a complex mixture of products.
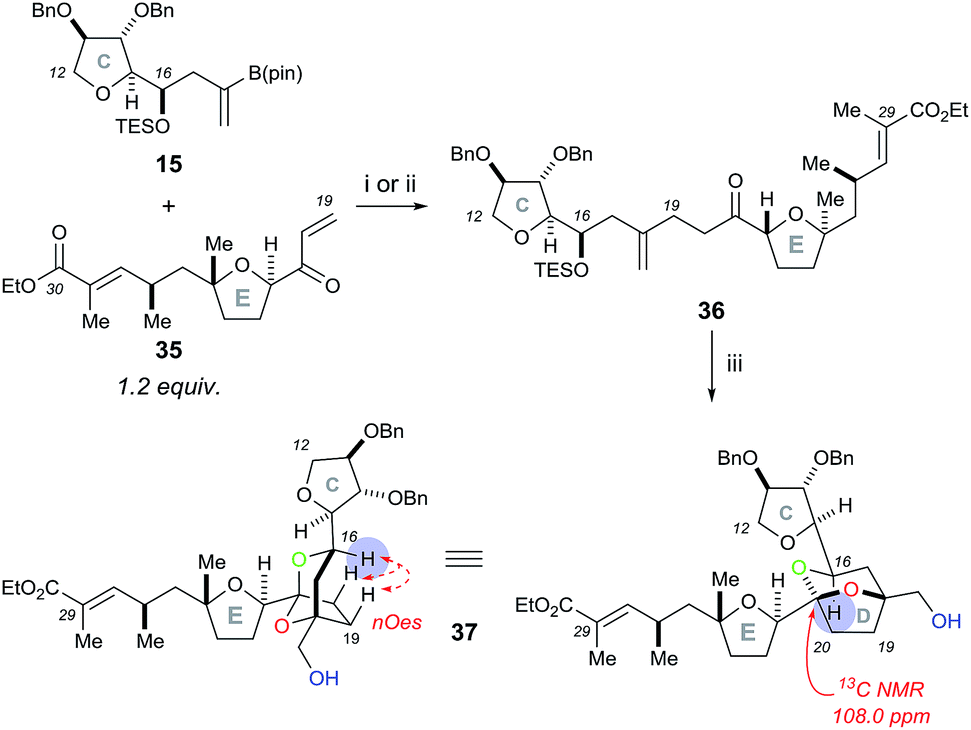 | ||
| Scheme 8 Rhodium-catalysed 1,4-addition reaction of the model C ring boronate 15 with E ring enone 35, followed by the Sharpless asymmetric dihydroxylation and acid-catalysed cyclisation sequence to access the CDE fragment 37. Reagents and conditions: (i) Rh(acac)(CO)2 (10 mol%), dppb (10 mol%), THF/H2O (6:1), 50 °C, 30%; (ii) [Rh(cod)OH]2 (15 mol%), THF/H2O (6:1), 50 °C, 53%; (iii) K2OsO2(OH)4, (DHQD)2PHAL, K3Fe(CN)6, K2CO3, t-BuOH/H2O (1:1), 0 °C then PPTS, CH2Cl2/MeOH (1:1), 40%. | ||
Conclusions
In conclusion, we have developed a novel route to the CDE fragment (C-12 to C-30) of PTX-4. The key C–C bond forming step was a rhodium catalysed 1,4-vinyl group addition to an enone which used a close to equimolar ratio of the two key components. Model studies revealed a reversal of ligand-facial selectivity during the SAD reaction of a 1,1-disubstituted homoallylic alcohol, resulting in the isolation of two different [5,6]-bicyclic ketals depending on the chiral ligand used. This methodology was then extended to incorporate the desired E ring fragment of PTX-4 in the synthesis of a CDE fragment of PTX-4. Further work is ongoing to utilise this methodology and complete the total synthesis of PTX-4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. W. R. is grateful to the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1) for studentships, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB and Vertex. We are grateful to T. Kwok and Y. Liu for assistance with the preparation of compound 29.Notes and references
- R. Halim and M. A. Brimble, Org. Biomol. Chem., 2006, 4, 4048–4058 RSC.
- (a) T. Yasumoto, M. Murata, Y. Oshima, M. Sano, G. K. Matsumoto and J. Clardy, Tetrahedron, 1985, 41, 1019–1025 CrossRef CAS; (b) K. Sasaki, J. L. C. Wright and T. Yasumoto, J. Org. Chem., 1998, 63, 2475–2480 CrossRef CAS PubMed.
- (a) H.-D. Chae, T.-S. Choi, B.-M. Kim, J. H. Hung, Y.-J. Bang and D. Y. Shin, Oncogene, 2005, 24, 4813–4819 CrossRef CAS PubMed; (b) D. Y. Shin, G. Y. Kim, N. D. Kim, J. H. Jung, S. K. Kim, H. S. Hang and Y. H. Choi, Oncol. Rep., 2008, 19, 517–526 CAS.
