
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Directed C(sp3)–H arylation of tryptophan: transformation of the directing group into an activated amide†
Lennart
Nicke
,
Philip
Horx
,
Klaus
Harms
 and
Armin
Geyer
*
and
Armin
Geyer
*
Philipps-Universität Marburg, Fachbereich Chemie, Hans Meerwein Straße, 35032 Marburg, Germany. E-mail: geyer@staff.uni-marburg.de
First published on 8th August 2019
Abstract
The 8-aminoquinoline (8AQ) directed C(sp3)–H functionalization was applied in the synthesis of β-arylated tryptophan derivatives. The laborious protecting group reorganization towards α-amino acids compatible for solid phase peptide synthesis (SPPS) was cut short by the transformation of the directing group into an activated amide, which was either used directly in peptide coupling or in the gram scale synthesis of storable Fmoc-protected amino acids for SPPS. In this work, directed C–H activation and nonplanar amide chemistry complement each other for the synthesis of hybrids between phenylalanine and tryptophan with restricted side chain mobility.
Introduction
Unnatural β-branched α-amino acids are promising tools for the synthesis of peptide ligands with conformational constriction in a topologically designed structure.1 The increased hydrophobic surface of β-branched α-amino acids can mediate molecular recognitions processes between bioactive peptides and their target receptors.2 Full control of χ-space3 is achieved in bicyclic peptidomimetics, which strongly influence the properties of the target peptides and proteins.4 The conformational design of open-chain β-branched α-amino acids, however, serves as a valuable platform in peptide ligand design.2 Consequently, the investigation of reliable synthetic routes towards β-branched α-amino acids has experienced a renaissance in the past couple of years.5 In particular, great attention has been payed to the synthesis of β,β-diaryl α-amino acids due to unique hydrophobic interaction patterns6 and promising medicinal applications of therapeutics containing these structural motifs (Fig. 1).7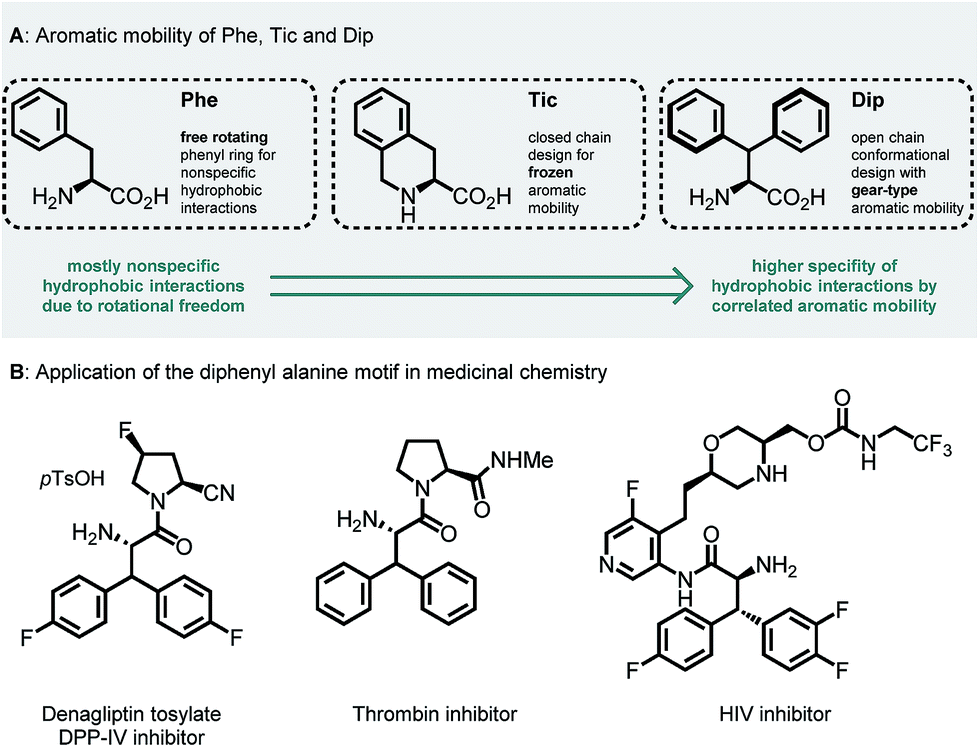 | ||
| Fig. 1 (A) Aromatic mobility and conformational design of Phe, Tic (L-tetrahydroisoquinoline carboxylate) and Dip (L-diphenyl alanine) amino acids.8 (B) Therapeutic applications of the β,β-diaryl alanine motif.7 | ||
In our investigation of peptide-receptor interactions, focusing on the influence of altered indole presentation on biological activity, we were in requirement of a χ1-constricted, conformationally anchored tryptophan building block for standard Fmoc-SPPS. We assumed that β-phenylated tryptophan will be suitable for this purpose. While the synthesis of β,β-diaryl α-amino acids bearing two identical aromatic groups was widely elaborated in earlier studies,9 the synthesis of these amino acids containing different aromatic groups is a more challenging endeavor. The construction of β,β-diaryl α-amino acids with different aryl substituents, ensuring full control over two vicinal stereocenters, has stimulated numerous synthetic studies in the field of asymmetric alkylation/conjugate addition,10 asymmetric hydrogenation11 and directed C–H activation.12 Most of these methods focus on the efficient construction of the carbon skeletons and offer remarkable stereocontrol, but neglect the use of versatile removable directing groups to give adequately protected amino acids for peptide synthesis based on the approach of Fmoc/Boc strategy. Thus, to the best of our knowledge, there is no example of a synthetic peptide bearing a nonsymmetrical β,β-diaryl alanine motif.13,14 Inspired by the seminal work of Corey,12a as well as Daugulis,12b Yu12c,d and Chen,15 we envisioned that diastereomeric β-phenyl tryptophans could be accessed employing sequential C–H activation of alanine. In necessity of a strong, bidentate directing group, we chose 8-aminoquinoline (8AQ). Phth-Ala-8AQ and an N-protected 3-iodoindole could be starting materials in a palladium-catalyzed, directed C–H activation to give access to an enantiomerically enriched tryptophan derivative.15b These compounds have been shown to be competent substrates in β-alkynylation16 and might also undergo arylation. Given the sensitivity of indoles towards oxidative conditions,12d we evaluated a mild method of 8-aminoquinoline cleavage. This effort has proven to be an unnerving challenge17 and the practitioner must choose from protocols only suitable for a limited class of substances. In spite of numerous reactions employing aminoquinoline-directed C–H activation chemistry,18 examples of its use in natural product or functional molecule synthesis are rare.19 The removal of 8-aminoquinoline often demands harsh reaction conditions, which represents a major drawback in synthetic applications.20,12b Studies addressing this concern have been published by Maulide, using an ozonolysis approach,21 Ohshima, using a Nickel-chelate assisted methanolysis22 and Mashima, utilizing epoxide opening for tandem esterification.23 A recent report by Chen et al. utilized IBX in an oxidative protocol for 8AQ cleavage to give various α-amino acids as amides in high yields, however, Trp formed a spiro-fused compound and could not be deprotected accordingly.24 Standard procedures of amide activation using Boc2O to generate a labile imide, show limited applicability, because they only tolerate substrates with low steric hindrance.25 More sterically demanding substrates usually require strain-releasing steps using hazardous TfN3 to achieve amide activation and directing group removal.26 From our point of view, an applicable method for directing group removal has to fulfil the following premises:
- high functional group tolerance
- preservation of stereochemical integrity
- compatibility with standard SPPS protecting groups
- useful yields in large-scale applications
Hence, we were intrigued by the structural similarity of 8-aminoquinoline amides and the widely used Dawson linker for C-terminal diversification of resin-bound peptides.27 A similar amide activation strategy could be feasible for 8-aminoquinoline amides, that opens new possibilities towards C-terminal amino acid modifications (Scheme 1).
