
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous cerium oxide-coated upconversion nanoparticles for tumor-responsive chemo-photodynamic therapy and bioimaging†
Tao
Jia‡
a,
Jiating
Xu‡
a,
Shuming
Dong
a,
Fei
He
*a,
Chongna
Zhong
a,
Guixin
Yang
a,
Huiting
Bi
a,
Mengshu
Xu
a,
Yingkui
Hu
a,
Dan
Yang
a,
Piaoping
Yang
 *a and
Jun
Lin
*b
*a and
Jun
Lin
*b
aKey Laboratory of Superlight Materials and Surface Technology, Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin, 150001, P. R. China. E-mail: hefei1@hrbeu.edu.cn; yangpiaoping@hrbeu.edu.cn
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130021, P. R. China. E-mail: jlin@ciac.ac.cn
First published on 31st July 2019
Abstract
Long-term tumor hypoxia has always been a huge obstacle for oxygen dependent photodynamic therapy (PDT) and anticancer drug chemotherapy. Herein, a hollow-structured biophotocatalyst was developed by coating mesoporous cerium oxide (mCeOx) on upconversion nanoparticles (UCNPs, NaGdF4:Yb,Tm@NaGdF4), and it can be initialized with a near-infrared (NIR) laser to achieve PDT with O2 compensation by decomposing the endogenous H2O2 in the tumor microenvironment. A NIR laser with a long wavelength has low phototoxicity to biotissue, and the core–inert shell structured UCNPs can efficiently convert the NIR photons into ultraviolet (UV) light, which can further trigger CeOx to produce reactive oxygen species (ROS). Moreover, the internal space of UCNPs@mCeOx is ideal for storing chemotherapeutic doxorubicin (DOX), and adequate O2 plays a key role in alleviating drug fastness via chemotherapy in hypoxic tumors, thereby strengthening the synergy between PDT and chemotherapy. After being injected into tumor-bearing mice intravenously, the nanomedicine was able to reach the tumor via an enhanced permeability and retention (EPR) effect. In addition, the capability to use the developed nanosystem in computed tomography (CT), magnetic resonance (MR) and upconversion luminescence (UCL) imaging was validated. Significantly, the NIR laser irradiated nanomedicine exhibits an excellent anticancer effect, implying promising theranostic applications.
Introduction
Cancer has become one of the main killers of human beings and causes millions of deaths each year.1,2 Traditional tumor treatments, including surgical resection, chemotherapy, and molecularly targeted therapy,3,4 can effectively treat some tumors and control tumor metastasis.5,6 However, these treatments still have many disadvantages, such as a high recurrence rate of surgical resection, whole-body adverse reactions from chemotherapy, and damage to normal tissues around tumors by ionizing radiation. Compared to traditional treatment methods, photodynamic therapy (PDT) utilizes light source targeting to selectively eliminate primary and recurrent tumors, thereby avoiding normal tissue damage, narrowing the scope of surgery, improving patient safety, activating immune function and reducing recurrence.7–9 In addition, this method could be implemented on various types of tumor cells and thus widely used. Therefore, PDT has become an emerging method for clinical treatment of tumors owing to its huge potential advantages.Suitable photosensitizers (PSs), excitation light sources, and oxygen (O2) are prerequisites for PDT.10–12 In the process of PDT, antitumor activity from reactive oxygen species (ROS) can oxidize lipids, amino acids and proteins in cell membranes or organelles (including the mitochondria, Golgi apparatus, and endoplasmic reticulum) resulting in irreversible cell damage.13–15 They are generated from molecular O2 by an excited photosensitizer upon local illumination for tumor cell ablation.16–18 However, solid tumors in the process of rapid proliferation, which results in insufficient blood supply and O2 sites in the tumor microenvironment (TME), will not only reduce the effect of traditional tumor treatment methods such as chemotherapy and radiotherapy, but will also have a significant impact on the therapeutic effect of PDT.19–21
At the same time, after the photosensitizer is exposed to light during the PDT process, the O2 at the tumor site will be rapidly consumed, causing the partial pressure of O2 to rapidly decline and fail to be supplemented, thus further limiting the killing effect of PDT.22–24 Therefore, improving the characteristics of hypoxic tumor sites is an urgent challenge to be addressed for PDT.
During the past few years, various strategies have been proposed for the use of microcarriers to deliver O2 to overcome tumor hypoxia.25–27 Chen et al. prepared a nano-pharmaceutical drug based on catalase (HAOP NP) that mainly included a PS, methylene blue (MB), a catalyst (CAT), cyclopentapeptide c(RGDfK) and quencher-linked polymers (BHQ-3) with a shell composition.28 The nano-pharmaceuticals could be specifically assimilated into tumor cells. Under the action of the CAT, H2O2 was converted into O2, which improves the efficiency of ROS production and overcomes the problem of tumor hypoxia. Luo's group used lipopolymers, hemoglobin (Hb) and indocyanine green (ICG) to prepare a nanodrug capable of carrying O2 that can increase the O2 content of tumor hypoxic tissue and strengthen the PDT therapeutic effect.29 Other researchers achieved the transportation of O2 by encapsulating Hb and a second-generation PS (ZnPc) in a micelle formed by a triblock copolymer.30,31 Hb as an O2 carrier provides a special method for overcoming tissue hypoxia, which makes use of the high reactivity and catalytic effect of manganese dioxide (MnO2) particles on endogenous hydrogen peroxide in the TME to produce O2. Separately, some researchers have combined hyperbaric O2 therapy with PDT to strengthen the therapeutic effect, further demonstrating the key role of tissue O2 in the efficacy of PDT therapy.32,33