- For selected references on synthetic approaches to the pectenotoxins, see: (a) M. Heapy, T. W. Wagner and M. M. Brimble, Synlett, 2007, 2359–2362 Search PubMed; (b) R. Halim, M. A. Brimble and J. Merten, Org. Lett., 2005, 7, 2659–2662 CrossRef CAS PubMed; (c) R. Halim, M. A. Brimble and J. Merten, Org. Biomol. Chem., 2006, 4, 1387–1399 RSC; (d) S. Carley and M. A. Brimble, Org. Lett., 2009, 11, 563–566 CrossRef CAS PubMed; (e) A. M. Heapy and M. A. Brimble, Tetrahedron, 2010, 66, 5424–5431 CrossRef CAS; (f) L. A. Paquette, X. Peng and D. Bondar, Org. Lett., 2002, 4, 937–940 CrossRef CAS PubMed; (g) X. Peng, D. Bondar and L. A. Paquette, Tetrahedron, 2004, 60, 9589–9598 CrossRef CAS; (h) D. Bondar, J. Liu, T. Müller and L. A. Paquette, Org. Lett., 2005, 7, 1813–1816 CrossRef CAS PubMed; (i) P. D. O'Connor, C. K. Knight, D. Friedrich, X. Peng and L. A. Paquette, J. Org. Chem., 2007, 72, 1747–1754 CrossRef PubMed; (j) S. D. Lotesta, Y. Hou and L. J. Williams, Org. Lett., 2007, 9, 869–872 CrossRef CAS PubMed; (k) R. V. Kolakowski and L. J. Williams, Tetrahedron Lett., 2007, 48, 4761–4764 CrossRef CAS; (l) S. Joyasawal, S. D. Lotesta, N. G. Akhmedov and L. J. Williams, Org. Lett., 2010, 12, 988–991 CrossRef CAS PubMed; (m) D. Vellucci and S. D. Rychnovsky, Org. Lett., 2007, 9, 711–714 CrossRef CAS PubMed; (n) P. M. Pihko and J. E. Aho, Org. Lett., 2004, 6, 3849–3852 CrossRef CAS PubMed; (o) J. A. Aho, E. Salomäki, K. Rissanen and P. M. Pihko, Org. Lett., 2008, 10, 4183–4186 CrossRef PubMed; (p) H. Helmboldt, J. A. Aho and P. M. Pihko, Org. Lett., 2008, 10, 4179–4182 CrossRef PubMed; (q) J. A. Aho, A. Piisola, K. S. Krishnan and P. M. Pihko, Eur. J. Org. Chem., 2011, 1682–1694 CrossRef CAS; (r) E. K. Kemppainen, G. Sahoo, A. Valkonen and P. M. Pihko, Org. Lett., 2012, 14, 1086–1089 CrossRef CAS PubMed; (s) G. C. Micalizio and W. R. Roush, Org. Lett., 2001, 3, 1949–1952 CrossRef CAS PubMed; (t) D. P. Canterbury and G. C. Micalizio, Org. Lett., 2011, 13, 2384–2387 CrossRef CAS PubMed; (u) O. Kubo, D. P. Canterbury and G. C. Micalizio, Org. Lett., 2012, 14, 5748–5751 CrossRef CAS PubMed; (v) N. F. O'Rourke, Mu A, H. N. Higgs, A. Eastman and G. C. Micalizio, Org. Lett., 2017, 19, 5154–5157 CrossRef PubMed; (w) G. Vassilikogiannakis, I. Alexopoulou, M. Tofi and T. Montagnon, Chem. Commun., 2011, 47, 259–261 RSC; (x) A. Kouridaki, T. Montagnon, M. Tofi and G. Vassilikogiannakis, Org. Lett., 2012, 14, 2374–2377 CrossRef CAS PubMed; (y) A. Kouridaki, T. Montagnon, D. Kalaitzakis and G. Vassilikogiannakis, Org. Biomol. Chem., 2013, 11, 537–541 RSC; (z) A. Kouridaki, M. Sofiadis, T. Montagnon and G. Vassilikogiannakis, Eur. J. Org. Chem., 2015, 7240–7243 CrossRef CAS ; for a review see ref. 1.
- (a) D. A. Evans, H. A. Rajapakse and D. Stenkamp, Angew. Chem., Int. Ed., 2002, 41, 4569–4573 ( Angew. Chem. , 2002 , 114 , 4751–4755 ) CrossRef CAS; (b) D. A. Evans, H. A. Rajapakse, A. Chiu and D. Stenkamp, Angew. Chem., Int. Ed., 2002, 41, 4573–4576 ( Angew. Chem. , 2002 , 114 , 4755–4758 ) CrossRef CAS.