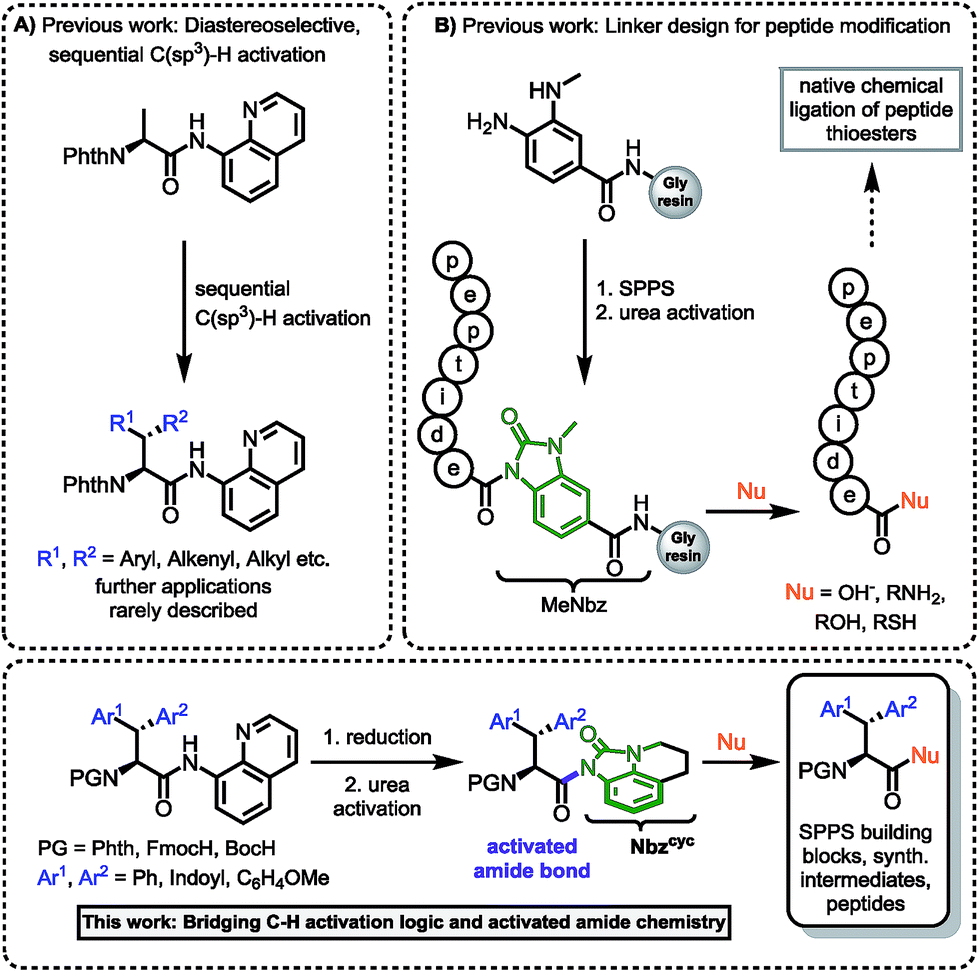 | ||
| Scheme 1 (A) Sequential C(sp3)–H activation.12a,b,13 (B) Linker design for C-terminal peptide modification.27 This work: Rationale of amidic weakening strategy applied for α-amino acids prepared by directed C–H activation. | ||
Results and discussion
To prove this concept, we subjected the common starting material Phth-Phe-8AQ to hydrogenation conditions to generate a 1,2,3,4-tetrahydroquinoline species. 5 mol% PtO2-catalyzed hydrogenation in DCM/AcOH gave rise to reduction product 1 in irreproducible yields with concomitant formation of non-identifiable byproducts (Scheme 2).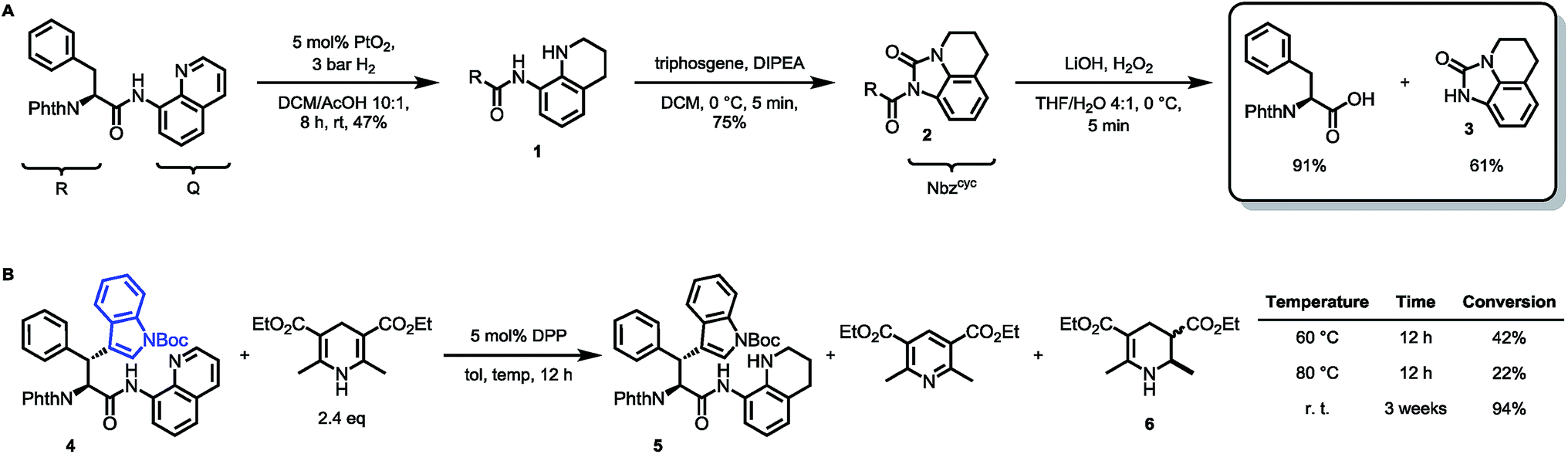 | ||
| Scheme 2 (A) Validation of 8-aminoquinoline cleavage by amidic weakening strategy. (B) Reaction outcome using Rueping's conditions. | ||
However, enough starting material was generated to perform a triphosgene amide activation, which has previously been used in custom-made directing group removal.28 Treatment of tetrahydroquinoline 1 with triphosgene in DCM in the presence of Hünig's base provided the acylurea 2 in 75% yield. In analogy to Dawson's linker (MeNbz: N-acyl benzimidazolinone) for C-terminal peptide modification, we suggest Nbzcyc (activated urea) as abbreviation to indicate a similar, cyclic (index: cyc) arrangement. We were pleased to find that exposure of the urea compound 2 to standard LiOH/H2O2 hydrolysis conditions gave a clean formation of Phth-Phe-OH without opening of the phthalimide protecting group or decarbonylation of the Nbzcyc group. As a bonus, simple acid/base extraction was enough to separate the pure free carboxylic acid from the cleaved urea 3. With this valid concept in hand, we started to develop an improved, chemoselective and high yielding procedure for quinoline reduction under conditions described by Rueping for the reduction of simpler quinolines using (chiral) phosphoric acids and Hantzsch ester as reducing agents.29 This hydrogenation method shows a high functional group tolerance which was displayed by the successful use of peptide catalysts for stereoinduction on substituted 8-aminoquinolines.30 Preliminary trials of reducing the 8-aminoquinoline moiety of Phth-Wrf(Boc)-8AQ314 (synthesis vide infra) with 2.4 eq. Hantzsch's ethyl ester (HEEt) in the presence of 5 mol% diphenylphosphate (DPP) in toluene at 60 °C for 12 h gave 42% conversion of the starting material to the corresponding transfer hydrogenation product. Encouraged by these results, we supposed that raising the reaction temperature might lead to higher conversion. Contradictory to our hypothesis, executing this reaction at 80 °C gave only 22% conversion of the starting material after full consumption of HEEt, whereas room temperature led to 94% conversion of the starting material after three weeks. With these surprising results in hand, we took an in-depth look in the reaction products, suspecting non-productive pathways to be present, resulting in consumption of