However, to obtain effective micro-carriers with O2 release and PDT functions, several barriers need to be overcome: the combination of an O2 carrier and PDT requires the combination of an O2 catalyst and an organic photosensitizer,34 leading to a very complicated carrier structure, and some photosensitive molecules that do not adsorb strongly on the surface of the carrier are easily shed in the blood. In addition, there are many reported organic photosensitizers that may suffer severe damage during light exposure and lose their long-lasting photodynamic effects.35,36 Consequently, it is necessary to find a photocatalyst with high optical stability to prevent photodamage.
Cerium oxide nanoparticles (NPs) have attracted increasing attention owing to their enzyme-like activity. They have superior redox properties and can be used as a catalyst with the ability to reversibly transform from Ce4+ to Ce3+ while converting H2O2 to H2O and to O2 even in the absence of light irradiation. Hence, cerium oxide NPs can achieve O2 compensation to continuously generate O2 and provide an O2 environment for PDT and chemotherapy, thus improving cancer treatment efficiency.37–39 As a semiconductor catalyst, cerium oxide has a relatively unusual specific surface area, which effectively enhances the light capturing ability of the material. The defects on its grain surface and abundant O2 vacancies further enhance its catalytic ability and the use of visible light.40–42 Furthermore, cerium oxide has a strong UV absorption and has a high refractive index and diffusibility, which are extremely pivotal for the degradation of contaminants and microorganisms.43–45 Therefore, combining photocatalytically effective cerium oxide and PDT is highly attractive for cancer therapy. When cerium oxide is applied for the treatment of PDT, the main hindrances include UV injury and the penetration distance in the tissue when UV light is directly irradiated.46–48
In this study, we designed a mesoporous core–shell structure by coating hollow cerium oxide on UCNPs (denoted as UCNPs@mCeOx), which is capable of achieving in situ O2 generation and NIR light-triggered PDT for efficient PDT simultaneously. UCNPs@mCeOx is able to efficiently and stably break down endogenous H2O2 to O2 at physiological pH or in a weakly acidic tumor microenvironment owing to its characteristics, such as its ability to act like a catalase,49–51 thus producing sufficient O2 to enhance the PDT effect and chemotherapy. Additionally, UCNPs are energy converters that can transform low-energy photons to high-energy photons and exhibit strong upconversion (UC) fluorescence.52–55 Compared to downconversion NPs, UCNPs can assimilate NIR light with a relatively high tissue penetration depth.56–58 Under these conditions, upon NIR 980 nm laser irradiation, Yb3+ and Tm3+-doped UCNPs (NaGdF4:Yb,Tm@NaGdF4, by coating a homogeneous NaGdF4 inert layer, the activator is far away from the surface of the NPs, which can considerably inhibit the annihilation of the luminescence caused by the surface defects of the material) can continuously convert NIR light into UV emission, which will trigger a cerium oxide-based photocatalytic reaction to generate an electron–hole (e−–h+) pair, and efficiently decompose H2O and O2 to hydroxyl radicals (˙OH) and superoxide radicals (˙O2−), inducing apoptosis in tumor cells.59–61 The sufficient O2 produced via the decomposition of H2O2 by cerium oxide significantly increases the yield of ˙O2−, thus improving the efficacy of PDT. Moreover, after the silica shell is etched, its hollow structure can be used as a carrier for the delivery of the chemotherapy drug DOX through the mesoporous channel.62,63 The surface water-solubility and surface potential of the NPs can be changed by modifying the surface of UCNPs@mCeOx with phosphoserine and mPEG to better combine with DOX and provide simultaneous treatment via PDT and chemotherapy. In addition, the developed nanosystem has multiple imaging capabilities, including use with UCL, MRI and CT, which have great potency in the cancer diagnosis field.
Results and discussion
The synthesis of mesoporous cerium oxide-coated nanoparticles UCNPs@mCeOx and the final PEG/UCP-DOX is shown in Scheme 1. As depicted, the core NPs doped with Yb3+ and Tm3+ were covered with an inert shell of NaGdF4 to form stable core–shell structured UCNPs, and the UCNPs@mSiO2 were obtained by coating mesoporous silica on the UCNPs. Subsequently, Ce(NO3)3 was added to the UCNPs@mSiO2 solution, and then hexamethylenetetramine was added to provide the basic conditions to hydrolyze Ce3+ to Ce(OH)3, which reacted with water and O2 at high temperatures to form Ce(OH)4. Followed by calcination and etching of the silica shell, the UCNPs@mCeOx were finally obtained. Upon modifying the surface of the CeOx shell with phosphoserine, the produced sample (abbreviated as UCP) had a hydrophilic surface. After the biocompatible polymer mPEG was covalently conjugated with phosphoserine, the chemotherapeutic drug DOX could be efficiently stored in the hollow structure to achieve the final PEG/UCP-DOX. The DOX loading process predominantly relied on a physical adsorption mechanism and proton-sensitive electrostatic binding interaction in the mesoporous channels.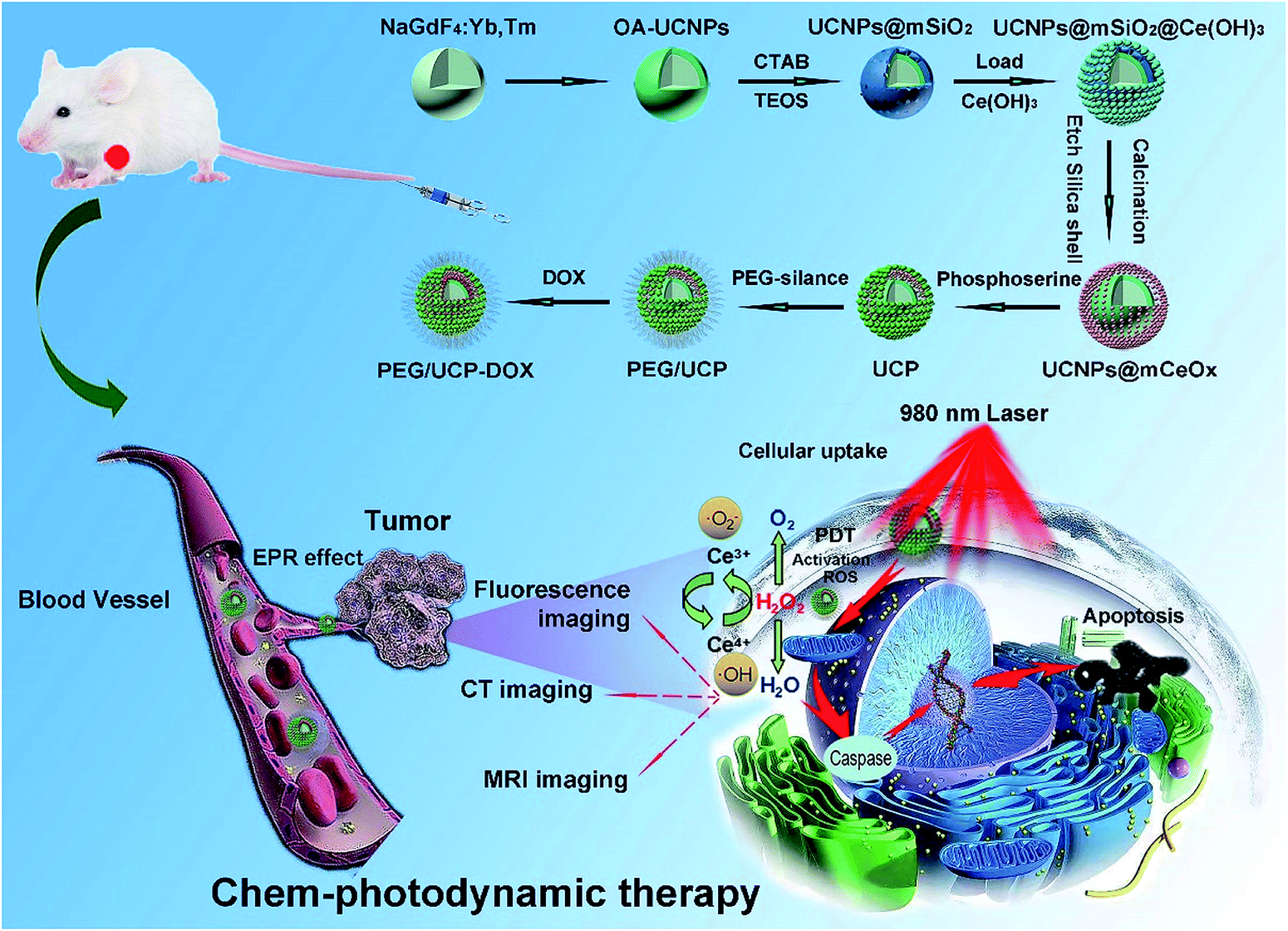 | ||
| Scheme 1 Schematic illustration for the formation and transport of PEG/UCP-DOX in blood vessels, EPR-mediated tumor accumulation, proposed PDT mechanism, and multiple imaging functions. | ||