- K. Fujiwara, Y. Suzuki, N. Koseki, Y. I. Aki, Y. Kikuchi, S. I. Murata, F. Yamamoto, M. Kawamura, T. Norikura, H. Matsue, A. Murai, R. Katoono, H. Kawai and T. Suzuki, Angew. Chem., Int. Ed., 2014, 53, 780–784 ( Angew. Chem. , 2014 , 126 , 799–803 ) CrossRef CAS PubMed.
- T. J. Donohoe and R. M. Lipiński, Angew. Chem., Int. Ed., 2013, 52, 2491–2494 ( Angew. Chem. , 2013 , 125 , 2551–2554 ) CrossRef CAS PubMed.
- A. Roushanbakhti, Y. Liu, P. Winship, M. Tucker, W. Akhtar, D. Walter, G. Wrigley and T. J. Donohoe, Angew. Chem., Int. Ed., 2017, 56, 14883–14887 ( Angew. Chem. , 2017 , 129 , 15079–15083 ) CrossRef CAS PubMed.
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229–4231 CrossRef CAS.
- F. Nicotra, L. Panza, G. Russo and L. Zucchelli, J. Org. Chem., 1992, 57, 2154–2158 CrossRef CAS.
- A. Hosomi and H. Sakurai, Tetrahedron Lett., 1976, 17, 1295–1298 CrossRef.
- B. M. Trost, T. A. Grese and D. M. T. Chan, J. Am. Chem. Soc., 1991, 113, 7350–7362 CrossRef CAS.
- (a) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS; (b) T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- J. Takagi, K. Takahashi, T. Ishiyama and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 8001–8006 CrossRef CAS PubMed.
- For selected references on anomalous asymmetric dihydroxylation of 1,1-disubstituted alkenes, see: (a) K. J. Hale, S. Manaviazar and S. A. Peak, Tetrahedron Lett., 1994, 35, 425–428 CrossRef CAS; (b) K. J. Hale and J. Cai, Tetrahedron Lett., 1996, 37, 4233–4236 CrossRef CAS; (c) K. J. Hale, S. Manaviazar and J. George, Chem. Commun., 2010, 46, 4021–4042 RSC; (d) S. Kowashi, T. Ogamino, J. Kamei, Y. Ishikawa and S. Nishiyama, Tetrahedron Lett., 2004, 45, 4393–4396 CrossRef CAS; (e) A. Nelson, P. O'Brien and S. Warren, Tetrahedron Lett., 1995, 36, 2685–2688 CrossRef CAS; (f) D. J. Krysan, Tetrahedron Lett., 1996, 37, 1375–1376 CrossRef CAS; (g) K. P. M. Vanhessche and K. B. Sharpless, J. Org. Chem., 1996, 61, 7978–7979 CrossRef CAS PubMed; (h) J. M. Gardiner and S. E. Bruce, Tetrahedron Lett., 1998, 39, 1029–1032 CrossRef CAS; (i) Y. Pang, C. Fang, M. J. Twiner, C. O. Miles and C. J. Forsyth, Angew. Chem., Int. Ed., 2011, 50, 7631–7635 CrossRef CAS PubMed; for selected references on anomalous asymmetric dihydroxylations on other alkenes, see: (j) N. H. Ertel, B. Dayal, K. Rao and G. Salen, Lipids, 1999, 34, 395–405 CrossRef CAS PubMed; (k) M. Iwashima, T. Kinsho and A. B. Smith, Tetrahedron Lett., 1995, 36, 2199–2202 CrossRef CAS; (l) D. L. Boger, J. A. McKie, T. Nishi and T. Ogiku, J. Am. Chem. Soc., 1996, 118, 2301–2302 CrossRef CAS; (m) P. Salvadori, S. Superchi and F. Minutolo, J. Org. Chem., 1996, 61, 4190–4191 CrossRef CAS PubMed; (n) D. L. Boger, J. A. McKie, T. Nishi and T. Ogiku, J. Am. Chem. Soc., 1997, 119, 311–325 CrossRef CAS; for a review on catalytic asymmetric dihydroxylation, see: (o) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- (a) K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H. L. Kwong, K. Morikawa and Z. M. Wang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS; (b) H. C. Kolb, P. G. Andersson and K. B. Sharpless, J. Am. Chem. Soc., 1994, 116, 1278–1291 CrossRef CAS.
- V. VanRheenen, R. C. Kelly and D. Y. Cha, Tetrahedron Lett., 1976, 17, 1973–1976 CrossRef.
- R. Itooka, Y. Iguchi and N. Miyaura, J. Org. Chem., 2003, 68, 6000–6004 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, compounds' characterisation data and NMR spectra. See DOI: 10.1039/c9sc01761e |
| This journal is © The Royal Society of Chemistry 2019 |