reducing agent. Indeed, high reaction temperatures favoured the progression of a Hantzsch ester disproportionation reaction, giving rise to a diastereomeric mixture of tetrahydropyridine 6 (compare scheme 2B) and the oxidized pyridine derivative, respectively. This side reaction was the underlying cause of rapid consumption of reducing agent, which was not observed in the reactions investigated by Rueping.32 We hypothesized, that the formation of the tetrahydropyridine is facilitated by the strong acid DPP, acting in a C-protonation step,33 followed by iminium reduction with another molecule of Hantzsch ester.34 This intriguing alternative reaction pathway prompted us to carry out a competition reaction between quinaldine, a commonly used substrate in Hantzsch ester mediated transfer hydrogenation, and Phth-Phe-8AQ (Scheme 3).
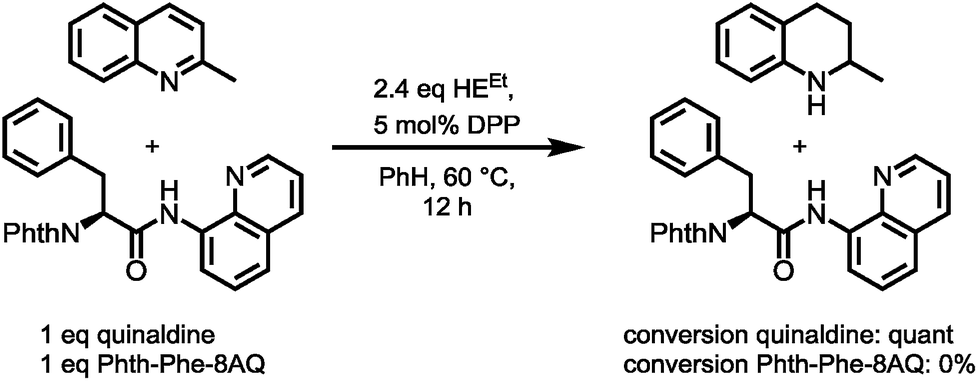 | ||
| Scheme 3 Competition experiment between quinaldine and Phth-Phe-8AQ. | ||
Only quinaldine was reduced in this competition experiment, so that 8-aminoquinoline amides were identified as a substrate with significantly lower reactivity. We reasoned that this finding is due to (1) the lack of beneficial effects of the methyl group participating in the stabilization of cationic intermediates and (2) the electron-donating properties of the amide nitrogen atom. We assumed that these electron-donating effects could potentially be lowered using oxygenophilic Lewis acids. Furthermore, the use of weaker Brønsted acids might decrease the Hantzsch ester's tendency to undergo disproportionation. The results of the reaction screening are surveyed in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Lewis acid | Brønsted acid | Temp. | Time | Conversion (NMR) |
| a no Hantzsch ester was used. b 0.25 eq. was used. c 2.4 eq. was used. n. r. = no reaction. | |||||
| 1 | — | DPP | 60 °C | 12 h | 18% |
| 2 | LiBr | DPP | 60 °C | 12 h | 78% |
| 3 | LiBr | HCOOH | 60 °C | 12 h | 86% |
| 4 | — | HCOOH | 60 °C | 24 h | n. r. |
| 5 | LiBr | — | 60 °C | 24 h | n. r. |
| 6a | LiBr | HCOOH | 60 °C | 24 h | n. r. |
| 7 | LiCl | HCOOH | 60 °C | 12 h | 85% |
| 8 | LiI | HCOOH | 60 °C | 12 h | 72% |
| 9 | LiBrb | HCOOHb | 60 °C | 12 h | 27% |
| 10 | LiBrc | HCOOHb | 60 °C | 12 h | 85% |
| 11 | LiBrc | HCOOH | 60 °C | 12 h | 90% |
| 12 | LiBr | HCOOH | 40 °C | 16 h | 99% |
| 13 | LiBr | HOAc | 40 °C | 16 h | 27% |
As shown in Table 1, the use of LiBr dramatically increases the conversion (entry 1 and 2). The use of weaker Brønsted acid HCOOH increases the conversion up to 86% (entry 3), but is not capable of catalyzing the reaction without Lewis acid (entry 4). As expected, using Lewis acid alone or omitting the Hantzsch ester shuts down the reaction completely (entry 5 and 6). Using LiCl or LiI did not result in an improvement (entry 7 and 8). Optimal results were obtained when 2.4 eq. HEtBu were used35 in THF in the presence of 1.0 eq. of lithium bromide and formic acid, respectively, at 40 °C.