The core NPs (OA-NaGdF4:Yb,Tm) and core–shell structured NPs (OA-UCNPs) were synthesized by the RE(oleate)3 thermal decomposition method in accordance with a prior study.64Fig. 1a–d present the TEM images of the OA-NaGdF4:Yb,Tm, OA-UCNPs, UCNPs@mSiO2, and UCNPs@mCeOx, respectively. Fig. 1a shows that the OA-NaGdF4:Yb,Tm NPs are composed of homogeneous and monodisperse particles (approximately 20 nm in diameter). To keep the activator away from the surface defects and the surface-related high-frequency vibrations of the material, a homogeneous inert layer of NaGdF4 was evenly coated on the core using an external extension growth method. As shown in the TEM photo (Fig. 1b), the dispersion and orderliness of OA-UCNPs were well maintained, and the average particle size was enhanced to 31 nm. Before coating the silica shell, the UCNPs were mixed with cetyltrimethylammonium bromide (CTAB) and stirred for approximately 12 h. The sol–gel technique was used to prepare the UCNPs@SiO2 nanospheres. Fig. 1c shows that the UCNPs@mSiO2 is composed of uniform particles with an average size of 46 nm. In contrast, careful observation showed the presence of worm-like tunnels on the siliceous shell. Then the cerium oxide was coated on the silica shell, and the shell was etched to obtain UCNPs@mCeOx. As shown in Fig. 1d, for UCNPs@mCeOx (approximately 48 nm in diameter), a hollow structure was evident with spherical UCNPs inside and an outer layer of CeOx composed of tiny particles. As presented in Fig. 1e, the elemental mapping provided evidence of the constituent elements and well-dispersed CeOx on the UCNPs. As displayed in Fig. 1f, the cross-sectional compositional line profiles validate the core–shell structure of the UCNPs@mCeOx. Notably, the dispersibility and uniformity of the UCNPs@mCeOx were well maintained after coating CeOx, which was confirmed by the SEM images shown in Fig. 1g–i. The energy-dispersive spectrum (EDS) (Fig. 1j) shows the basic elements of UCNPs@mCeOx, verifying the successful coating of CeOx, which is consistent with Fig. 1e. The particle size change for the prepared samples was also confirmed by dynamic light scattering (DLS) measurements. Such a size could promote better aggregation of the nanomedicine at a tumor site via an EPR effect.65–67 As displayed in Fig. S1,† the particle size of UCNPs@mCeOx changed from 82 nm to 103 nm owing to the polymer coating, the etching of the mesoporous silica shell and the adsorbed water molecules on the NP surface. Moreover, as shown in Fig. S2,† after CeOx modification, the zeta potential of the UCNPs@SiO2 NPs changed from negatively charged (−17 mV) to positively charged (+1.64 mV), confirming the successful surface modification of UCNPs. The UCNPs@mCeOx was then modified with phosphoserine and further modified with mPEG to form PEG/UCP, which strengthened its water-solubility and can prolong the blood recurrence period in a living system. After it was further modified with PEG, the zeta potential of the final PEG/UCP sample changed to −3.17 mV. The hydrodynamic size of the polymer after modification was approximately 110 nm, showing excellent stability in physiological solutions. We also studied whether pure cerium oxide can produce ROS in the absence of a laser. As presented in Fig. S3,† even after prolonged mixing of NPs in DPBF solution, the absorption curve of DPBF hardly changed, confirming that the NPs did not produce ROS in the absence of the laser. Furthermore, Fig. 7b shows the cell viability of HeLa cells incubated with different concentrations of PEG/UCP, and almost no change was found compared with the control group (natural growth), implying that PEG/UCP did not enable the PDT effect without laser irradiation. The colloidal stabilities of the NPs in PBS (pH 7.4), FBS, serum and culture medium were also investigated. After the PEG/UCP NPs were shocked softly in different solutions for various times (0, 6, 12, 24, and 48 h) at 37 °C, the particle size was measured using dynamic light scattering (DLS). As presented in Fig. S4,† the particle size of PEG/UCP did not show any significant change in the different solutions for various periods, indicating the excellent stability of the PEG/UCP in biological mimicking systems.
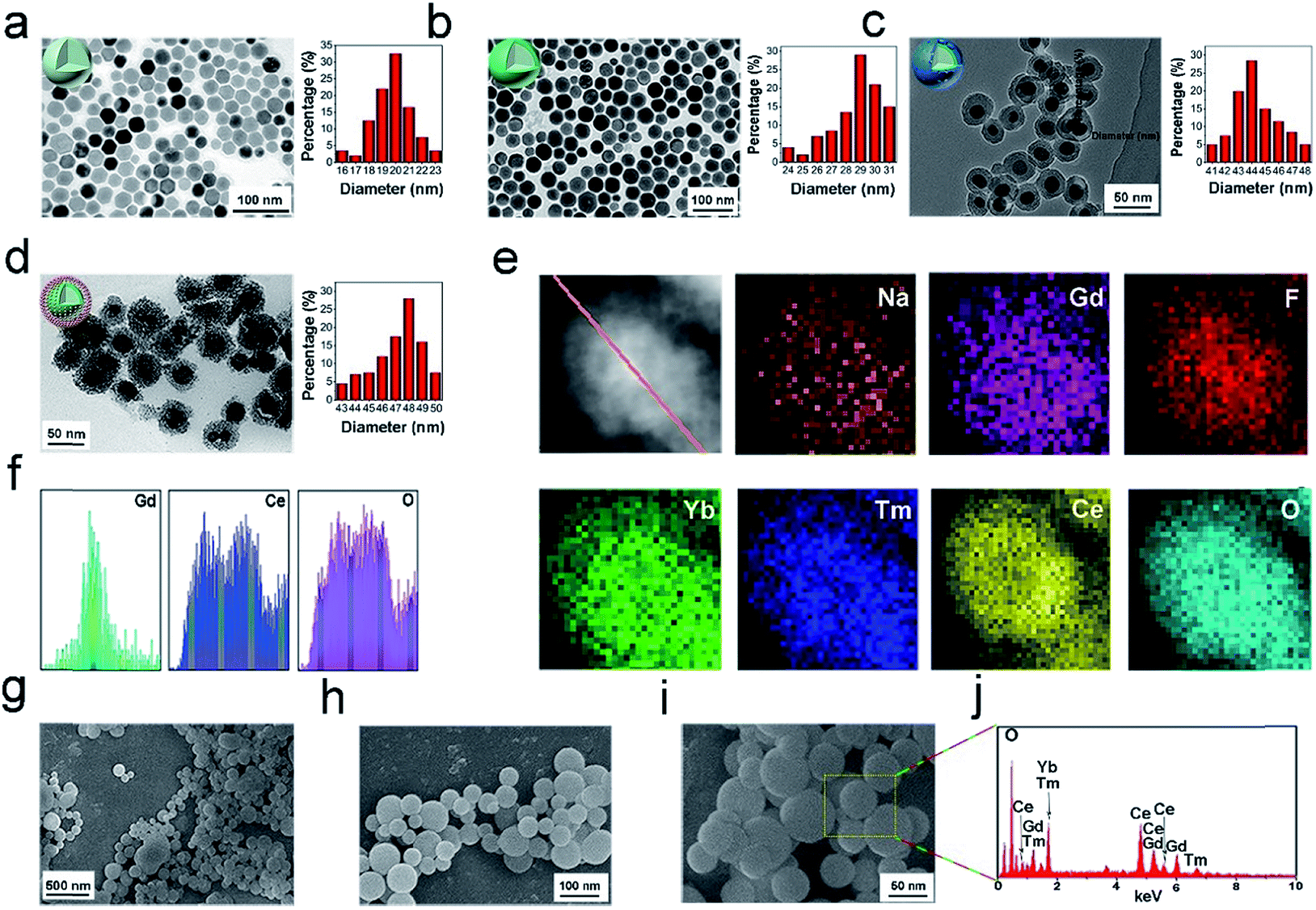 | ||
| Fig. 1 TEM images and morphology sketches of OA-NaGdF4:Yb,Tm core NPs (a), core–shell OA-UCNPs (b), UCNPs@mSiO2 (c) and UCNPs@mCeOx (d). Scanning transmission electron microscopy (STEM), elemental mapping images of UCNPs@mCeOx (e) and the corresponding cross-sectional compositional lines of Gd, Ce, and O (f). SEM images of UCNPs@mCeOx with magnification scales of 500 nm (g), 100 nm (h) and 50 nm (i) and EDS spectrum of UCNPs@mCeOx (j). | ||
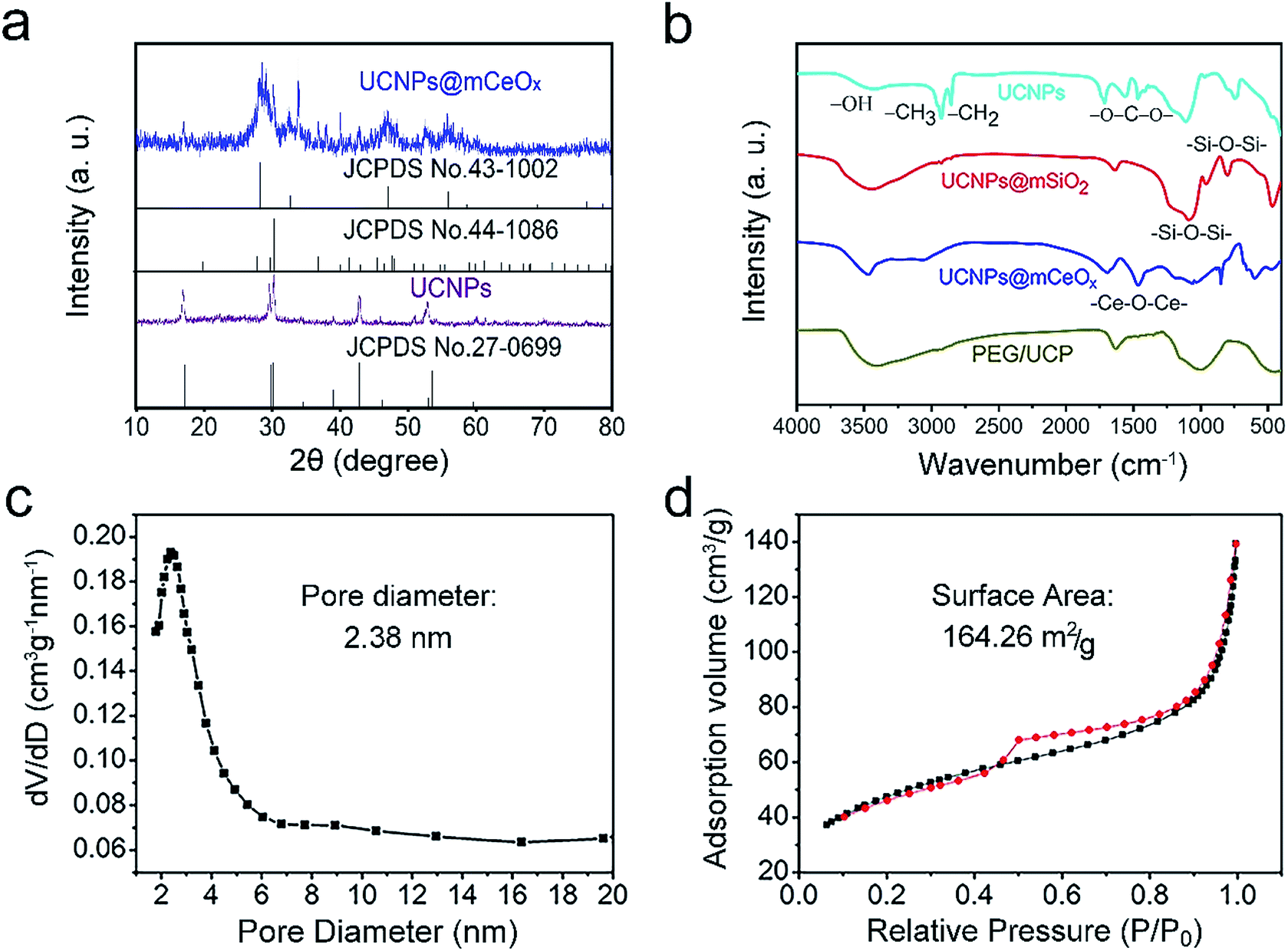 | ||
| Fig. 2 XRD patterns of UCNPs and UCNPs@mCeOx (the standard lines of β-NaGdF4, CeO2 and Ce2O3 are supplied for comparison) (a) and FT-IR spectra of OA-UCNPs, UCNPs@mSiO2, UCNPs@mCeOx and PEG/UCP (b). The pore-size distribution (c) and N2 adsorption/desorption isotherm (d) of UCNPs@mCeOx. | ||
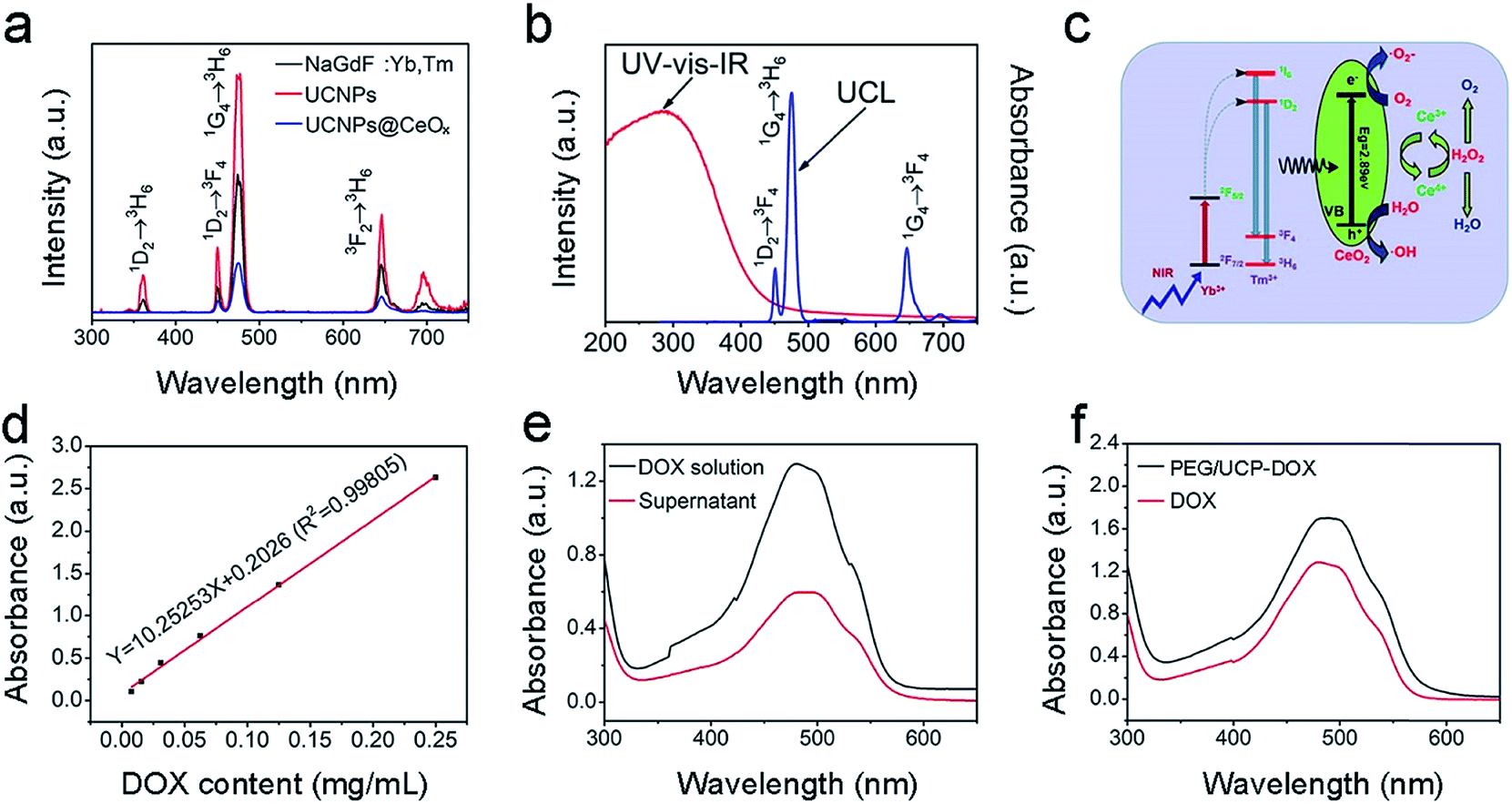 | ||
| Fig. 3 Upconversion emission spectra of the NaGdF4:Yb,Tm core NPs, UCNPs, and UCNPs@mCeOx (a). The UV-vis-IR DRS and UCL of the UCNPs@mCeOx (b). Schematic illustration of the UC, photocatalysis, and enzyme-like catalysis mechanism of the UCNPs@mCeOx (from left to right) (c). The standard curve for DOX solution detected at 480 nm (d). The absorption spectra of the initial DOX solution and supernatant obtained after coating the drug (e). UV-vis absorption spectra of the DOX molecules and the final PEG/UCP-DOX (f). | ||
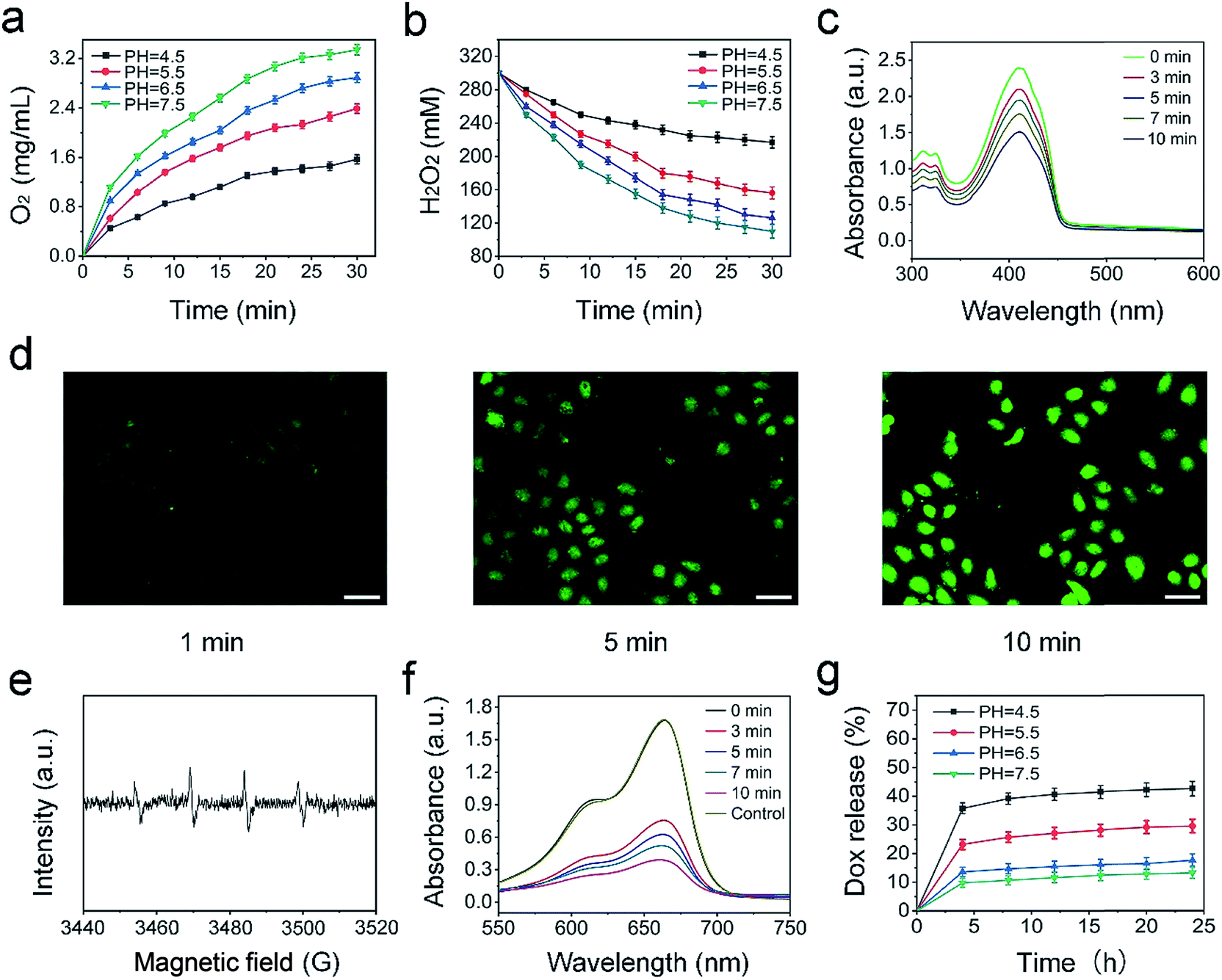 | ||
| Fig. 4 Production of O2 (a) and decomposition of H2O2 (b) catalyzed by UCNPs@mCeOx aqueous solution in 30 min. NPs were observed under 980 nm laser irradiation. The absorbance of DPBF solutions with UCNPs@mCeOx at different time periods under 980 nm laser irradiation (0.72 W cm−2) (c). The CLSM images of HeLa cells with oxidized DCF fluorescence at different illumination times (d). ESR spectra for detection of hydroxyl radicals versus magnetic field (e). UV-vis absorption spectra of MB solution with UCNPs@mCeOx at different light times (f). Release rate of DOX in PBS under different pH conditions as a function of the release time (g). | ||