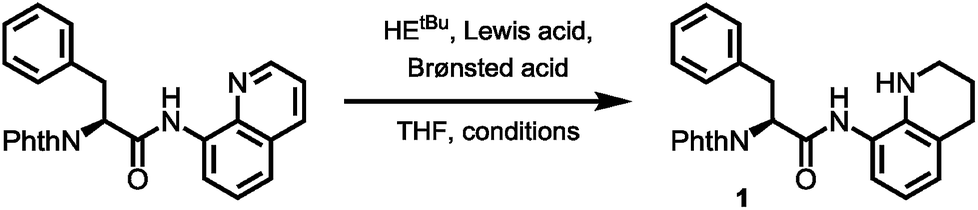
With optimized conditions for 8AQ reduction in hand, the arylation of alanine to generate a suitably protected tryptophan was undertaken. Owing to the high steric demand of N-protected 3-iodoindoles, C–H arylation was deemed challenging. However, after screening of silver additives (AgTFA, AgOAc, AgBF4, Ag2CO3 and Ag2CO3/(BnO)2PO2H), we found that AgTFA gave a clean indoylation at 60 °C using 10 mol% Pd(OAc)2 in tert-amyl alcohol to provide 83% of the desired tryptophan 7 with complete retention of the α-stereocenter (99% ee). The methylene arylation proceeded in a substrate-controlled, highly diastereoselective fashion (dr > 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and good yields (71%). The presence of an Nα-phthalimide protecting group significantly increased α-epimerization in subsequent reactions. Generally, the manageability of the large-scale synthesis was improved, when the phthalimide protecting group was removed directly after C–H arylation. The phthalimide deprotection was accomplished using excess ethylenediamine to give the free amine in 82% yield. Treatment of the amine with FmocCl resulted in the formation of the Fmoc carbamate 9 in nearly quantitative yield. The following reduction of the quinoline directing group was accomplished using our optimized reaction conditions gave 66% of the tetrahydroquinoline, which was treated with triphosgene to generate the acylurea 10 in 63% yield. With the activated amide in hand, LiOH/H2O2 mediated hydrolysis was carried out. We were rewarded with the clean amide hydrolysis needing as little as five minutes to give 96% of the target SPPS building block 11 without noteworthy cleavage of the Fmoc protecting group. Remarkably, all the reaction steps of the sequence shown in Scheme 4 could be conducted in gram scale, so that in the end, 1.10 g (1.83 mmol) of the building block was obtained as a white solid, easily storable and ready to use for peptide synthesis.
:1) and good yields (71%). The presence of an Nα-phthalimide protecting group significantly increased α-epimerization in subsequent reactions. Generally, the manageability of the large-scale synthesis was improved, when the phthalimide protecting group was removed directly after C–H arylation. The phthalimide deprotection was accomplished using excess ethylenediamine to give the free amine in 82% yield. Treatment of the amine with FmocCl resulted in the formation of the Fmoc carbamate 9 in nearly quantitative yield. The following reduction of the quinoline directing group was accomplished using our optimized reaction conditions gave 66% of the tetrahydroquinoline, which was treated with triphosgene to generate the acylurea 10 in 63% yield. With the activated amide in hand, LiOH/H2O2 mediated hydrolysis was carried out. We were rewarded with the clean amide hydrolysis needing as little as five minutes to give 96% of the target SPPS building block 11 without noteworthy cleavage of the Fmoc protecting group. Remarkably, all the reaction steps of the sequence shown in Scheme 4 could be conducted in gram scale, so that in the end, 1.10 g (1.83 mmol) of the building block was obtained as a white solid, easily storable and ready to use for peptide synthesis.
 | ||
| Scheme 4 Building block synthesis of Fmoc-Wsf(Boc)-OH (11). | ||
For the synthesis of the β-diastereomer, it was necessary to reverse the order of introduction of the aromatic residues,15b which was achieved by starting from phenylalanine. The β-indoylation of Phth-Phe-8AQ was expected to be a challenging step, because the aryl halide decomposes at elevated temperature. However, conducting the reaction at temperatures not higher than 80 °C favoured the C–H arylation pathway. At these temperatures, the reaction rate is only moderate and was elevated using as much as five equivalents of the halide. Luckily, the majority of unconsumed halide could be reisolated (98%) and used in further reactions without any loss of efficiency. More importantly, the arylated amino acid 4 could be isolated in 65% yield, again with excellent diastereoselectivity (dr > 25:1) and no loss in enantiomeric excess (97% ee). The next steps were conducted in the same manner as for diastereomer 11 shown in Scheme 4 giving yields in comparable ranges. Notably, for this respective diastereomer, the tetrahydroquinoline product was isolated by filtration directly from the reaction mixture in 72% yield. Again, the final hydrolysis step yielded excellent 97% of the SPPS building block Fmoc Wrf(Boc) OH (12) (Scheme 5).
 | ||
| Scheme 5 Key arylation and completion of Fmoc-Wrf(Boc)-OH (12) synthesis. | ||
With both building block syntheses completed, we were interested in elucidating the origin of amidic weakening by our developed method. In a rapidly growing field of cross-coupling36 and transamidation chemistry25a,37 using activated, nonplanar amides as valuable synthons, we suspected the Nbzcyc to exhibit comparable features. Fortunately, we were able to crystallize a racemic sample of Phth-Wrf(Boc)-8AQ (rac-4) and D-configured Phth-ala-Nbzcyc (13). X-ray analysis of these single crystals (Fig. 2) revealed distinct Winkler–Dunitz parameters38 regarding an increased amide twist τ of the activated urea (τ = 6.9°) compared to the amino-quinoline amide (τ = 3.3°). Nitrogen pyramidalization (χN) was found to rise to 11.7° in the activated urea form, whereas χN was found to be 2.3° for the 8AQ amide rac-4. Furthermore, the respective C–N bond length between the carbon atom of the carbonyl and the nitrogen atom of the directing group was enlarged from 1.35 Å to 1.40 Å in the activated urea form, resulting in a lowered efficiency of amidic resonance.
 | ||
| Fig. 2 The crystal structures of rac-4 and Phth-ala-Nbzcyc (13) show distinct differences concerning amide twist, C–N bond length and nitrogen pyramidalization, explaining the easy removal of Nbzcyc. | ||
To gain a profound insight in the induced amidic weakening, theoretical investigations were applied. Szostak et al. showed that the resonance energy (ER) of amides correlates to their reactivity in nucleophilic substitution reactions.37,39 We therefore calculated the resonance energy for 8AQ amide rac-4 and urea 13, using crystal structures (Fig. 2) as starting points for calculations. Following the established COSNAR method,40 which is readily applied for nonplanar amides,41 a decrease of 9.6 kcal mol−1 in comparison to the parent aminoquinoline was observed. This decline in resonance energy correlates well to the increase in reactivity towards nucleophilic substitution. Additionally, the bond length from C–N increases while the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond contracts, further supporting the weakening in amidicity.
O bond contracts, further supporting the weakening in amidicity.
The Winkler–Dunitz distorsion parameters of the calculated structures are compared to the X-ray structures in Table 2.
| Parameter | rac-4 (X-ray) | rac-4 (Calcd) | Urea 13 (X-ray) | Urea 13 (Calcd) |
|---|---|---|---|---|
| a Relative to dimethyl acetamide. | ||||
| τ [°] | 3.3 | 3.0 | 6.9 | 8.4 |
| χN [°] | 2.3 | 0.3 | 11.7 | 6.3 |
| χC [°] | 3.1 | 3.4 | 1.7 | 0.9 |
| τ + χN [°] | 5.6 | 3.3 | 18.6 | 14.7 |
| CO [Å] |
1.22 | 1.22 | 1.21 | 1.21 |
| C–N [Å] | 1.35 | 1.37 | 1.40 | 1.41 |
| E R [kcal mol−1] | — | 16.6 | — | 7.0 |
| Amidicitya | — | 91% | — | 38% |
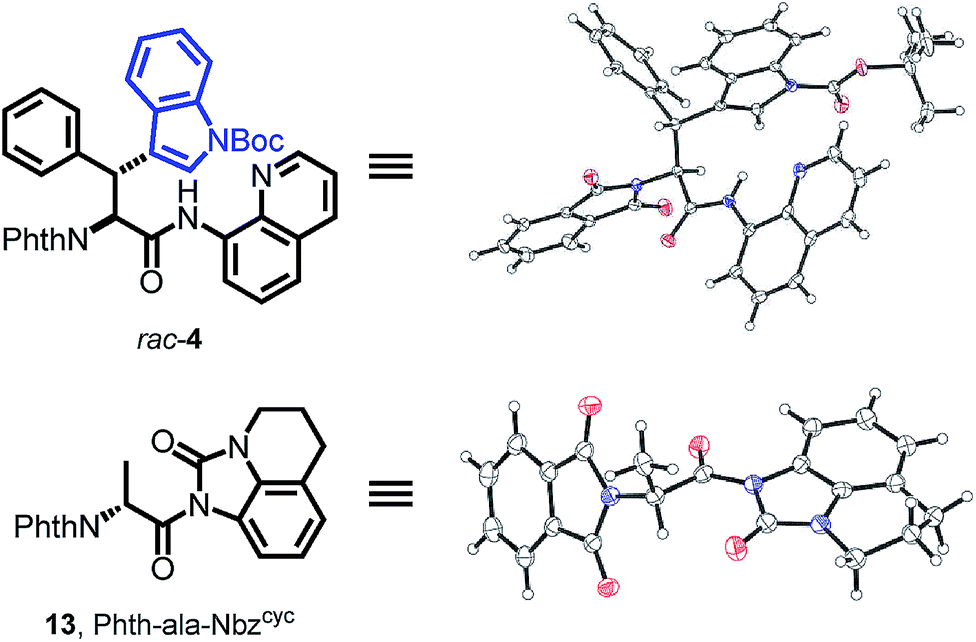
To prove the overall applicability of the concept, we synthesized the chimeric amino acid Boc-Wsy(Boc,Me)-Nbzcyc (15) and probed the urea cleavage using nucleophiles for common synthetic precursors. Considering the utility of Dawson's linker,27b,c Nbzcyc ureas could provide rapid access to several carboxylic acid derivatives. The esterification of activated amide 15 using K2CO3 in MeOH led to a loss of stereointegrity at the acidic α-position (dr 4:1), making a milder approach using Hünig's base imperative to give 90% of the methyl ester 16 with no detectable erosion42 of the α-stereocenter. Next, we examined the potential of the activated amide 15 to function in an active ester fashion for coupling of dipeptides. The formation of a dipeptide 17 was achieved using glycine methyl ester as the nucleophile in DMF with the aid of Hünig's base at 50 °C for 24 h, which was isolated in 82% yield. Finally, treatment of the activated amide with NaBH4 resulted in the clean formation of the protected amino alcohol 18 in 91% yield (Scheme 6).
 | ||
| Scheme 6 Diverse synthetic applications. | ||
Based on the results of dipeptide formation, that proved the ureas capability of acylating primary amines, we performed the acylation of a resin-bound Leu-enkephalin precursor 19. Treatment of the resin-bound peptide with Boc-Wsy(Boc,Me)-Nbzcyc led to the formation of the Leu-enkephalin derivate 20 (Scheme 7A). Furthermore, Fmoc-Wrf(Boc)-OH and Fmoc-Wsf(Boc)-OH were used as a tryptophan substitute to replace the buried Trp6 residue in standard automated SPPS to generate two modified 20mer Trp cage (TC5b) mutants 21 and 22 (Scheme 7B).43
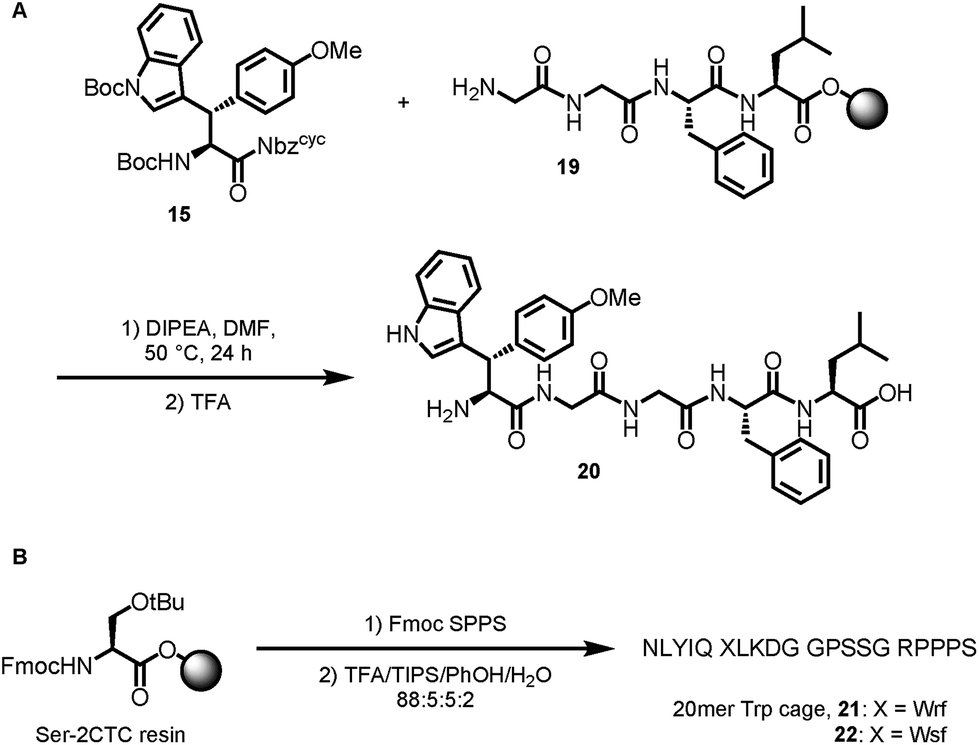 | ||
| Scheme 7 Applications in peptide synthesis. | ||
The Trp cage miniprotein was chosen because the side chain rotamer of the buried Trp6 indole group is similar to Wrf. Therefore, we hypothesized that the incorporation of Wrf into TC5b would lead to less disturbance of the densely arranged cage fold. The indole presentation in Wsf, however, should destabilize the cage fold, because the antiperiplanar orientation between Cα–Cβ would force the phenyl ring to occupy the indole cavity in the Trp cage motif. Both diastereomeric peptides 21 and 22 were studied by NMR spectroscopy and compared to the native Trp cage miniprotein TC5b. The chemical shift deviations of key residues for cage fold indication44 were used to determine the folded fraction of the two modified miniproteins (Fig. 3).