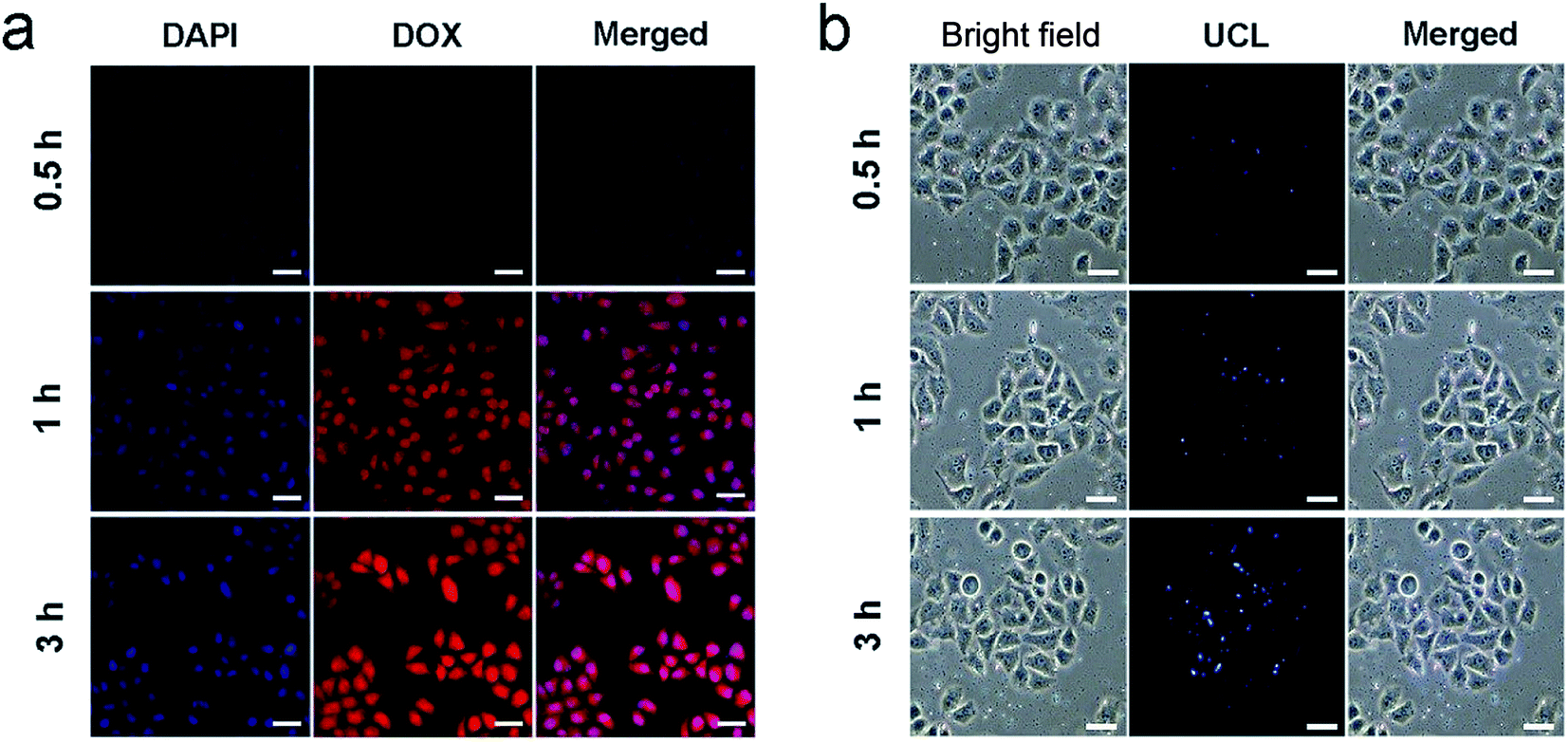 | ||
| Fig. 5 CLSM images of HeLa cells incubated with PEG/UCP-DOX for 0.5, 1, and 3 h at 37 °C. Scale bar: 50 μm. (a) UCLM images of HeLa cells incubated with PEG/UCP NPs at 37 °C for 0.5, 1, and 3 h. Scale bar: 50 μm (b). | ||
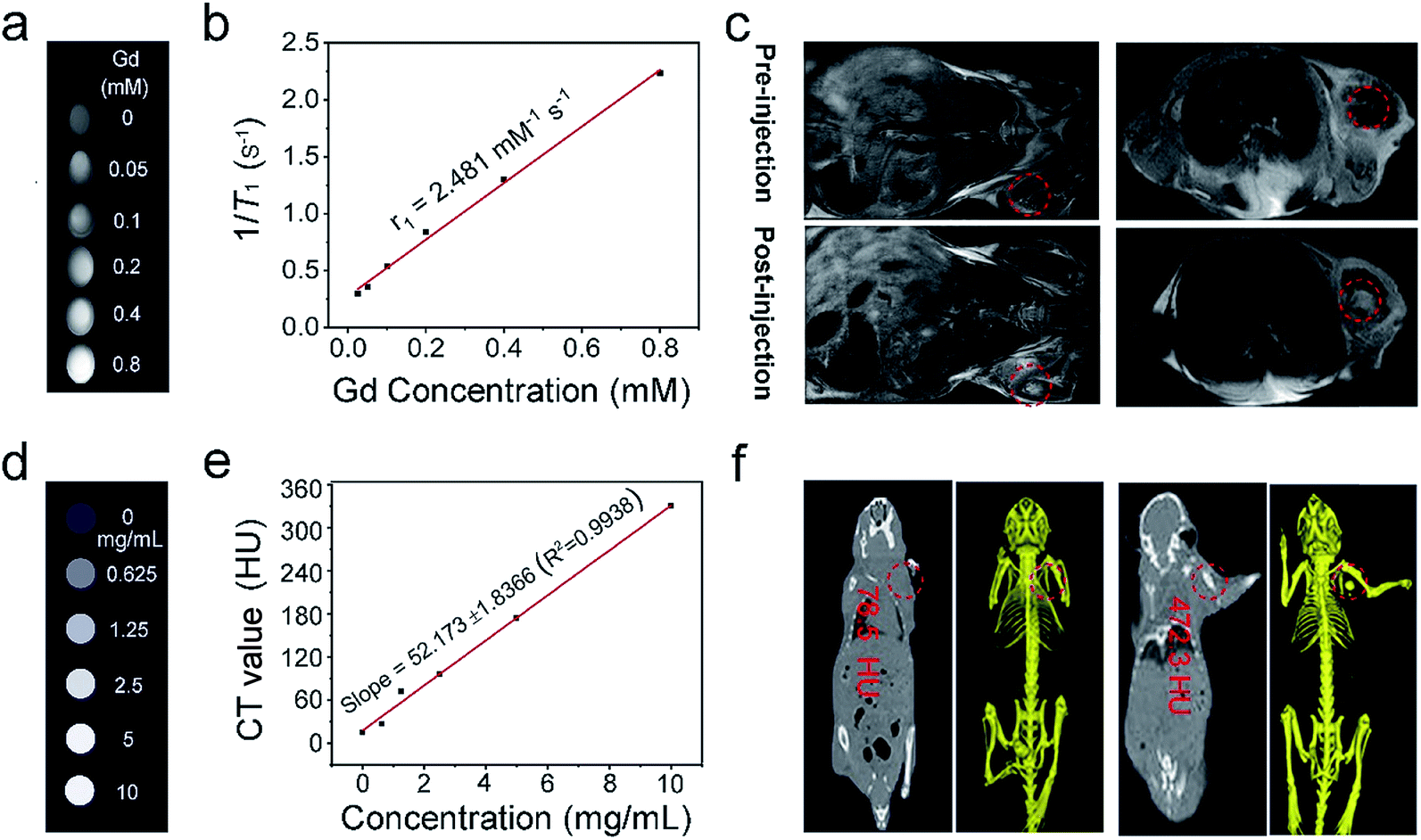 | ||
| Fig. 6 In vitro T 1-weighted MR images for various Gd3+ concentrations of PEG/UCP (a). Relaxation rate 1/T1 as a function of the Gd3+ molar concentration (b). In vivo T1-weighted MR images of tumor-bearing mice before and after injection of PEG/UCP (c). In vitro CT images of PEG/UCP at different concentrations of Gd3+ (d). The CT values of PEG/UCP at different concentrations (e). In vivo CT images of a tumor-bearing mouse before and after injection (f). | ||
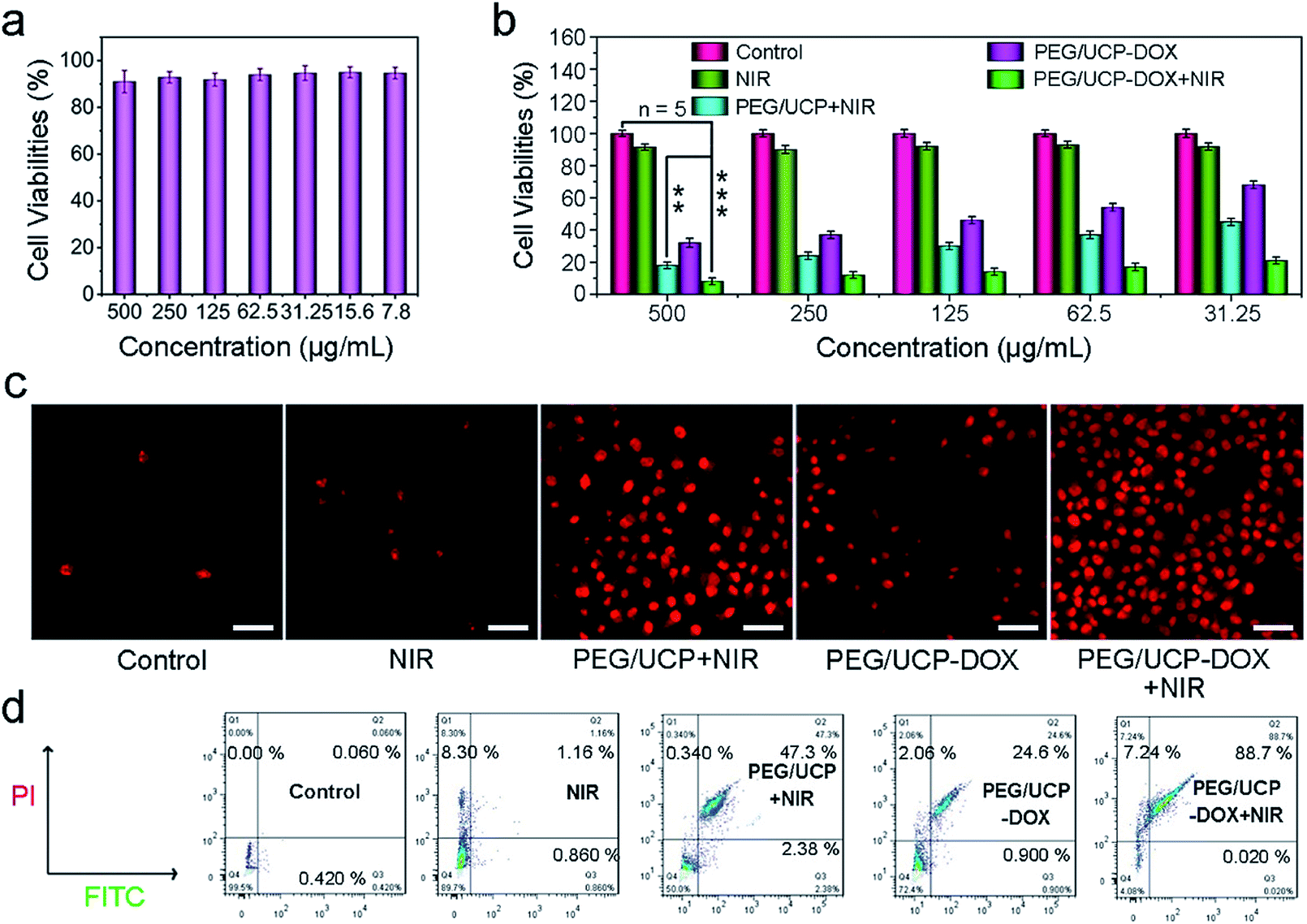 | ||
| Fig. 7 Cell viabilities of L929 fibroblast cells with incubation of different concentrations of PEG/UCP (a). Cell viability versus incubated particle concentration (7.8, 15.6, 31.2, 125, 250, and 500 μg mL−1) for HeLa cells under different conditions (control, NIR, UCNPs@mCeOx, PEG/UCP-DOX, and PEG/UCP-DOX + NIR) mean ± s.d, n = 5. P values were calculated using Tukey's post-test (***p < 0.001) (b). In vitro HeLa cell viabilities after incubation with the culture (control), 980 nm laser irradiation (NIR), PEG/UCP + NIR, PEG/UCP-DOX, and PEG/UCP-DOX + NIR. All cells were labeled with PI. Scale bar: 100 μm (c). Apoptosis of cells induced by NIR, PEG/UCP + NIR, PEG/UCP-DOX and PEG/UCP-DOX + NIR (d). | ||
The XRD patterns for the OA-UCNPs and UCNPs@mCeOx, and the standard lines for hexagonal NaGdF4 (JCPDS no. 27-0699), CeO2 (JCPDS no. 43-1002) and Ce2O3 (JCPDS no. 44-1086) are shown in Fig. 2a. As shown, the diffraction peak for pure β-NaGdF4 remains well formed for these samples. Comparing UCNPs@mCeOx with UCNPs reveals that there are several sharp peaks at approximately 2θ = 28.5°, 2θ = 31.5°, 2θ = 47.5° and 2θ = 58.6° assigned to CeOx, which implies the existence of crystalline CeOx. The FT-IR spectra (Fig. 2b) of OA-UCNPs, UCNPs@mSiO2, UCNPs@mCeOx and PEG/UCP were measured to confirm the successful modification. As displayed in Fig. 2b, OA-UCNPs show a band associated with carboxyl groups at 1463 and 1564 cm−1, and the wide band at 3450 cm−1 is ascribed to the O–H telescopic vibration. The strong bands at 2924 and 2854 cm−1 are mainly derived from symmetric and asymmetric stretching vibrations of –CH2. For UCNPs@mSiO2, the broad peaks at 3432 and 947 cm−1 indicate that there is considerable OH (–OH groups and Si–OH) on the surface, and the peaks at 802 and 1087 cm−1 are associated with Si–O–Si vibrations. The FT-IR spectrum of UCNPs@mCeOx is analogous to that of UCNPs@mSiO2 because of the identical functional groups on the particle surface, apart from a vibration band of Ce–O–Ce at 1400 cm−1. After surface modification of UCNPs@mCeOx with PEG, an intensive C–O stretching vibration in PEG was found in contrast to the vibrations for UCNPs@mCeOx. For PEG/UCP-DOX, the additional bands at 1250–1750 cm−1 are related with the loaded DOX numerators (Fig. S5†). The pore size distribution profile and the N2 adsorption/desorption isotherm of UCNPs@mCeOx are displayed in Fig. 2c and d.
The sample exhibits a representative type IV isotherm, indicating a mesoporous silica channel. Additionally, the sample has a Brunauer–Emmett–Teller (BET) surface area of 164.26 m2 g−1, an average pore size of 2.83 nm, and a total pore volume of 0.42 cm3 g−1. After it was modified with L-O-phosphoserine and mPEG-NH2, the corresponding values for the sample decreased to 156.64 m2 g−1, 2.24 nm, and 0.35 cm3 g−1, respectively (Fig. S6a and b†). The final PEG/UCP-DOX had a lower BET surface area of 107.26 m2 g−1, an average pore diameter of 2.16 nm and a smaller pore volume of 0.29 m2 g−1 (Fig. S6c and d†), which may be due to the DOX loading.
In general, the cerium chemical valence state on the periphery of the UCNPs@mCeOx is a blended valence state. In Fig. S7,† the X-ray photoelectron spectroscopy (XPS) spectrum shows the trivalent and tetravalent Ce ions (Ce3+/Ce4+ = 1/3).
This special valence combination endows UCNPs@mCeOx with catalase-like activity; thus, Ce4+ and Ce3+ can react as follows:
| H2O2 + 2Ce4+ → 2H+ + O2 + 2Ce3+ | (1) |
| H2O2 + 2Ce3+ + 2H+ → 2Ce4+ + 2H2O | (2) |
The UC emission spectra for NaGdF4:Yb,Tm, UCNPs and UCNPs@mCeOx excited by the 980 nm NIR laser (a power density of 0.72 W cm−2) are displayed in Fig. 3a. The emissions at 361, 453, 475, and 640–670 were assigned to 1D2 → 3H6, 1D2 → 3F4, 1G4 → 3H6 and 1G4 → 3F4 Tm3+ ion transitions, respectively. The NaGdF4 inert layer was coated on the core particles to achieve ultra-bright UC under low excitation power. Obviously, the UC emission intensity of the UCNPs is much stronger than that of pure core NPs. Thus, the NaGdF4 layer can protect the activators of Tm3+ from surface-related high-frequency non-radiative decay vibrations, thus improving the emission intensity of the core NPs. Compared to that of UCNPs that have a core–shell structure, the UC emission intensity of UCNPs@mCeOx drastically decreased in the UV region, which was perhaps caused by the efficient energy transfer between the UCNPs and the CeOx shell. In Fig. 3b, the ultraviolet-visible-infrared (UV-vis-IR) diffuse reflectance spectrum of UCNPs@mCeOx demonstrates a high UV absorption. When excited by 980 nm light, only the blue UC emission (1G4 → 3H6 transition) was present in the emission spectrum of UCNPs@mCeOx. Compared to that of the UCNPs, the decrease in the UV range (1I6 → 3F4 transition) of UCNPs@mCeOx showed that the emitted UV light excites the photo-generated electrons in the valence band to the conduction band of mCeOx, resulting in the formation of a photo-induced electron–hole (e−–h+) pair (Fig. 3b).
Compared to that of the UCNPs, the luminescence intensity of UCNPs@mCeOx is considerably reduced perhaps owing to the quenching effect of cerium oxide and the strong absorption of cerium oxide in the UV region. In accordance with theory, the luminescence intensity of a luminescent donor decreases with the combination of energy receptors of the photosensitizer. This is consistent with the lifetime measurement shown in Fig. S8.† As displayed, the decay lifetime of Tm3+ at 475 nm decreased from 964.14 μs to 277.74 μs after coating with CeOx, confirming the effective energy transfer between UCNPs and CeOx. Fig. 3d and e show the DOX standard profile, and the absorption spectra of the DOX original solution and the supernatant of PEG/UCP-DOX at 480 nm, respectively. On the basis of the Lambert–Beer method (A = Kbc), the DOX loading rate was calculated to be 47.52%. In Fig. 3f, the UV-vis absorption spectrum of PEG/UCP-DOX shows a distinct absorption band at 460–500 nm, which is mainly caused by DOX loading.
To assess the catalytic capability of the UCNPs@mCeOx, the O2 produced by depleting H2O2 at different time periods was measured upon 980 nm laser irradiation (Fig. 4a and b). The quantity of O2 produced reaches 2.97 mg L−1 in H2O2 solution under physiological pH conditions (pH 7.5) within 30 min, and the O2 production is decreased to 2.06 mg L−1 and 1.32 mg L−1 at pH values of 5.5 and 4.5, respectively. These results agree with prior reports.68,69 Moreover, the catalytic capability of UCNPs@mCeOx in the absence of laser irradiation was also measured. Fig. S9† shows that the O2 production decreased slightly, while the rates of H2O2 decomposition became lower than the corresponding values in the presence of NIR light irradiation. This result can be explained by the fact that NIR light separates electrons and holes. The holes have a strong oxidation capability that can cause H2O2 to decompose to O2, resulting in an increased decomposition of H2O2 and an enhanced production of O2. Although some O2 will change to superoxide radicals (˙O2−), the trend of increased production of O2 and decomposition of H2O2 is predominant overall. In addition, it is noteworthy that at pH 6.5, the O2 production still reaches 2.73 mg L−1, indicating that endogenous H2O2 can be efficiently converted to O2 in the weakly acidic microenvironment of a tumor site upon laser irradiation. Fig. 4c shows the change in absorbance of DPBF solutions of UCNPs@mCeOx at different time periods upon 980 nm laser irradiation. Clearly, the absorption peak for DPBF decreased at 350–470 nm, demonstrating that the ROS yield increases gradually with irradiation time. Furthermore, the intracellular ROS production of PEG/UCP as measured with 2,7-dichlorofluorescein-diacetate (DCFH-DA) was studied. DCFH-DA could be absorbed by the cells and did not fluoresce. After intracellular esterase hydrolysis to DCFH, non-fluorescent DCFH could be oxidized by ROS to form 2,7-dichlorofluorescein (DCF), which emitted green fluorescence upon 488 nm light irradiation. Fig. 4d shows the CLSM photos of HeLa cells with fluorescence of DCF, and the more intense green fluorescence illustrates the efficient ROS production in PEG/UCP upon 980 nm light irradiation.
The NIR light excited photocatalytic performance of UCNPs@mCeOx was studied. The hydroxyl radicals and superoxide radicals generated in aqueous solutions upon 980 nm laser irradiation were detected via electron spin resonance (ESR) (Fig. 4e). The spin trap dimethyl pyrroline N-oxide (DMPO) is a capture agent that is used to react with generated hydroxyl radicals and superoxide radicals to form different detection signals. The four peaks assigned to ˙OH were trapped by DMPO, which illustrated the production of ˙OH (Fig. 4e), and the sextet ESR signal indicated the existence of superoxide radicals (˙O2−) (Fig. S10†). Furthermore, MB is a dye that can be decolorized by ˙OH and was chosen to test the generation of ˙OH at an absorbance wavelength of 662 nm. In Fig. 4f, with the extension of the 980 nm laser irradiation time, a significant decrease occurred, proving that UCNPs@mCeOx can produce ˙OH upon NIR light irradiation. Moreover, the stability of MB under 980 nm laser irradiation was studied, and the pure MB solution exhibited almost no change after irradiation with the 980 nm light for 30 min, certifying that MB has excellent stability under irradiation. Additionally, the drug release properties of PEG/UCP-DOX were studied. As presented in Fig. 4g, the PEG/UCP-DOX has a pH-responsive release behavior in PBS. The DOX release progressively increased upon increasing the solution acidity, and finally reached a value of approximately 42% at pH 4.5 after 25 h, which illustrated that the release of DOX from the nanosystem was favorable under acidic conditions. The results show the possibility of DOX release in the acidic tumor microenvironment.
To study the cell uptake behavior of the resulting nanosystem, HeLa cells were cultured with PEG/UCP-DOX at 37 °C for 0.5, 1 and 3 h, respectively, and the related CLSM images are shown in Fig. 5a. 4′,6-Diamidino-2-phenylindole (DAPI) has the ability to emit blue fluorescence to label the cell nucleus. The loaded DOX can radiate red fluorescence when stimulated by a 488 nm laser, and the corresponding merged channels of DAPI and DOX are subsequently shown. As indicated, after 0.5 h of incubation, there was a small section of red fluorescence, indicating that a small proportion of PEG/UCP-DOX NPs were internalized by the HeLa cells. With an increase in incubation time, the red fluorescence signal became more intense, demonstrating that more NPs were internalized by the cells. These results confirmed that the NPs can be efficiently assimilated by tumor cells. It is worth noting that the ingestion of the nanomedicine did not produce morphological changes in the cells, confirming the excellent biocompatibility of the as-synthesized nanosystem. To verify that the acquired sample could be used as a UCL imaging contrast agent, HeLa cells were cultured with PEG/UCP for 0.5, 1 and 3 h, separately. As demonstrated in Fig. 5b, upon irradiation with 980 nm light, the particles in the cells showed upconverted blue emission, and the intensity became brighter as the incubation time increased. Interestingly, no fluorescence signal was detected outside the cell, indicating that the as-prepared particles were internalized in the cell instead of just being dyed on the membrane surface. The cell uptake of free DOX was also studied. As presented in Fig. S11a and b,† the red fluorescence intensity increased with the extension of incubation time, and free DOX was mainly located in cell nuclei, which was due to the rapid diffusion mechanism of small molecules. Furthermore, the UCL signal was mainly situated in the cytoplasm, confirming that the particles passed through the cell membrane under endocytic and lysosomal effects. The above results imply that PEG/UCP is an efficient UCL imaging contrast agent with a negligible background.
It is known that Gd3+-doped nanomaterials can positively enhance the properties of T1-MRI signals; thus, we assume that NaGdF4-based UCNPs should have favorable T1 MRI imaging performance.70 This was performed in a non-invasive way to obtain high function information and soft tissue comparison. MRI has superior space resolution (tens of microns) and high penetration depth. The signal was positively enhanced over a broad concentration range from 0 to 0.8 mM (Fig. 6a), and the longitudinal relaxivity (r1) value was calculated to be 2.481 mM−1 s−1 (Fig. 6b). The in vivo T1-weighted MRI performance of PEG/UCP was also detected. As shown in Fig. 6c, the mouse injected with the sample in the tumor showed significant T1-weighted signal enhancement compared to the tumor without injection, demonstrating that PEG/UCP can be used as a T1 MRI contrast agent. It is known that CT imaging provides high credibility because it supplies high resolution 3D structural details and deep tissue penetration.71,72 Yb3+-doped materials have been widely investigated as CT imaging contrast agents. Fig. 6d presents in vitro CT imaging of PEG/UCP. As shown, in accordance with the sample concentration increase, the CT signal significantly strengthened. Additionally, the CT value positively increased relative to the concentration with a steep slope of 52.17 (Fig. 6e). In vivo CT imaging is shown in Fig. 6f. The CT value for the tumor site after injection of the NPs with saline was 472.3 Hounsfield units (HU), which is significantly higher than that without injection (78.5 HU).
Prior to actual application, non-cancerous L929 fibroblast cells were used to characterize the in vitro safety of PEG/UCP to normal cells. A typical 3,4,5-dimethylthiazol-2-yl-2,5-diphenyl tetrazolium bromide (MTT) cell survey was carried out on the L929 fibroblast cells. As Fig. 7a shows, the activity of the L929 cells was maintained above 90% after incubation with DMEM for 24 h with different concentrations of PEG/UCP, showing no significant toxic effects over a wide concentration range. This superior biocompatibility shows its potential as a photoactive drug for bio-applications. Additionally, the sample blood compatibility was assessed to ensure its in vivo application (Fig. S12a†). Obviously, there was no appreciable hemolysis phenomenon when different amounts of PEG/UCP were added, indicating a high blood compatibility. And the materials' coagulation was also tested, and the results are shown in Fig. S12b.† As displayed, the coagulation times of PBS (as a control) and different concentration samples (PEG/UCP) were evaluated by activated partial thromboplastin time (APTT), prothrombin time (PT) and thrombin time (TT). It can be clearly seen that with the increase of sample concentration, there is no obvious extension of coagulation time of APTT, PT and TT, demonstrating that the designed nanoparticles have a negligible effect on coagulation time. The in vitro cytotoxicity to HeLa cells under different treatment conditions was assessed by the MTT measurement method. Media containing PEG/CUP and PEG/UCP-DOX NPs were placed in specific wells with different treatments and then incubated for an additional 24 h. As shown in Fig. 7b, the cancer cells seeded with PEG/UCP-DOX with the 980 nm laser irradiation could be substantially inhibited in the entire concentration range. The control group (PEG/UCP) showed negligible toxicity to the cancer cells, while PEG/UCP + NIR exhibited remarkable anti-cancer activity, implying that pure CeOx cannot generate a ROS-induced PDT effect, although it can produce O2 in the absence of NIR light.
The key function of NIR light was also confirmed by the MTT test using the DOX loaded sample. As shown in Fig. 7b, the HeLa cells treated with PEG/UCP-DOX + NIR (t test, P = 1.8 × 10−4versus control) exhibited much lower viability than those treated with PEG/UCP-DOX (t test, P = 2.1 × 10−3versus control). In addition, PI is capable of staining red dead cells with a red emission and was used to prominently label the dead cells under different conditions to illustrate the cell killing efficacy. As Fig. 7c points out, there were almost no red cells treated only with culture medium and only the 980 nm laser, illustrating that NIR irradiation alone has no significant killing efficacy on HeLa cells. Approximately 70% of red cells were observed in the PEG/UCP + NIR treatment group compared to that with only NIR irradiation. This result demonstrates that PDT has a significant effect. For the cells cultivated with PEG/UCP-DOX, almost half of the cells were killed, which means that the nanomaterial successfully released the loaded DOX. As expected, the bottom right image of the group treated with PEG/UCP-DOX + NIR shows the largest number of dead cells. CLSM photos and in vitro viability assays confirmed that PEG/UCP is effective at delivering DOX to cancer cells and exerting excellent anticancer effects. In addition, the related flow-cytometry apoptosis assay (sample concentration: 50 μg mL−1) and vitamin C dyeing experiments certify the cell apoptosis. As shown in Fig. 7d, the percentages of cell apoptosis for treatment with NIR, PEG/UCP + NIR, PEG/UCP-DOX and PEG/UCP-DOX + NIR were 1.4%, 49.7%, 25.5, and 88.7%, respectively, which was consistent with the above MTT results. Vitamin C was used to react with ROS generated by PEG/UCP nanoparticles, which could be utilized to detect the cell cytotoxicity. As displayed in Fig. S13a and b,† with the extension of irradiation time, more and more cells died, certifying the consumption of vitamin C and generation of ROS. The corresponding in vivo UCL images of pork tissue (Fig. S14a†) and a tumor-bearing mouse (Fig. S14b†) indicate that the up-conversion fluorescence of the sample excited by the 980 nm laser could be clearly visualized at a depth of approximately 8 mm, implying the feasibility of its clinical application as an imaging probe.
To clarify the advantages of mCeOx compared with other mesoporous PDT catalysts, we designed UCNPs@mSiO2@mTiO2 (on the basis of the synthesis of UCS, APTES was used to aminate the surface, and 2 mL of ethanol solution containing 20% TBOT was added dropwise to UCS-NH2 and stirred under reflux for 2 h. Then, the obtained NPs were added into a 150 mL reactor and refluxed at 180 °C for 24 h to obtain a crystalline TiO2 shell. After it was treated with ethanol several times, the final mesoporous UCNPs@mSiO2@mTiO2 (denoted as UCST) was obtained. The subsequent modification with PEG and the loading of the drug are also similar to those of the mCeOx based samples). The corresponding XRD patterns, TEM image and EDS spectrum are provided in Fig. S15a–c.† The pore size distribution profile (Fig. S15d†) and N2 adsorption/desorption isotherm (Fig. S15e†) indicate that UCST is a mesoporous material. As displayed in Fig. S15f,† the absorption curve for DPBF gradually decreased with the extended irradiation time, showing that UCST has the ability to generate ROS. Moreover, a typical MTT test was performed on L929 fibroblast cells to verify the in vitro viability of PEG-modified UCST (denoted as PEG/UCST), and it showed no significant toxic effects over a wide concentration range. The in vitro cytotoxicity to HeLa cells subjected to different conditions was assessed by using the MTT method, and Fig. S15i† shows the CLSM images of HeLa cells with the fluorescence of DCF. As shown, the more intense green fluorescence illustrates efficient ROS production in PEG/UCST upon 980 nm laser irradiation. Compared to that indicated by Fig. S15h,†Fig. 7b indicates that PEG/UCP exhibits better PDT (lower cell viability of HeLa cells) at the same sample concentration because cerium oxide has the catalytic ability to decompose endogenous H2O2 into H2O and O2, thereby increasing the level of intracellular O2 to enhance PDT. Moreover, the forbidden bandwidth of cerium oxide is at 2.89 eV, while that of titanium dioxide is at 3.2 eV, which means that cerium oxide can absorb more UV photon energy to generate electron–hole pairs, implying a better PDT effect.