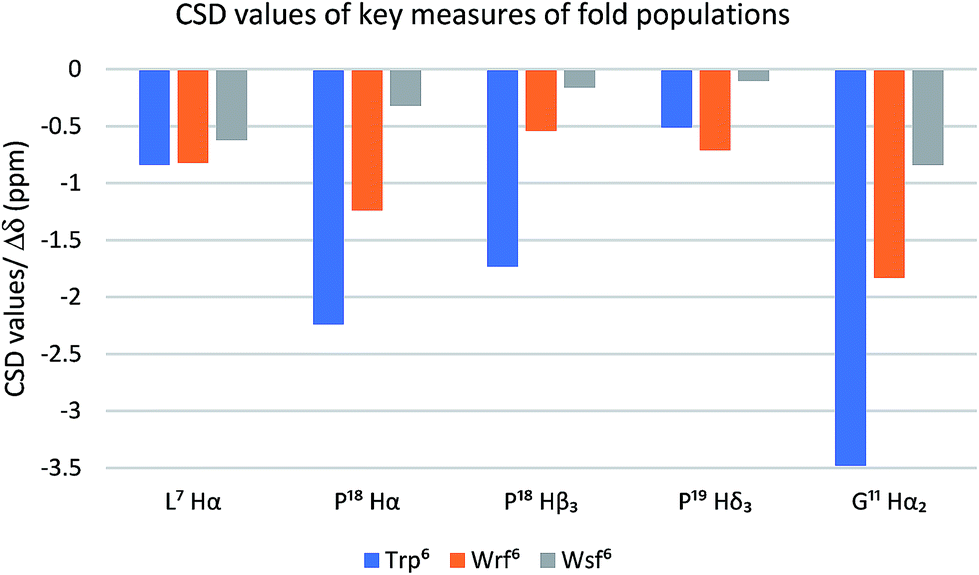 | ||
| Fig. 3 The Trp cage CSD values (blue bars) serve as a quantitative folding reference for the two diastereomeric peptides containing Wrf (orange bars) and Wrf (grey bars). | ||
NOE patterns suggest that the N-terminal α-helical environment is retained for both diastereomers of the peptide. However, according to the CSD values only the Wrf6 cage mutant 21 assumes a folded state (75%), whereas the Wsf6 mutant 22 shows a Trp cage folding population of only 28%. The strong influence of the restricted side chain mobility and the preferred orientation of the indole moiety on peptide and protein folding is highlighted by these two examples.
Conclusion
In conclusion, we showed that the close-to-inert amide bond of the widely used directing group 8-aminoquinoline for C–H activation could be addressed via amidic weakening through a two-step synthetic procedure, including a highly chemo-selective Hantzsch ester mediated reduction of the pyridyl moiety, followed by urea cyclization. These findings paved the way to the gram-scale synthesis of highly sterically congested β-branched α-amino acids in an orthogonally protected fashion for solid phase peptide synthesis, bridging the rapidly growing fields of directed C–H activation and nonplanar amide chemistry. The altered amide geometry was quantified through X-ray and computational analysis of respective 8-aminoquinoline amides and derived urea compounds to give a better understanding of the induced reactivity in nucleophilic substitution and addition reactions. Ultimately, the generated chimeric amino acids Wrf, Wsf and Wsy were used in solid phase peptide synthesis to give derivatives of Leu-enkephalin and the miniprotein Trp cage, posing the first example of incorporating the optically pure, nonsymmetrical β,β-diaryl alanine motif in peptidic environments. Further investigations on the influence of these amino acids concering the folding and biological activity of peptides are currently under way in our laboratory. We hope that this work will stimulate the use of unsymmetric β-branched α-amino acids to access novel designs of peptide ligands with tailor-made biological features.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Paultheo von Zezschwitz for access to chiral HPLC equipment. Furthermore, we thank Magnus Rueping, Erli Sugiono and Christian Heinrich Müller for helpful discussion.Notes and references
- V. J. Hruby, F. Al-Obeidi and W. Kazmierski, Biochem. J., 1990, 268, 249–262 CrossRef CAS PubMed.
- V. J. Hruby, G. Li, C. Haskell-Luevano and M. Shenderovich, Biopolymers, 1997, 43, 219–266 CrossRef CAS PubMed.
- S. E. Gibson, N. Guillo and M. J. Tozer, Tetrahedron, 1999, 55, 585–615 CrossRef CAS.
- (a) P. Tremmel and A. Geyer, Angew. Chem., Int. Ed., 2004, 43, 5789–5791 CrossRef CAS PubMed; (b) B. Eckhardt, W. Grosse, L.-O. Essen and A. Geyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18336–18341 CrossRef CAS PubMed; (c) C. Priem, A. Wuttke, M. Berditsch, A. S. Ulrich and A. Geyer, J. Org. Chem., 2017, 82, 12366–12376 CrossRef CAS PubMed.
- J. Michaux, G. Niel and J.-M. Campagne, Chem. Soc. Rev., 2009, 38, 2093–2116 RSC.
- (a) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC; (b) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- (a) D. E. Patterson, J. D. Powers, M. LeBlanc, T. Sharkey, E. Boehler, E. Irdam and M. H. Osterhout, Org. Process Res. Dev., 2009, 13, 900–906 CrossRef CAS; (b) M. Nilsson, M. Hämäläinen, M. Ivarsson, J. Gottfries, Y. Xue, S. Hansson, R. Isaksson and T. Fex, J. Med. Chem., 2009, 52, 2708–2715 CrossRef CAS PubMed; (c) B. Baum, L. Muley, A. Heine, M. Smolinski, D. Hangauer and G. Klebe, J. Mol. Biol., 2009, 391, 552–564 CrossRef CAS PubMed; (d) J. A. McCauley, S. Crane, C. Beaulieu, D. J. Bennett, C. J. Bungard, R. K. Chang, T. J. Greshock, L. Hao, K. Holloway, J. J. Manikowski, D. Mckay, C. Molinaro, O. M. Moradei, P. G. Nantermet, C. Nadeau, S. Tummanapalli, W. Shipe, S. K. Singh, V. L. Truong, V. P. Sivalenka, D. C. Williams and C. M. Wiscount, WO 2014043019 A1, 2014.
- R. W. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 2054–2070 CrossRef PubMed.
- Selected examples: (a) H. G. Chen, V. G. Beylin, M. Marlatt, B. Leja and O. P. Goel, Tetrahedron Lett., 1992, 33, 3293–3296 CrossRef CAS; (b) M. P. Sibi, P. K. Deshpande, A. J. La Loggia and J. W. Christense, Tetrahedron Lett., 1995, 36, 8961–8964 CrossRef CAS; (c) D. E. Patterson, S. Xie, L. A. Jones, M. H. Osterhout, C. G. Henry and T. D. Roper, Org. Process Res. Dev., 2007, 11, 624–627 CrossRef CAS; (d) R. B. Appell, L. T. Boulton, E. D. Daugs, M. Hansen, C. H. Hanson, J. Heinrich, C. Kronig, R. C. Lloyd, D. Louks, M. Nitz, C. Praquin, J. A. Ramsden, H. Samuel, M. Smit and M. Willets, Org. Process Res. Dev., 2013, 17, 69–76 CrossRef CAS.
- (a) B.-H. Zheng, C.-H. Ding, X.-L. Hou and L.-X. Dai, Org. Lett., 2010, 12, 1688–1691 CrossRef CAS PubMed; (b) J. Wang, S. Zhou, D. Lin, X. Ding, H. Jian and H. Liu, Chem. Commun., 2011, 47, 8355–8357 RSC; (c) F.-S. He, J.-H. Jin, Z.-T. Yang, X. Yu, J. S. Fossey and W.-P. Deng, ACS Catal., 2016, 6, 652–656 CrossRef CAS; (d) X.-Z. Zhang, Y.-H. Deng, X. Yan, K.-Y. Yu, F.-X. Wang, X.-Y. Ma and C.-A. Fan, J. Org. Chem., 2016, 81, 5655–5662 CrossRef CAS PubMed.
- C. Molinaro, J. P. Scott, M. Shevlin, C. Wise, A. Ménard, A. Gibb, E. M. Junker and D. Lieberman, J. Am. Chem. Soc., 2015, 137, 999–1006 CrossRef CAS PubMed.
- (a) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS PubMed; (b) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188–5191 CrossRef CAS PubMed; (c) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed; (d) G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338–3351 CrossRef CAS PubMed.
- Examples of the use of Dip in peptides: (a) K.-H. Hsieh, T. R. LaHann and R. C. Speth, J. Med. Chem., 1989, 32, 898–903 CrossRef CAS PubMed; (b) J. R. Holder, R. M. Bauzo, Z. Xiang and C. Haskell-Luevano, J. Med. Chem., 2002, 45, 3073–3081 CrossRef CAS PubMed; (c) A. Kwiatkowska, D. Sobolewski, A. Prahl, L. Borovikova, J. Slaninova and B. Lammek, Eur. J. Med. Chem., 2009, 44, 2862–2867 CrossRef CAS PubMed; (d) G. E. Flaten, G. Kottra, W. Stensen, G. Isaksen, R. Karstad, J. S. Svendsen, H. Daniel and J. Svenson, J. Med. Chem., 2011, 54, 2422–2432 CrossRef CAS PubMed; (e) T. Doi, T. Muraoka, T. Ohshiro, D. Matsuda, M. Yoshida, T. Takahashi, S. Omura and H. Tomoda, Bioorg. Med. Chem. Lett., 2012, 22, 696–699 CrossRef CAS PubMed; (f) Y. Itoh, M. Ishikawa, R. Kitaguchi, K. Okuhira, M. Naito and Y. Hashimoto, Bioorg. Med. Chem. Lett., 2012, 22, 4453–4457 CrossRef CAS PubMed; (g) Y. C. Koay, N. L. Richardson, S. S. Zaiter, J. Kho, S. Y. Nguyen, D. H. Tran, K. W. Lee, L. K. Buckton and S. R. McAlpine, ChemMedChem, 2016, 11, 881–892 CrossRef CAS PubMed; (h) B. E. Haug, K. A. Camilio, L. T. Eliassen, W. Stensen, J. S. Svendsen, K. Berg, B. Mortensen, G. Serin, J.-F. Mirjolet, F. Bichat and Ø. Rekdal, J. Med. Chem., 2016, 59, 2918–2927 CrossRef CAS PubMed.
- Examples of β-methylated Trp in peptides: (a) Z. Huang, Y.-B. He, K. Raynor, M. Tallent, T. Reisine and M. Goodman, J. Am. Chem. Soc., 1992, 114, 9390–9401 CrossRef CAS; (b) C. Haskell-Luevano, L. W. Boteju, H. Miwa, C. Dickinson, I. Gantz, T. Yamada, M. E. Hadley and V. J. Hruby, J. Med. Chem., 1995, 38, 4720–4729 CrossRef CAS PubMed; (c) C. Haskell-Luevano, K. Toth, L. Boteju, C. Job, A. M. de L. Castrucci, M. E. Hadley and V. J. Hruby, J. Med. Chem., 1997, 40, 2740–2749 CrossRef CAS PubMed; (d) G. G. Bonner, P. Davis, D. Stropova, S. Edsall, H. I. Yamamura, F. Porreca and V. J. Hruby, J. Med. Chem., 2000, 43, 569–580 CrossRef CAS PubMed.
- (a) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed; (b) B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Chem. Sci., 2014, 5, 3952–3957 RSC; (c) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635–645 CrossRef CAS PubMed.
- B. Wang, G. He and G. Chen, Sci. China: Chem., 2015, 58, 1345–1348 CrossRef CAS.