To assay the O2 generation capability from H2O2 with PEG/UCP, 4-hydroxy-3-methoxyphenylacetic acid (HVA) was chosen as the H2O2 probe to react with hydrogen peroxide in the presence of peroxidase to produce a strongly fluorescent substance excited by 312 nm light. As presented in Fig. S16a,† the luminescence gradually weakened with reaction time, which means that H2O2 gradually decomposed. Meanwhile, as the O2 probe, bis(triphenylphosphine) ruthenium(II) dicarbonyl chloride ([Ru(dpp)3]Cl2) was chosen to test the production of O2, and it was oxidized by dissolved O2 and faded when excited by the 488 nm laser. In Fig. S16b,† the progressively attenuated emission intensity in the [Ru(dpp3)Cl2] channel shows evidence of production of O2.
Inspired by the in vitro results, U14 tumor (murine cervical carcinoma) xenografts were injected into female Kunming mice for treatment. In order to study the in vivo performances of the PEG/UCP NPs, U14 tumor-bearing mice that had an i.v. injection of PEG/UCP NPs were imaged using a Bruker In-Vivo F PRO small-animal imaging system. As shown in Fig. S17a,† the fluorescence signal of the PEG/UCP NPs rapidly increased owing to the EPR effect at the tumor sites with the maximum at ≈12 h, indicating that 12 h post injection of the PEG/UCP NPs is the optimal time for in vivo PDT treatment. Moreover, to assess the residual amount of the PEG/UCP NPs in vivo, the major organs were collected from the U14-tumor-bearing mice at different time intervals post injection of the PEG/UCP NPs and then imaged. Fig. S17b† clearly shows that PEG/UCP NPs were highly accumulated in the tumor 12 h post injection compared to the accumulation in the other major organs. Although the liver and lung exhibited high fluorescence intensity because of the macrophage uptake in the reticuloendothelial system, the PEG/UCP NPs steadily decreased over time 48 h postinjection. Next, the Ce level in the major organs at different time intervals after i.v. injection was measured using ICP-MS. As presented in Fig. S17c,† the trends and biodistribution are consistent with FL imaging. The above results show that the designed nanoparticles can be utilized in biosystems to guide PDT therapy.
The therapeutic effect was evaluated by injecting PEG/UCP-DOX intravenously and the equivalent dose of DOX was 2 mg kg−1. Fig. 8a shows the average and relative mice tumor volume for every group during the 14 day treatment period. As expected, PEG/UCP-DOX + NIR showed excellent tumor inhibition performance. Similar to the in vitro results, the tumor growth of the PEG/UCP-DOX + NIR treated group (t test, P = 3.6 × 10−4versus control) was more suppressed than that of the PEG/UCP-DOX treated group (t test, P = 3.5 × 10−3versus the control), suggesting the key role of the NIR light irradiation. To determine the biodistribution of PEG/UCP, the concentration of Ce in the tumors was determined by ICP-MS after injection, and the accumulation of PEG/UCP NPs in the tumors was also tested. As demonstrated in Fig. S18a,† high concentrations of Ce were observed in the tumors after 12 h owing to the EPR effect, and 12 to 48 h was the relatively slow clearance period of PEG/UCP according to Fig. S18b.† After 2 weeks of treatment, tumor growth was slightly inhibited for the group treated with only 980 nm laser irradiation. As displayed in Fig. 8b, the images of tumors from representative mice also confirmed that the PEG/UCP-DOX + NIR-treated group had the smallest tumor volume, proving its superior anti-tumor effect. Meanwhile, Fig. 8c shows clearly that the weights of the tumors after PDT significantly reduced in contrast to those for the control group, while the tumor weights for the PEG/UCP-DOX + NIR-treated group (PDT plus chemotherapy) were the lowest, indicating that the combination of the two therapies had a better effect than treatment with only PEG/UCP-DOX (t test, P = 2.3 × 10−3versus PEG/UCP-DOX). In Fig. 8d, the H&E stained images show the most serious cell necrosis in the PEG/UCP-DOX + NIR treated group, which agrees with the results shown in Fig. 7c. The H&E staining photos of the liver, lung, kidney, heart and spleen (Fig. S19†) confirmed that there was no significant organ damage in any of the groups, revealing the outstanding in vivo biocompatibility of the as-prepared nanomedicine. In addition, complete blood count assessment and serum biochemistry assay were performed after two weeks of treatment, as shown in Table S1,† and the biochemical results including those for alanine transaminase, creatinine, blood urea nitrogen, total protein, and aspartate transaminase, which are closely related to the function of the liver and kidney, were checked. There was no obvious injury to the liver or kidney when compared with the control group. Furthermore, the complete blood tests showed that there was no evident interference of physiological regulation of haem or the immune response in the PEG/UCP-treated mice. In summary, the obtained nanoparticles exhibited high safety when used for in vivo antitumor application.
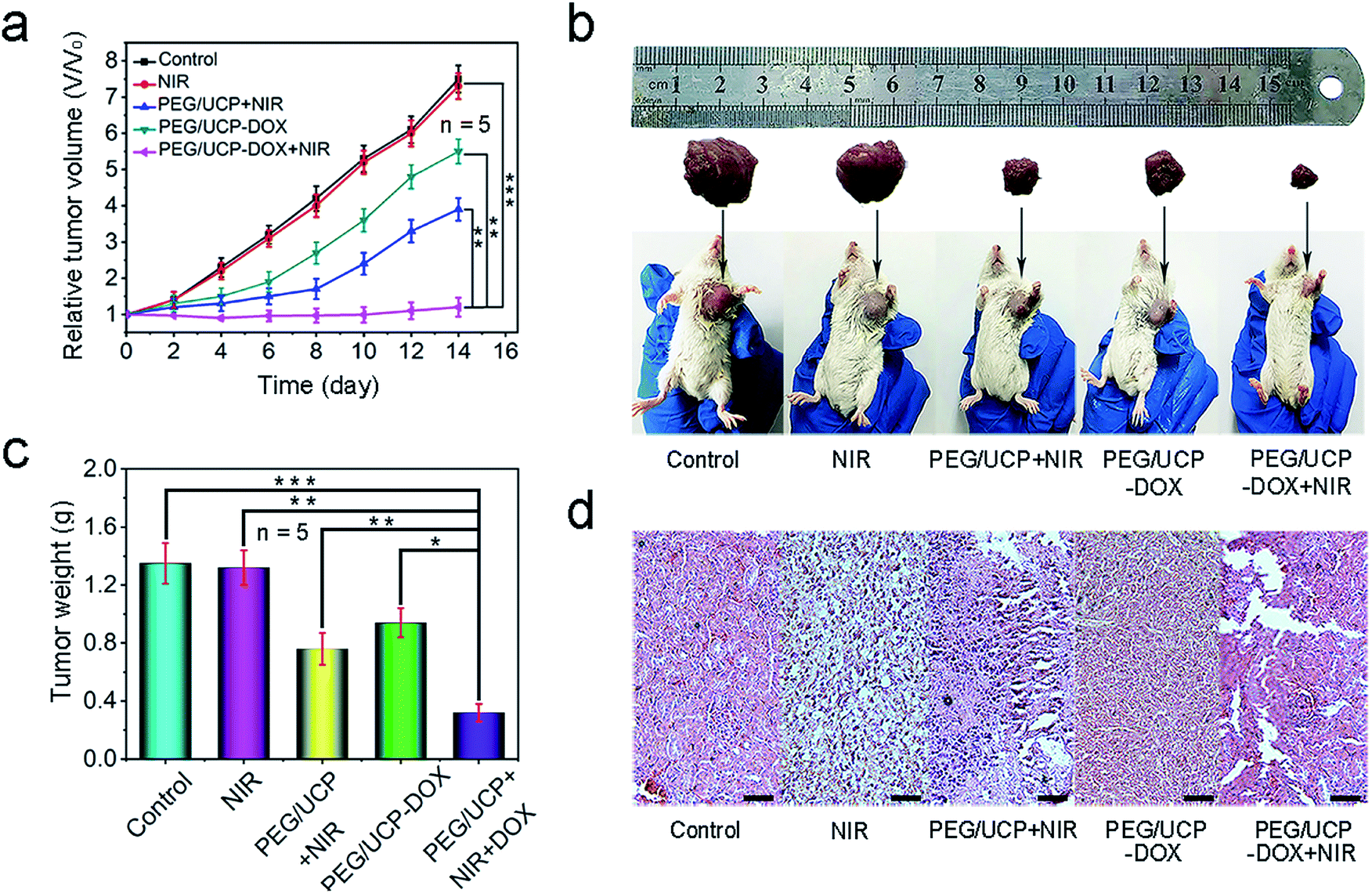 | ||
| Fig. 8 The relative tumor volume achieved from the mice with different treatments. Error bars are based on the standard errors of the mean (n = 5) (a). Photographs of representative mice and excised tumors (b), and the tumor weights with different treatments. Error bars are based on the standard errors of the mean (n = 5) (c). H&E stained images of tumor tissues obtained after 14 days of treatment. P values were calculated using Tukey's post-test (***p < 0.001, **p < 0.01, or *p < 0.05). Scale bar: 100 μm (d). | ||
Conclusions
In summary, we successfully developed a mesoporous core–shell-structured nanoplatform that is comprised of a UCNP core and a porous cerium oxide shell, as a one-piece biophotocatalyst to realize a synergistic therapeutic mode for efficient PDT and chemotherapy. The core–shell NPs with high efficiency and stable luminescence can continuously convert 980 nm NIR laser UV light, thereby exciting the photocatalytic action of cerium oxide for PDT. These H2O2 responsive agents achieved sustained self-sufficiency of O2 during PDT to overcome the obstacles of tumor hypoxia upon NIR irradiation. Tumor cell targeting features (EPR) resulted in a high PDT selectivity for tumor cells and avoided damage to normal cells, which significantly reduced the side effects, and the suitable nanoscale size could also improve passive tumor targeting via the EPR effect. In addition, the inorganic UCNPs@mCeOx with high photostability can achieve stable long-lasting photodynamic effects under NIR irradiation in contrast to commonly used organic photosensitizers that suffer severe photodamage during light irradiation. The as-developed nanoplatform is applicable for UCL, MRI and CT imaging, which is of great significance for multimodal treatment. Moreover, the huge hollow structure effectively loaded the chemotherapeutic drug DOX, thereby achieving synergistic treatment that combines PDT with chemotherapy, thus improving the treatment efficiency and providing a more obvious anti-tumor effect.Experimental section
Chemicals and reagents
Yb2O3 (99.99%), Gd2O3 (99.99%), Tm2O3 (99.99%), sodium fluoride (NaF), hydrochloric acid (HCl), tetraethyl orthosilicate (TEOS), ammonium nitrate (NH4NO3), Ce(NO3)3·6H2O, trifluoroacetic acid (CF3COOH), oleic acid (OA), DMPO, DOX, MB and CTAB were obtained from Shanghai Chemical Co. Ltd. DAPI, DCFH-DA, hexamethylenetetramine, 1-octadecene (ODE), MTT, methoxy PEG amine (mPEG-NH2) (Mw = 2000), HVA and [Ru(dpp)3]Cl2 were purchased from Sigma-Aldrich. L-O-Phosphoserine was purchased from Tokyo Chemical Industry. All the reagents were of analytical grade and were used without additional treatment.Synthesis of NaGdF4:Yb,Tm@NaGdF4 (denoted as UCNPs)
In short, 1 mmol of RE(oleate)3 (RE = 79%Gd + 20%Yb + 1%Tm) in a mixture of ODE and OA (both volumes were 15 mL), and NaF (5 mmol) were mixed under stirring. Then, the mixture was continuously heated from 30 °C to 110 °C and this temperature was maintained for 0.5 h in a vacuum environment. Then the vacuum was turned off, and N2 purging was performed for several minutes, followed by a temperature increase to 300 °C, which was maintained for 90 min. Subsequently, the temperature of the mixture was allowed to naturally cool to 30 °C, and the product was successively centrifuged using cyclohexane, ethanol and cyclohexane, and then ultrasonically dispersed uniformly into 5 mL of cyclohexane. Afterwards, 1 mmol of CF3COONa and 0.5 mmol of Gd(CF3COO)3 were mixed in cyclohexane (5 mL) with the core NPs; then 15 mL of both OA and ODE were added to a three-necked flask, and later the temperature was continuously increased to 120 °C and maintained for 30 min to remove residual gas impurities in vacuo. Subsequently, the temperature of the mixture was increased to 310 °C and maintained for 60 min. Then, the temperature of the mixture was slowly decreased to 30 °C, and the product was centrifuged with cyclohexane and ethanol, separately, and dispersed into 5 mL of cyclohexane.Synthesis of UCNPs@mSiO2
The method of coating mesoporous silica is based on previous literature.73 In general, UCNPs stored from the previous step in 5 mL of cyclohexane were transferred to an Erlenmeyer flask and diluted to approximately 16 mL with cyclohexane. Then, 0.1 g of CTAB was placed in a beaker under atmospheric conditions, and 20 mL of deionized water was added. Then the system was sonicated until transparent, followed by addition of 2 mL of UCNPs dropwise.After it was heated for approximately 10 min, the solution was stirred at room temperature overnight until transparent; then 6 mL of ethanol, 40 mL of water and 300 μL of NaOH (2 M) were mixed into the system, and it was heated to 70 °C. Then 200 μL of TEOS solution was added dropwise and the temperature maintained for 10 min. After centrifuging three times with ethanol, the final centrifugation product was added to 50 mL of ethanol and 0.3 g of NH4NO3, and the mixture was stirred for 2 h at 60 °C, and then centrifuged with deionized water and ethanol in that order. Finally the UCNPs@mSiO2 was obtained.