- (a) C. P. Ting and T. J. Maimone, Angew. Chem., Int. Ed., 2014, 53, 3115–3119 CrossRef CAS PubMed; (b) D. P. Affron, O. A. Davis and J. A. Bull, Org. Lett., 2014, 16, 4952–4959 CrossRef PubMed.
- O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed.
- (a) T. Kinsinger and U. Kazmaier, Org. Lett., 2018, 20, 7726–7730 CrossRef CAS PubMed; (b) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507–7510 CrossRef CAS PubMed; (c) W. R. Gutekunst and P. S. Baran, J. Org. Chem., 2014, 79, 2430–2452 CrossRef CAS PubMed; (d) L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS PubMed; (e) L. M. Chapman, J. C. Beck, C. R. Lacker, L. Wu and S. E. Resiman, J. Org. Chem., 2018, 83, 6066–6085 CrossRef CAS PubMed; (f) J. C. Beck, C. R. Lacker, L. M. Chapman and S. E. Reisman, Chem. Sci., 2019, 10, 2315–2319 RSC.
- Selected examples: (a) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965–3972 CrossRef CAS PubMed; (b) A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maitia, Adv. Synth. Catal., 2017, 359, 1301–1307 CrossRef CAS; (c) P. Hu and T. Bach, Synlett, 2015, 262, 2853–2857 Search PubMed; (d) B. Gopalakrishnan, S. A. Babu and R. Padmavathi, Tetrahedron, 2015, 71, 8333–8349 CrossRef CAS.
- M. Berger, R. Chauhan, C. A. B. Rodrigues and N. Maulide, Chem.–Eur. J., 2016, 22, 16805–16808 CrossRef CAS PubMed.
- T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157–3161 CrossRef CAS.
- Y. Nishii, T. Hirai, S. Fernandez, P. Knochel and K. Mashima, Eur. J. Org. Chem., 2017, 34, 5010–5014 CrossRef.
- Z. Zhang, X. Li, M. Song, Y. Wan, D. Zheng, G. Zhang and G. Chen, J. Org. Chem., 2019 DOI:10.1021/acs.joc.9b01362.
- (a) O. Verho, M. P. Lati and M. Oschmann, J. Org. Chem., 2018, 83, 4464–4476 CrossRef CAS PubMed; (b) B. Wang, Y. Liu, R. Jiao, Y. Feng, Q. Li, C. Chen, L. Liu, G. He and G. Chen, J. Am. Chem. Soc., 2016, 138, 3926–3932 CrossRef CAS PubMed; (c) B. Mondal, B. Roy and U. Kazmaier, J. Org. Chem., 2016, 81, 11646–11655 CrossRef CAS PubMed; (d) Q. Yang and S.-D. Yang, ACS Catal., 2017, 7, 5220–5224 CrossRef CAS; (e) D. Antermite, D. P. Affron and J. A. Bull, Org. Lett., 2018, 20, 3948–3952 CrossRef CAS PubMed.
- (a) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef CAS PubMed; (b) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed.
- (a) J. B. Blanco-Canosa, B. Nardone, F. Albericio and P. E. Dawson, J. Am. Chem. Soc., 2015, 137, 7197–7209 CrossRef CAS PubMed; (b) C. A. Arbour, T. D. Kondasinghe, H. Y. Saraha, T. L. Vorlicek and J. L. Stockdill, Chem. Sci., 2018, 9, 350–355 RSC; (c) C. A. Arbour, R. E. Stamatin and J. L. Stockdill, J. Org. Chem., 2018, 83, 1797–1803 CrossRef CAS PubMed.
- G. He, S.-Y. Zhang, W. A. Nack, R. Pearson, J. Rabb-Lynch and G. Chen, Org. Lett., 2014, 16, 6488–6491 CrossRef CAS PubMed.
- (a) M. Rueping, T. Theissmann and A. P. Antonchick, Synlett, 2006, 7, 1071–1074 CrossRef; (b) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683–3686 CrossRef CAS PubMed.
- C. R. Shugrue and S. J. Miller, Angew. Chem., Int. Ed., 2015, 54, 11173–11176 CrossRef CAS PubMed.
- Naming of the SPPS building block: Fmoc-Wsf(Boc)-OH; W is the single letter code for tryptophan, having higher priority than the second side chain of phenylalanine (f). The middle letter is displaying the configuration (R or S) of the side chain.
- Personal communication with E. Sugiono and M. Rueping.
- G. Barbe and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 18–19 CrossRef CAS PubMed.
- M. Rueping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 3781–3783 CrossRef CAS PubMed.
- For large scale preparations, 2.6 eq. HE was used. The ester residue was chosen varying on the Rf value of the substrate to give easier chromatographic separations.
- (a) L. Hie, N. F. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed; (b) G. Meng and M. Szostak, Eur. J. Org. Chem., 2018, 20, 2352–2365 CrossRef.
- (a) E. L. Baker, M. M. Yamano, Y. Zhou, S. M. Anthony and N. K. Garg, Nat. Commun., 2016, 7, 11554 CrossRef PubMed; (b) Y. Liu, S. Shi, M. Achtenhagen, R. Liu and M. Szostak, Org. Lett., 2017, 19, 1614–1617 CrossRef CAS PubMed.
- F. K. Winkler and J. D. Dunitz, J. Mol. Biol., 1971, 59, 169–182 CrossRef CAS PubMed.
- V. Pace, W. Holzer, G. Meng, S. Shi, R. Lalancette, R. Szostak and M. Szostak, Chem.–Eur. J., 2016, 22, 14494–14498 CrossRef CAS PubMed.
- (a) A. Greenberg and C. A. Venanzi, J. Am. Chem. Soc., 1993, 115, 6951–6957 CrossRef CAS; (b) A. Greenberg, D. T. Moore and T. D. DuBois, J. Am. Chem. Soc., 1996, 118, 8658–8668 CrossRef CAS.
- S. A. Glover and A. A. Rosser, J. Org. Chem., 2012, 77, 5492–5502 CrossRef CAS PubMed.
- Using 1H-NMR of the crude product.
- (a) J. W. Neidigh, R. M. Fesinmeyer and N. H. Andersen, Nat. Struct. Biol., 2002, 9, 425–430 CrossRef CAS PubMed; (b) B. Barua, J. C. Lin, V. D. Williams, P. Kummler, J. W. Neidigh and N. H. Andersen, Protein Eng., Des. Sel., 2008, 21, 171–185 CrossRef CAS PubMed.
- K. A. Graham, A. Byrne, R. Son and N. H. Andersen, Biopolymers, 2019, 110, e23260 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1903900 and 1903901. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03440d |
| This journal is © The Royal Society of Chemistry 2019 |