Synthesis of mesoporous cerium oxide (mCeOx) coated UCNPs
The as-fabricated UCNPs@mSiO2 (∼50 mg) and 0.111 g of Ce(NO3)3·6H2O were uniformly dispersed in 50 mL of aqueous solution, and agitated at a constant speed for 1 h at normal atmospheric temperature (25 °C). Then 0.05 g hexamethylenetetramine was added, and the mixture was agitated in an oil bath maintained at 90 °C for 2 h. Subsequently, the mixture was collected by centrifugation, and the product was rinsed with deionized water to remove possible impurities, and then dried overnight at 60 °C. The product was then treated at 600 °C for 2 h. The silica template was etched using Na2CO3 (2 M) and stirred for approximately 7 h at 60 °C; then the product was cleaned several times with deionized water. Finally, the hollow mCeOx coated UCNPs (denoted as UCNPs@mCeOx) were obtained.Modification of L-O-phosphoserine on UCNPs@mCeOx
First, 15 mg of UCNPs@mCeOx was uniformly dispersed in deionized water (20 mL). Then 20 mg of L-O-phosphoserine was added to deionized water and the system was stirred constantly at 4 °C for 2 h. Then the product was collected by centrifugation and rinsed several times with deionized water to wash away unbound L-O-phosphoserine. The obtained UCNPs@mCeOx-L-O-phosphoserine was denoted as UCP.PEGylation of UCP
UCP (20 mg) and mPEG-NH2 (20 mg) were dispersed in 20 mL of deionized water under ultrasound treatment, and 6 mg of EDC was added to increase the coupling efficiency between the hydroxyl and carboxyl groups; the mixed solution was stirred for approximately 12 h. Later the sample was collected by centrifugation and rinsed repeatedly with deionized water to remove unbound mPEG-NH2. The resulting product was dried at 60 °C, and the final PEG/UCP was obtained.DOX loading and release from PEG/UCP-DOX
In a darkroom, PEG/UCP (20 mg) was uniformly dispersed into deionized water with DOX (10 mL, 0.25 mg mL−1) and stirred overnight at normal atmospheric temperature. After centrifugation with distilled water, PEG/UCP-DOX was collected, and the supernatant was removed for UV-vis spectroscopy at 480 nm. The mixture was retained for testing the DOX release processes. 10 mL of PBS was added and stirred in a water bath at room temperature, and the supernatant was kept for further UV-vis analysis to calculate the DOX content. The loading rate of the material and the DOX content in the PBS were measured using UV-vis spectroscopy at 480 nm.Characterization
Samples for X-ray diffraction (XRD) measurements were prepared by depositing a sample solution on a glass slide and vacuum drying it at 80 °C. N2 adsorption/desorption measurement was performed on a Micromeritics Tristar 3000 instrument. Transmission electron microscopy (TEM) images were obtained using an FEI Tecnai G2 S-twin transmission electron microscope. Ultraviolet-visible absorption spectra were obtained using a TU-1901 dual-beam UV-vis spectrophotometer.MB detection of hydroxyl radicals
A MB solution (10 mg L−1) was configured, and 5 mg of UCP/PEG was mixed. The mixed solution was then sonicated to disperse it uniformly and irradiated with 980 nm light for 0, 3, 5, 7 and 10 min, respectively. After irradiation and centrifugation, the supernatant was collected to calculate the absorbance of the supernatant within the range of 550–750 nm using UV-vis spectroscopy.In vitro and in vivo X-ray CT imaging
The sample was diluted to half strength, gradually reduced, and added to rows for CT imaging. The mice were anesthetized by intraperitoneal injection with 10% chloral hydrate (0.04 mL per g per mice). Subsequently, 100 μL of PEG/UCP physiological saline solution was injected intratumorally into a tumor-bearing mouse for scanning. All the female Kunming mice were bought from Second Affiliated Hospital, Harbin Medical University. All the animal experiments were conducted in line with the specifications of The National Regulation of China for Care and Use of Laboratory Animals and approved by the Harbin Science and Technology Bureau.In vitro and in vivo T1-weighted MR imaging
The in vitro MR imaging tests were executed in 0.5 T MRI magnets. The PEG/UCP was diluted to different concentrations (layer by layer in half). T1 measurements were recorded versus the repetition time (TR) on a Huantong 1.5 TMR scanner with a non-linear fit to the change in mean signal intensity for every well. Finally, the r1 relaxation value was determined by curve fitting the 1/T1 relaxation time (s−1) to the particle concentration (mg mL−1). In vivo MR imaging tests were employed on Huantong 1.5 TMR.Hemolytic detection of PEG/UCP
In order to ensure in vivo compatibility of the synthesized material, it was necessary to detect hemolysis. In general, the obtained normal human red blood cells were cleaned with physiological saline and then centrifuged 5–7 times. Then, the centrifuged blood cells were blended with PBS buffer solution (10 mL). And 1 mg sample (PEG/UCP) was separately dissolved in 4 mg of deionized water as the positive control and in PBS solution as the negative control, and in 2 mL of a series of concentrations of the substance suspension (15.6, 31.25, 62.5, 125, 250, and 500 μg mL−1). Then the centrifuge tube was stirred and kept still for the remaining 1 h. Finally, the sample was centrifuged and the supernatant was collected to detect the absorbance.In vitro cellular uptake and UCL microscopy (UCLM) test
To study the cellular uptake procedure on the HeLa cell culture using confocal laser scanning microscopy (CLSM), HeLa cells (7000–8000 per well) were cultivated in 6-well plates in a humidified incubator (5% CO2, 37 °C) overnight (approximately 12 h) to acquire monolayer cells. Thereafter, 1 mL of PEG/UCP solution (1 mg mL−1) was added to the wells and cultivated for different periods, separately. Afterwards, the cells were cleaned three times with PBS and dyed with 1 mL of DAPI for 10 min. Then, 1 mL of glutaraldehyde (2.5%) was added for 10 min to fix the cell morphology, and then the cells were further rinsed three times with PBS. Finally, fluorescence photos of the cells were recorded using Leica TCS SP8 apparatus. For UCLM observation, slides were prepared by an identical method except that photos were recorded with an inverted fluorescence microscope (Nikon Ti-S), and the samples were irradiated using an external continuous wavelength 980 nm laser.Detection of intracellular hydrogen peroxide decomposition and O2 production
The HeLa cells were cultured with 1 mL of HVA (1 mg mL−1) solution for 2 h, and 1 mL of PEG/UCP (1 mg mL−1) was added and cultured for different time periods (0 min, 20 min and 40 min). Subsequently, the cells were washed with PBS three times and dyed with DAPI (1 mL) for 10 min. Then 1 mL of glutaraldehyde (2.5%) was added to fix the cell morphology and the cells were washed with PBS several times. Then, the fluorescence photos of the cells were recorded using Leica TCS SP8 apparatus.Detection of intracellular O2 production
After the HeLa cells were treated with 50 μM Ru(dpp3)Cl2 for 4 h, 1 mL of PEG/UCP and DOX-EPG/UCP (1 mg mL−1) were mixed into the wells and cultivated for different periods (0 min, 20 min and 40 min); then they were washed three times with PBS and dyed with 1 mL of DAPI for 10 min. Subsequently, 1 mL of glutaraldehyde was added to fix the cell morphology and they were washed with PBS several times. After that, fluorescence photos of the cells were recorded using Leica TCS SP8 apparatus.In vitro biocompatibility
L929 cells were used to detect the biocompatibility in vitro. After the L929 cells (7000–8000 per well) were cultured with DMEM in a 96-well plate overnight at 37 °C in a 5% CO2 atmosphere to attach them to the bottom, materials with different concentrations (7.8, 15.6, 31.25, 62.5, 125, 250, and 500 μg mL−1) were added and co-cultured for 12 h. After that, 20 μL of MTT (5 mg mL−1) was added to each well. After 4 h of cultivation, 150 μL of DMSO was added into the medium, and the absorbance at 490 nm was measured after 10 min.In vitro cytotoxicity
A particular MTT test was used to assess the in vitro cytotoxicity. HeLa cells (7000–8000 per well) were cultivated in a 96-well plate with DMEM in a humidified incubator (5% CO2, 37 °C) for 12–24 h. Then PEG/UCP and PEG/UCP-DOX were dispersed in the medium at a concentration gradient of 0, 31.2, 62.5, 125, 250 and 500 μg mL−1, separately. Then, the cells were treated with the control (PEG/UCP), NIR irradiation, PEG/UCP-DOX, PEG/UCP + NIR, and PEG/UCP-DOX + NIR (pump power: 0.72 W cm−2). The material was added and cultivated for 2 h to complete cell uptake followed by irradiation. Thereafter, 20 μL of MTT solution (5 mg mL−1) was added to each well. After 6 h of incubation, the medium was discarded, and DMSO (150 μL) was dropped into the wells. After 10 min of incubation, the absorbance of the solution at 490 nm was measured. The percentage of viable cells in the experimental group and the percentage of viable cells in the control group were used to express cytotoxicity. The in vitro viability of PEG/UCP on the L929 fibroblasts was evaluated by a similar MTT assay. Specimen concentrations were 7.8, 15.6, 31.2, 62.5, 125, 250 and 500 μg mL−1.In vivo toxicity
U14 cells were injected in the left armpit of each female mouse. When the tumor size was approximately 6–8 mm, the mice were divided into 5 groups (n = 5 per group). One group served as a control group that was injected with physiological saline. The remaining groups were treated with the control, NIR, PEG/UCP-DOX, PEG/UCP + NIR and PEG/UCP-DOX + NIR. After 12 h of injection, the tumor site was irradiated with a 980 nm laser (0.72 W cm−2) for 10 min. The tumor size and body weight were surveyed and recorded before treatment.Histological examination
After continuous treatment for 14 days, the mice were subjected to histological analysis. Less than 1 cm × 1 cm of tissue was removed from the heart, lung, spleen, kidney, liver, and tumor organs of five representative mice. Then, buffered formalin was used to continuously dehydrate the excised tissue with different concentrations of xylene and ethanol. Finally, the above dehydrated tissue was embedded in liquid paraffin and sectioned for hematoxylin and eosin (H&E) staining. The stained sections were inspected using optical microscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (NSFC; 51772059, 51502050, 51720105015, 51828202, 51702070, 51602073, and 51602072), a General Financial Grant from the China Postdoctoral Science Foundation (2016M600241), a Special Financial Grant from the China Postdoctoral Science Foundation (2017T00228), the Science and Technology Development Planning Project of Jilin Province (20170414003GH), the CAS Croucher Funding Scheme for Joint Laboratories (CAS18204), the Key Research Program of Frontier Sciences of the Chinese Academy of Sciences (YZDY-SSW-JSC018), and the Fundamental Research Funds for the Central Universities is greatly acknowledged.Notes and references
- V. J. N. Bykov, S. E. Eriksson, J. Bianchi and K. G. Wiman, Nat. Rev. Cancer, 2018, 18, 89–102 CrossRef CAS PubMed.
- H. Chen, W. Zhang, G. Zhu, J. Xie and X. Chen, Nat. Rev. Mater., 2017, 2, 17024 CrossRef CAS PubMed.
- W. Fan, B. Yung, P. Huang and X. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- T. C. Chua, N. Engl. J. Med., 2010, 363, 943–953 CrossRef PubMed.
- R. P. DeMatteo, K. V. Ballman, C. R. Antonescu, R. G. Maki, P. W. T. Pisters, G. D. Demetri, M. E. Blackstein, C. D. Blanke, M. von Mehren, M. F. Brennan, S. Patel, M. D. McCarter, J. A. Polikoff, B. R. Tan, K. Owzar and A. I. A. G. Stud, Lancet, 2009, 373, 1097–1104 CrossRef CAS.
- S. Kopetz, G. J. Chang, M. J. Overman, C. Eng, D. J. Sargent, D. W. Larson, A. Grothey, J.-N. Vauthey, D. M. Nagorney and R. R. McWilliams, J. Clin. Oncol., 2009, 27, 3677–3683 CrossRef PubMed.
- S. L. Gai, G. X. Yang, P. P. Yang, F. He, J. Lin, D. Y. Jin and B. G. Xing, Nano Today, 2018, 19, 146–187 CrossRef CAS.
- W. Hou, Y. Liu, Y. Jiang, Y. Wu, C. Cui, Y. Wang, L. Zhang, I. T. Teng and W. Tan, Nanoscale, 2018, 10, 10986–10990 RSC.
- T. Y. Hulchanskyy, I. Roy, L. N. Goswami, Y. Chen, E. J. Bergey, R. K. Pandey, A. R. Oseroff and P. N. Prasad, Nano Lett., 2007, 7, 2835–2842 CrossRef PubMed.
- K. Deng, C. Li, S. Huang, B. Xing, D. Jin, Q. Zeng, Z. Hou and J. Lin, Small, 2017, 13, 1702299 CrossRef PubMed.
- J. Mou, T. Q. Lin, F. Q. Huang, H. R. Chen and J. L. Shi, Biomaterials, 2016, 84, 13–24 CrossRef CAS PubMed.
- J. Xu, W. Han, P. Yang, T. Jia, S. Dong, H. Bi, A. Gulzar, D. Yang, S. Gai, F. He, J. Lin and C. Li, Adv. Funct. Mater., 2018, 28, 1803804 CrossRef.
- E. Ju, K. Dong, Z. Chen, Z. Liu, C. Liu, Y. Huang, Z. Wang, F. Pu, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2016, 55, 11467–11471 CrossRef CAS PubMed.
- R. Lv, D. Yang, P. Yang, J. Xu, F. He, S. Gai, C. Li, Y. Dai, G. Yang and J. Lin, Chem. Mater., 2016, 28, 4724–4734 CrossRef CAS.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- Z. Feng, P. Tao, L. Zou, P. Gao, Y. Liu, X. Liu, H. Wang, S. Liu, Q. Dong, J. Li, B. Xu, W. Huang, W.-Y. Wong and Q. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 28319–28330 CrossRef CAS PubMed.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- L. Xia, X. Kong, X. Liu, L. Tu, Y. Zhang, Y. Chang, K. Liu, D. Shen, H. Zhao and H. Zhang, Biomaterials, 2014, 35, 4146–4156 CrossRef CAS PubMed.
- Y. Liu, Y. Liu, W. Bu, C. Cheng, C. Zuo, Q. Xiao, Y. Sun, D. Ni, C. Zhang, J. Liu and J. Shi, Angew. Chem., Int. Ed., 2015, 54, 8105–8109 CrossRef CAS PubMed.
- C. Zhang, W.-H. Chen, L.-H. Liu, W.-X. Qiu, W.-Y. Yu and X.-Z. Zhang, Adv. Funct. Mater., 2017, 27, 1700626 CrossRef.
- W. Zhu, Z. Dong, T. Fu, J. Liu, Q. Chen, Y. Li, R. Zhu, L. Xu and Z. Liu, Adv. Funct. Mater., 2016, 26, 5490–5498 CrossRef CAS.
- W. Fan, W. Bu, B. Shen, Q. He, Z. Cui, Y. Liu, X. Zheng, K. Zhao and J. Shi, Adv. Mater., 2015, 27, 4155–4161 CrossRef CAS PubMed.
- Y. Liu, W. Y. Zhen, L. H. Jin, S. T. Zhang, G. Y. Sun, T. Q. Zhang, X. Xu, S. Y. Song, Y. H. Wang, J. H. Liu and H. J. Zhang, ACS Nano, 2018, 12, 4886–4893 CrossRef CAS PubMed.
- Z. Ma, X. Jia, J. Bai, Y. Ruan, C. Wang, J. Li, M. Zhang and X. Jiang, Adv. Funct. Mater., 2017, 27, 1604258 CrossRef.
- C. S. Jin, J. F. Lovell, J. Chen and G. Zheng, ACS Nano, 2013, 7, 2541–2550 CrossRef CAS PubMed.
- C. Liu, D. Wang, S. Zhang, Y. Cheng, F. Yang, Y. Xing, T. Xu, H. Dong and X. Zhang, ACS Nano, 2019, 13, 4267–4277 CrossRef CAS PubMed.
- D. W. Zheng, B. Li, C. X. Li, J. X. Fan, Q. Lei, C. Li, Z. S. Xu and X. Z. Zhang, ACS Nano, 2016, 10, 8715–8722 CrossRef CAS PubMed.
- D. Hu, Z. Chen, Z. Sheng, D. Gao, F. Yan, T. Ma, H. Zheng and M. Hong, Nanoscale, 2018, 10, 17283–17292 RSC.
- Z. Luo, H. Tian, L. Liu, Z. Chen, R. Liang, Z. Chen, Z. Wu, A. Ma, M. Zheng and L. Cai, Theranostics, 2018, 8, 3584–3596 CrossRef CAS PubMed.
- S. Cui, D. Yin, Y. Chen, Y. Di, H. Chen, Y. Ma, S. Achilefu and Y. Gu, ACS Nano, 2013, 7, 676–688 CrossRef CAS PubMed.
- S. Wang, F. Yuan, K. Chen, G. Chen, K. Tu, H. Wang and L.-Q. Wang, Biomacromolecules, 2015, 16, 2693–2700 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS PubMed.
- Y. Cheng, H. Cheng, C. Jiang, X. Qiu, K. Wang, W. Huan, A. Yuan, J. Wu and Y. Hu, Nat. Commun., 2015, 6, 8785 CrossRef CAS PubMed.
- S. Gao, P. Zheng, Z. Li, X. Feng, W. Yan, S. Chen, W. Guo, D. Liu, X. Yang, S. Wang, X.-J. Liang and J. Zhang, Biomaterials, 2018, 178, 83–94 CrossRef CAS PubMed.
- S. S. Lucky, N. M. Idris, Z. Li, K. Huang, K. C. Soo and Y. Zhang, ACS Nano, 2015, 9, 191–205 CrossRef CAS PubMed.
- T. Nomoto, S. Fukushima, M. Kumagai, K. Miyazaki, A. Inoue, P. Mi, Y. Maeda, K. Toh, Y. Matsumoto, Y. Morimoto, A. Kishimura, N. Nishiyama and K. Kataoka, Biomater. Sci., 2016, 4, 826–838 RSC.
- Q. Q. Bao, P. Hu, Y. Y. Xu, T. S. Cheng, C. Y. Wei, L. M. Pan and J. L. Shi, ACS Nano, 2018, 12, 6794–6805 CrossRef CAS PubMed.
- C. Yao, W. Wang, P. Wang, M. Zhao, X. Li and F. Zhang, Adv. Mater., 2018, 30, 1704833 CrossRef PubMed.
- D. D. Zhao, Y. C. Pi, Q. Shao, Y. G. Feng, Y. Zhang and X. Q. Huang, ACS Nano, 2018, 12, 6245–6251 CrossRef CAS PubMed.
- T. Dayakar, K. V. Rao, K. Bikshalu, V. Malapati and K. K. Sadasivuni, Biosens. Bioelectron., 2018, 111, 166–173 CrossRef CAS PubMed.
- N. Thanh-Son, G. Postole, S. Loridant, F. Bosselet, L. Burel, M. Aouine, L. Massin, P. Gelin, F. Morfin and L. Piccolo, J. Mater. Chem. A, 2014, 2, 19822–19832 RSC.
- L. Zhao, C. Li, Y. Wang, H. Wu, L. Gao, J. Zhang and G. Zeng, Catal. Sci. Technol., 2016, 6, 6076–6086 RSC.
- A. Incel, T. Guner, O. Parlak and M. M. Demir, ACS Appl. Mater. Interfaces, 2015, 7, 27539–27546 CrossRef CAS PubMed.
- C. Karunakaran, S. Narayanan and P. Gomathisankar, J. Hazard. Mater., 2010, 181, 708–715 CrossRef CAS PubMed.
- Y.-Q. Xu, S.-Y. Wu, L.-N. Wu and L.-J. Zhang, Mater. Sci. Semicond. Process., 2018, 75, 253–262 CrossRef CAS.
- K. Kasinathan, J. Kennedy, M. Elayaperumal, M. Henini and M. Malik, Sci. Rep., 2016, 6, 38064 CrossRef CAS PubMed.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- M. Zhang, W. Zhu, S. Xun, H. Li, Q. Gu, Z. Zhao and Q. Wang, Chem. Eng. J., 2013, 220, 328–336 CrossRef CAS.
- S. Bhagat, N. V. S. Vallabani, V. Shutthanandan, M. Bowden, A. S. Karakoti and S. Singh, J. Colloid Interface Sci., 2018, 513, 831–842 CrossRef CAS PubMed.
- B.-H. Chen and B. S. Inbaraj, Crit. Rev. Biotechnol., 2018, 38, 1003–1024 CrossRef CAS PubMed.
- A. Pratsinis, G. A. Kelesidis, S. Zuercher, F. Krumeich, S. Bolisetty, R. Mezzenga, J. C. Leroux and G. A. Sotiriou, ACS Nano, 2017, 11, 12210–12218 CrossRef CAS PubMed.
- L.-S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H.-H. Yang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- L. Sun, R. Wei, J. Feng and H. Zhang, Coord. Chem. Rev., 2018, 364, 10–32 CrossRef CAS.
- W. You, D. Tu, W. Zheng, X. Shang, X. Song, S. Zhou, Y. Liu, R. Li and X. Chen, Nanoscale, 2018, 10, 11477–11484 RSC.
- Z. H. Zhang, L. L. Shi, C. W. Wu, Y. Su, J. W. Qian, H. P. Deng and X. Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 29505–29514 CrossRef CAS PubMed.
- Z. Yu, Y. Ge, Q. Sun, W. Pan, X. Wan, N. Li and B. Tang, Chem. Sci., 2018, 9, 3563–3569 RSC.
- Z. Yu, W. Pan, N. Li and B. Tang, Chem. Sci., 2016, 7, 4237–4244 RSC.
- Z. Yu, Q. Sun, W. Pan, N. Li and B. Tang, ACS Nano, 2015, 9, 11064–11074 CrossRef CAS PubMed.
- H. He, S. Ji, Y. He, A. Zhu, Y. Zou, Y. Deng, H. Ke, H. Yang, Y. Zhao, Z. Guo and H. Chen, Adv. Mater., 2017, 29, 1606690 CrossRef PubMed.
- K. Liu, R. Xing, Q. Zou, G. Ma, H. Mohwald and X. Yan, Angew. Chem., Int. Ed., 2016, 55, 3036–3039 CrossRef CAS PubMed.
- Q. Liu, X. Zou, Y. Shi, B. Shen, C. Cao, S. Cheng, W. Feng and F. Li, Nanoscale, 2018, 10, 12573–12581 RSC.
- S. Wang, X. Liu, S. Chen, Z. Liu, X. Zhang, X. J. Liang and L. Li, ACS Nano, 2018, 29, 10377–10385 Search PubMed.
- L. Yao, J. Zhou, J. Liu, W. Feng and F. Li, Adv. Funct. Mater., 2012, 22, 2667–2672 CrossRef CAS.
- J. Xu, A. Gulzar, Y. Liu, H. Bi, S. Gai, B. Liu, D. Yang, F. He and P. Yang, Small, 2017, 13, 1701841 CrossRef PubMed.
- X. Tan, S. Luo, L. Long, Y. Wang, D. Wang, S. Fang, Q. Ouyang, Y. Su, T. Cheng and C. Shi, Adv. Mater., 2017, 29, 1704196 CrossRef PubMed.
- W. Tang, Z. Yang, S. Wang, Z. Wang, J. Song, G. Yu, W. Fan, Y. Dai, J. Wang, L. Shan, G. Niu, Q. Fan and X. Chen, ACS Nano, 2018, 12, 2610–2622 CrossRef CAS PubMed.
- Y. Wang, X. Huang, Y. Tang, J. Zou, P. Wang, Y. Zhang, W. Si, W. Huang and X. Dong, Chem. Sci., 2018, 9, 8103–8109 RSC.
- L. Alili, M. Sack, A. S. Karakoti, S. Teuber, K. Puschmann, S. M. Hirst, C. M. Reilly, K. Zanger, W. Stahl, S. Das, S. Seal and P. Brenneisen, Biomaterials, 2011, 32, 2918–2929 CrossRef CAS PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Nanoscale, 2011, 3, 1411–1420 RSC.
- F. Chen, W. Bu, S. Zhang, J. Liu, W. Fan, L. Zhou, W. Peng and J. Shi, Adv. Funct. Mater., 2013, 23, 298–307 CrossRef CAS.
- S. Lu, D. Tu, P. Hu, J. Xu, R. Li, M. Wang, Z. Chen, M. Huang and X. Chen, Angew. Chem., Int. Ed. Engl., 2015, 54, 7915–7919 CrossRef CAS PubMed.
- Y.-M. Wang, W. Liu and X.-B. Yin, Chem. Sci., 2017, 8, 3891–3897 RSC.
- J. Xu, F. He, Z. Cheng, R. Lv, Y. Dai, A. Gulzar, B. Liu, H. Bi, D. Yang, S. Gai, P. Yang and J. Lin, Chem. Mater., 2017, 29, 7615–7628 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Dynamic light scattering (DLS) of UCNPs@mSiO2, UCNPs@mCeOx and PEG/UCP; the changes of zeta potentials for nanoparticles obtained for each step; the in vitro detection of ROS with UCNPs@mCeOx; FT-IR spectra of the DOX loaded PEG/UCP-DOX samples; Brunauer–Emmett–Teller (BET) test and the corresponding pore size distribution curves of PEG/UCP and PEG/UCP-DOX samples; the XPS spectra of typical element Ce 2p; decay curves for 1G4–3H6 emission (475 nm) of Tm3+ in UCNPs and UCNPs@mCeOx; the test of O2 production rate and H2O2 decomposition rate catalyzed by UCNPs@mCeOx in the absence of NIR light irradiation; EPR spectra of ˙O2− in the UCNPs@mCeOx aqueous solution; the in vitro UCL imaging of PEG/UCP in pork tissue and a mouse; hemolytic percentage of PEG/UCP to human red blood cells; the corresponding characterization and supplementary data of UCNPs@mSiO2@mTiO2; the confocal laser scanning microscopy (CLSM) images of the degradation of H2O2 and production of O2 at various time periods; ICP-MS of Ce in tumor-bearing mice after injecting PEG/UCP; and H&E stained images of the liver, lung, kidney, heart and spleen achieved from different groups after 14 days of treatment. See DOI: 10.1039/c9sc01615e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |