
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
s-Block cooperative catalysis: alkali metal magnesiate-catalysed cyclisation of alkynols†
Michael
Fairley
a,
Laia
Davin
a,
Alberto
Hernán-Gómez
a,
Joaquín
García-Álvarez
 *b,
Charles T.
O'Hara
*a and
Eva
Hevia
*a
*b,
Charles T.
O'Hara
*a and
Eva
Hevia
*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: charlie.ohara@strath.ac.uk
bDepartamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, E-33071 Oviedo, Spain
First published on 14th May 2019
Abstract
Mixed s-block metal organometallic reagents have been successfully utilised in the catalytic intramolecular hydroalkoxylation of alkynols. This success has been attributed to the unique manner in which these reagents can overcome the challenges of the reaction: namely OH activation and coordination to and then addition across a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond. In order to optimise the reaction conditions and to garner vital catalytic system requirements, a series of alkali metal magnesiates were enlisted for the catalytic intramolecular hydroalkoxylation of 4-pentynol. In a prelude to the main investigation, the homometallic magnesium dialkyl reagent MgR2 (where R = CH2SiMe3) was utilised. This reagent was unsuccessful in cyclising the alcohol into 2-methylenetetrahydrofuran 2a or 5-methyl-2,3-dihydrofuran 2b, even in the presence of multidentate Lewis donor molecules such as N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA). Alkali metal magnesiates MIMgR3 (when MI = Li, Na or K) performed the cyclisation unsatisfactorily both in the absence/presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA) or PMDETA. When higher-order magnesiates (i.e., MI2MgR4) were employed, in general a marked increase in yield was observed for MI = Na or K; however, the reactions were still sluggish with long reaction times (22–36 h). A major improvement in the catalytic activity of the magnesiates was observed when the crown ether molecule 15-crown-5 was combined with sodium magnesiate Na2MgR4(TMEDA)2 furnishing yields of 87% with 2a
C bond. In order to optimise the reaction conditions and to garner vital catalytic system requirements, a series of alkali metal magnesiates were enlisted for the catalytic intramolecular hydroalkoxylation of 4-pentynol. In a prelude to the main investigation, the homometallic magnesium dialkyl reagent MgR2 (where R = CH2SiMe3) was utilised. This reagent was unsuccessful in cyclising the alcohol into 2-methylenetetrahydrofuran 2a or 5-methyl-2,3-dihydrofuran 2b, even in the presence of multidentate Lewis donor molecules such as N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA). Alkali metal magnesiates MIMgR3 (when MI = Li, Na or K) performed the cyclisation unsatisfactorily both in the absence/presence of N,N,N′,N′-tetramethylethylenediamine (TMEDA) or PMDETA. When higher-order magnesiates (i.e., MI2MgR4) were employed, in general a marked increase in yield was observed for MI = Na or K; however, the reactions were still sluggish with long reaction times (22–36 h). A major improvement in the catalytic activity of the magnesiates was observed when the crown ether molecule 15-crown-5 was combined with sodium magnesiate Na2MgR4(TMEDA)2 furnishing yields of 87% with 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2b ratios of 95:5 after 5 h. Similar high yields of 88% with 2a:2b ratios of 90:10 after 3 h were obtained combining 18-crown-6 with potassium magnesiate K2MgR4(PMDETA)2. Having optimised these systems, substrate scope was examined to probe the range and robustness of 18-crown-6/K2MgR4(PMDETA)2 as a catalyst. A wide series of alkynols, including terminal and internal alkynes which contain a variety of potentially reactive functional groups, were cyclised. In comparison to previously reported monometallic systems, bimetallic 18-crown-6/K2MgR4(PMDETA)2 displays enhanced reactivity towards internal alkynol-cyclisation. Kinetic studies revealed an inhibition effect of substrate on the catalysts via adduct formation and requiring dissociation prior to the rate limiting cyclisation step.
:2b ratios of 95:5 after 5 h. Similar high yields of 88% with 2a:2b ratios of 90:10 after 3 h were obtained combining 18-crown-6 with potassium magnesiate K2MgR4(PMDETA)2. Having optimised these systems, substrate scope was examined to probe the range and robustness of 18-crown-6/K2MgR4(PMDETA)2 as a catalyst. A wide series of alkynols, including terminal and internal alkynes which contain a variety of potentially reactive functional groups, were cyclised. In comparison to previously reported monometallic systems, bimetallic 18-crown-6/K2MgR4(PMDETA)2 displays enhanced reactivity towards internal alkynol-cyclisation. Kinetic studies revealed an inhibition effect of substrate on the catalysts via adduct formation and requiring dissociation prior to the rate limiting cyclisation step.
Introduction
Since first reported by Wittig in 1951,1 alkali metal magnesiates have evolved from mere curiosities to a new family of versatile organometallic reagents which finds widespread applications in organic synthesis.2–7 By engaging metal–metal cooperativities, these bimetallic systems can offer superior chemo- and regioselectivities and/or functional group tolerances to those of their monometallic counterparts.8 Most reactivity studies have focused on using these reagents as metallating reagents (via Mg–H or Mg–X exchange processes) as well as anionic transfer agents to unsaturated organic molecules.9,10 Some key breakthroughs in the field which highlight the synergistic power of these bimetallic partnerships include Knochel's Turbo Grignard reagents RMgCl·LiCl (R = alkyl) which allow the functionalisation of a wide range of organic molecules via Mg–halogen exchange2 or Mulvey and O'Hara's template metallation, where mixed Na/Mg macrocyclic bases enable remote di-magnesiation of aromatic molecules.4,5 Despite the increasing interest that s-block metal catalysis is currently attracting,9,11–30 catalytic applications of these bimetallic systems have hardly been researched.31 Breaking new ground in this area, we recently reported sodium tris(alkyl)magnesiate NaMg(CH2SiMe3)332 as an efficient precatalyst for hydroamination of a variety of carbodiimides and isocyanates.33,34 Operating synergistically, this bimetallic precatalyst displays an enhanced catalytic ability compared to those found for its homometallic components NaCH2SiMe3 and Mg(CH2SiMe3)2, allowing hydroaminations to take place under very mild reaction conditions (mostly at ambient temperature) in nearly quantitative yields.
Going beyond these proof of concept studies, here we investigate the catalytic ability of alkali metal magnesiates to promote intramolecular hydroalkoxylation reactions of alkynyl alcohols. Being 100% atom efficient,35 this is a highly desirable route to provide straightforward access to a series of synthetically significant O-heterocycles such as hydrofurans, pyrans, benzofurans, etc. that are fundamentally important to many natural products36 and pharmaceutical37 compounds. However, the large bond enthalpy of typical O–H bonds and the modest reactivity of electron-rich olefins with nucleophiles can make these transformations especially challenging.38
A catalogue of catalysts has been developed for the cyclisation of alkynols, including various transition metal complexes as well as alkaline earth and f-block compounds. Transition metals previously used to promote this transformation are mostly precious metals39–48 but a few others such as copper,49 iron50 and tungsten51,52 have also been investigated. Different metal complexes have been found to offer different product selectivities, as there are two distinct cyclisation routes for hydroalkoxylation of alkynols (Scheme 1). For instance, tungsten, ruthenium and molybdenum complexes have been used to produce endocyclised enol ether products. This occurs due to the proposed formation of an η2-vinylidene metal complex intermediate (Scheme 1, I), which is then susceptible to nucleophilic addition to generate a Fischer oxacarbene (Scheme 1, II) that ultimately yields the endocyclised product (Scheme 1, III).53–56 However more commonly, where no electronically biased intermediate is formed, exocyclisation occurs due to the more favourable approach of the nucleophile with respect to the π-system.57,58
 | ||
| Scheme 1 Conversion of 4-pentynol to respective five-membered exo- and six-membered endocyclised products. The latter conversion proceeds via vinylidene (I) and Fischer oxacarbene (II) intermediates. | ||
Besides the numerous examples of transition metal catalysts, the use of f- and s-block catalysts for this reaction has been investigated. In terms of f-block chemistry, Marks has utilised lanthanide amide catalysts,59,60 while Otero and Lara-Sánchez have employed heteroscorpionate rare earth catalysts.61 For s-block catalysis, Hill62 has used calcium, strontium and barium amides (Scheme 2).
 | ||
| Scheme 2 Hydroalkoxylation of 4-pentynol catalysed by heavier alkaline earth metal amides.62 | ||
These particular studies highlight that transformations proceed selectively via exocyclisation, although with alkaline earth metal amides two different product isomers were observed, an internal and external alkene. Since Hill's initial study, several papers focusing on alkali metal and alkaline earth catalysts have been published. Liu has shown that potassium tert-butoxide is an effective and selective catalyst for cyclisation of aromatic alkynylamines and alkynols,63 albeit with the need for elevated temperatures and highly polar solvents. Utilising similar reaction conditions, Wang employed potassium carbonate to prepare indole/pyrrole-fused 1,4-oxazines.64 Baire has also recently reported that cycloisomerisation of cis-6-hydroxy- and cis-6-acyloxyhex-2-en-4-ynals to 2-acylfurans and 2-(1-acyloxyalkenyl)furans is achievable using calcium catalysis.65 In contrast, magnesium-based reagents have not followed suit by showing very little promise to catalyse these types of transformations; whereas the catalytic properties of s-block bimetallic complexes remain unexplored in this context.
Expanding the scope of s-block cooperative catalysis, here we report the first catalytic applications of alkali metal magnesiates to promote cyclisation reactions, focusing on intramolecular hydroalkoxylation of alkynols. Benefitting from the enhanced metallating and nucleophilic abilities of these synergistic systems, we envisaged that they could play a dual role in overcoming the main challenges encountered in these transformations, facilitating not only OH activation, but also the required addition across CC bonds. Combining kinetic experiments with reactivity studies, here we provide informative mechanistic insights on how cooperative effects can be maximised as well as on the key role that each metal plays in these novel magnesiate-catalysed transformations.
Results and discussion
Intrigued by the lack of a magnesium amide-catalysed reaction from the series of alkaline earth metals investigated by Hill, our first reaction used the neutral bis(alkyl)magnesium reagent Mg(CH2SiMe3)2, containing thermally stable monosilyl groups, as a potential pre-catalyst. Using 4-pentynol (1) (as previously employed by Marks and Hill) for benchmarking with a catalytic quantity of the magnesium reagent, no reaction was observed even when the mixture was heated to 75 °C for 36 h (Table 1, entry 1).|
|
||||
|---|---|---|---|---|
| Entry | Pre-catalyst (R = CH2SiMe3) | Time, h | Yieldb (%) | Isomer ratio (2a:2b) |
| a Reactions were performed in a Young's cap NMR tube, using 0.5 mmol substrate (4-pentynol) (1) and 0.025 mmol (5 mol%) pre-catalyst. b Reactions were performed in a Young's cap NMR tube, using 0.6 mmol (1.2 eq.) substrate (4-pentynol) (1) and 0.025 (5 mol%) pre-catalyst. This additional 0.1 mmol (20 mol%/0.2 eq.) 1 was employed to convert the pre-catalyst to the ‘active catalyst’ (vide infra). c Calculated from 1H NMR spectroscopic data by integration against an internal standard (10 mol% 1,2,3,4-tetraphenylnaphthalene). d A stoichiometric quantity of crown ether co-catalyst used according to the alkali metal [i.e., 10 mol% 18-c-6 for 5 mol% K2MgR4(PMDETA)2]. e Due to low yields obtained and the inherent error within the measurement, an isomer ratio is not reported. | ||||
| 1 | MgR2a | 36 | 0 | — |
| 2 | MgR2 + 18-c-6 (±PMDETA)a,d | 36 | 0 | — |
| 3 | KRa | 36 | 11 | 99:1 |
| 4 | KR + 18-c-6c | 16 | 70 | 93:7 |
| 5 | LiMgR3a | 36 | 3 | —d |
| 6 | NaMgR3a | 36 | 12 | 97:3 |
| 7 | NaMgR3(TMEDA)a | 36 | 13 | 96:4 |
| 8 | KMgR3a | 36 | 16 | 98:2 |
| 9 | KMgR3(PMDETA)a | 36 | 20 | 94:6 |
| 10 | KMgR3 + 18-c-6a,d | 10 | 77 | 81:19 |
| 11 | Li2MgR4(TMEDA)2b | 36 | 3 | —e |
| 12 | Na2MgR4(TMEDA)2b | 36 | 83 | 91:9 |
| 13 | K2MgR4(PMDETA)2b | 22 | 91 | 86:14 |
| 14 | Na2MgR4(TMEDA)2 + 15-c-5b,d | 5 | 87 | 95:5 |
| 15 | K2MgR4(PMDETA)2 + 18-c-6b,d | 3 | 88 | 90:10 |
| 16 | K2MgR4(TMEDA)2 + 18-c-6b,d | 3 | 94 | 90:10 |

This raises the question, “why is magnesium unable to mediate this cyclisation whereas calcium can?” Major contributory factors are likely due to the significant difference in ionic radii between the two metal cations and the low π-philicity of magnesium.66 DFT calculations were carried out by Marks investigating the quantity of alkynol molecules which could coordinate to the active lanthanide catalyst when La(HMDS)3 is added as a pre-catalyst.67 Interestingly these calculations showed that the most likely catalytically active species is a La ion coordinated to three alkynoxide ligands, binding via the oxide anion as well as forming a close-range π-interaction with the alkyne unit. An additional substrate molecule coordinates to the metal via its O atom, affording a coordinatively saturated species. Furthermore, it appears that these La–π-interactions activate the alkyne for insertion into the lanthanum–oxygen bond and additionally position the alkynoxide perfectly for cyclisation. Bearing this in mind, the lack of reactivity observed when using Mg(CH2SiMe3)2 could be due to the magnesium centre not being large enough to simultaneously bind to the oxide anion and be in close proximity to the π-density of the carbon–carbon triple bond.
We next focused on testing homometallic alkali metal organometallics, which, being significantly more polar than their Mg counterparts, have recently found applications in hydrophosphination catalysis.68 Li(CH2SiMe3) and Na(CH2SiMe3) were employed in the same reaction; however, the conversions were poor (<5%) (see ESI†). Using the heavier congener K(CH2SiMe3) (Table 1, entry 3) also results in low conversion over a 36 h period (11%). Although the conversion is again low, the fact that the reaction was more successful than those with Li(CH2SiMe3), Na(CH2SiMe3) and Mg(CH2SiMe3)2 can perhaps be attributed to the larger ionic radius of potassium versus the other metals allowing it to interact with the carbon–carbon triple bond, activating it for cyclisation.
Next, we investigated the catalytic ability of a lower-order (1:1, alkali metal:Mg ratio) magnesiate. It was envisaged that a bimetallic magnesiate could function cooperatively, i.e., promote a dual-activation where a magnesium alkoxide forms, thus preparing one end of the substrate for cyclisation while the alkali metal could then interact with the π-system of the carbon–carbon triple bond. As alluded to previously, this latter feature is essential as the magnesium alone cannot provide this interaction. LiMg(CH2SiMe3)3 (Table 1, entry 5) was the first magnesiate tested but it struggled to promote the reaction, presumably again due to size limitations of the lithium centre (i.e., it is sterically blocked by the ligand set). Moving to sodium (Table 1, entry 6), disappointingly, this sodium magnesiate only promoted the reaction modestly [about as effectively as K(CH2SiMe3)]. When the potassium magnesiate KMg(CH2SiMe3)3 (Table 1, entry 8) was employed, the yield (16%) was comparable to that observed for K(CH2SiMe3) (11%). So, in summary, lower order magnesiate reagents are generally poor at catalysing the hydroalkoxylation/cyclisation reaction.
In the hope of improving this situation, higher-order (2:1, alkali metal:Mg ratio) magnesiate reagents were considered. The Lewis basic proligands, TMEDA (N,N,N′,N′-tetramethylethylenediamine) and PMDETA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine), were employed to completely fill the coordination sphere of the alkali metals. Again, starting with the lithium derivative, the higher-order magnesiate (Table 1, entry 11) performed as poorly as the lower-order lithium magnesiate in the reaction (3%). However, when Na2MgR4(TMEDA)2 was employed (Table 1, entry 12), pleasingly a substantially more successful conversion of the alcohol to the cyclised product (83%) was obtained within 36 h. The reaction efficiency can be improved further when the potassium reagent K2MgR4(PMDETA)2 is used (91% after 22 h, Table 1, entry 13). These two higher-order magnesiates display greatly enhanced catalytic activity compared to that of homometallic K(CH2SiMe3). In addition, they continue to have a high isomer selectivity with up to 90% of the external alkene isomer being produced. The remarkable alkali metal effect observed between Li2MgR4(TMEDA) and K2MgR4(PMDETA)2 is even more striking when considering that both mixed-metal species are isostructural.69,70 At this juncture we reasoned that addition of TMEDA or PMDETA in these reactions influenced the marked increase in reactivity. To test whether these multidentate Lewis bases were the key drivers in increasing the yields, we tested the lower-order NaMgR3(TMEDA) and KMgR3(PMDETA) (Table 1, entries 7 and 9), but found that adding these multidentate ligands did not enhance reaction performance. With the higher-order magnesiates, the reaction rate using K2MgR4(PMDETA)2 is unaffected when the bidentate Lewis base TMEDA is used in place of its tridentate analogue PMDETA (Table 2, entry 16). Hence K2MgR4(TMEDA)2 displayed the same reaction time and selectivity as its PMDETA variant, demonstrating the reaction rate to be unaffected by the choice of Lewis base.
| Entrya,c | Substrate (5 mol% cat., C6D6) | Yieldb |
|---|---|---|
| a Reactions were performed in a Young's cap NMR tube, using 0.6 mmol (1.2 eq.) substrate (3, 5, 7, or 9) and 0.025 mmol (5 mol%) pre-catalyst with 0.1 mmol (20 mol%/0.2 eq.) 1 consumed in converting the pre-catalyst to the ‘active catalyst’ (vide infra). b Calculated from 1H NMR spectroscopic data by integration against an internal standard (10 mol% 1,2,3,4-tetraphenylnaphthalene). c A stoichiometric quantity of crown ether co-catalyst used according to the alkali metal [i.e., 10 mol% 18-c-6 for 5 mol% K2MgR4(PMDETA)2]. | ||
| 1 |

|
99% (14:9:77) |
| 2 |

|
86% |
| 3 |

|
89% |
| 4 |
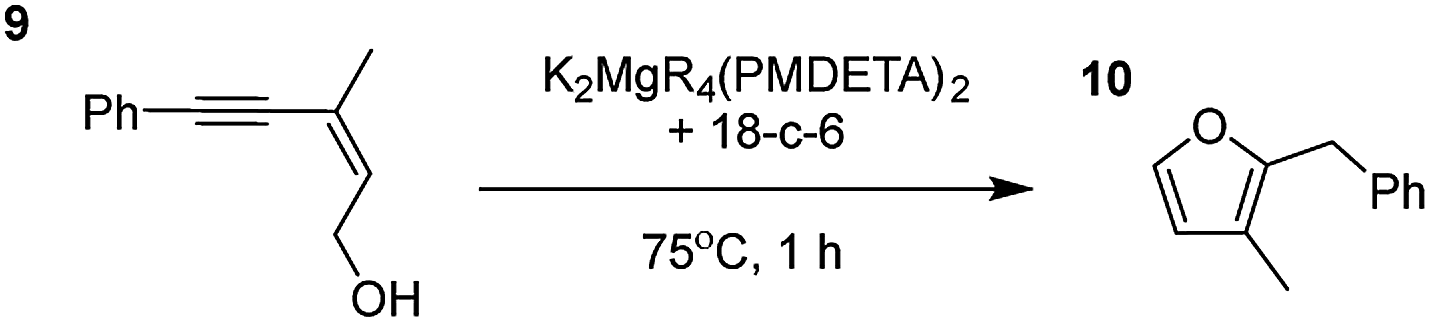
|
90% |
To establish whether or not both metals need to be present for the magnesiate to function, a catalytic amount of crown ether was added as a co-catalyst. As a crown ether of appropriate size is often able to sequester an alkali metal, the intention here was to ascertain, if the alkali metal was captured (non-cooperative), that the remaining anionic Mg(OR)3− or Mg(OR)42− fragment, of the now solvent-separated complex, was not able to perform the catalysis on its own. Surprisingly, in contrast, the results obtained in these reactions showed a dramatic improvement in reactivity!
Adding catalytic quantities of 15-crown-5 or 18-crown-6 to the sodium and potassium systems respectively dramatically increased the activity of the magnesiate catalysts, with the sodium higher-order magnesiate now nearing completion in 5 h (Table 1, entry 14) and the potassium analogue in 3 h (Table 1, entry 15), in reactions retaining good selectivity. This reactivity enhancement was also seen with both the monometallic and lower order potassium magnesiate (Table 1, entries 4 and 10 respectively).
Due to the significant difference in completion rates of these two reactions (5 h vs. 3 h; Table 1, entries 14 and 15), the idea of a completely solvent-separated Mg(OR)42− dianion can be discounted as it would be expected that similar rates would be observed if this dianion was a catalytically active species. Although the crown ether is often expected to sequester the alkali metal there are examples of structures where the alkali metal is still able to maintain π-interactions with the anionic moiety whilst coordinated by the crown ether.71–78 Thus it appears that under these conditions, the addition of the crown ether must tune the coordination environment of K, modifying its Lewis acidity to generate a more catalytically competent species (vide infra). Therefore, these results suggest that the alkali metal is intimately involved and plays a key role in the cyclisation reaction. To ascertain whether the 18-crown-6 is catalytically active, when it is combined with only Mg(CH2SiMe3)2 the reaction fails (entry 2).
Summarising Table 1, it is clear that the organomagnesium compound used in this study is unable to catalytically promote cyclisation of alkynols. Homometallic alkali metal compounds can only poorly promote the reaction; however, when working cooperatively with a Mg compound the two metals can perform more efficiently by dual-activating the substrate. There are also clear trends that higher-order magnesiates perform better than lower-order magnesiates and that when comparing alkali metals K > Na ≫ Li presumably due to a sequential decrease in size and reduction in π-philicity. Perhaps the most surprising outcome of the optimisation study was that introducing a crown ether into higher-order magnesiates appears to greatly enhance the rate of the cyclisation, whilst maintaining a high selectivity. The reaction times and selectivity of our system (Table 1, entry 15) compare favourably with those of other catalytic systems. In terms of typical reaction times (3 h; Table 1, entry 15), this is similar to that observed by Marks (2.5 h) where only isomer 2a is produced by employing La[N(SiMe3)2]3.38 Perhaps most pertinent to our study, Hill has employed heavy group 2 amides [Ae{N(SiMe3)2}2 where Ae = Ca, Sr, Ba], where reaction times ranged from 2.5–6 h, exhibiting a 2a isomer selectivity of 90–97% (Scheme 2).62 Therefore to re-emphasize, our results show that by pairing a magnesium complex with a potassium one, it has been possible to overcome the severe limitations imposed by a magnesium homometallic catalyst on its own. Through cooperativity, the surprising effect was the generation of a potassium magnesiate catalyst which is even more active and equally as selective as the homometallic heavier group 2 congeners.
Having established the optimal conditions for cyclisation of 4-pentynol, the scope of the reaction was evaluated. Terminal and internal alkynes were tested to assess the range and robustness of the catalytic system (Tables 2 and 3). Interestingly, by increasing the alkynol chain length by one carbon atom (5-hexynol, 3, Table 2, entry 1), the major product is isomerised alkyne (4c), where the carbon–carbon triple bond has moved from a terminal to an internal location (Table 2, entry 1). It can be envisaged that isomerisation occurs due to the angle of ring closure being unfavourable, and hence it will occur prior to a cyclisation process. Related to these findings, Tsurugi has reported that organomagnesium complexes can promote alkyne isomerisations of related compounds.79 Where the alkynol substrate is already held in a manner that is partially aligned to that of the cyclised product, the cyclisation progresses much more readily (Table 2, entry 2). This occurs even at ambient temperature in 1 h. Interesting selectivity is observed when it comes to cyclisation of Z-enynols (7 and 9). In Table 2 entry 3, with a terminal enynol (7) employing the optimised conditions we see the production of an aromatic furan product (8). This product differs from the product previously reported using heavier alkaline earth59,60,80 or lanthanide amides.38,57,58 With these catalytic systems the non-aromatic product, 3-methyl-2-methylene-2,5-dihydrofuran, is observed, and it is only when we move to a transition metal catalyst,44 we see formation of the aromatic furan product.
| Entrya,c | Substrate [Ar–CC–(CH2)3OH] |
Product(s) | Conditions [5 mol% K2MgR4(PMDETA)2, C6D6] | Yield (and product ratio)b |
|---|---|---|---|---|
| a Reactions were performed in a Young's cap NMR tube, using 0.5 mmol substrate (11, 13, 15, 17, 19, 21, and 23) and 0.025 mmol (5 mol%) pre-catalyst with 0.1 mmol (20 mol%/0.2 eq.) 1 consumed in converting the pre-catalyst to the ‘active catalyst’ (vide infra). b Calculated from 1H NMR spectroscopic data by integration against an internal standard (10 mol% ferrocene). c A stoichiometric quantity of crown ether co-catalyst used according to the alkali metal [i.e., 10 mol% 18-c-6 for 5 mol% K2MgR4(PMDETA)2]. | ||||
| 1 |

|

|
75 °C, 1 h | 84% (10:90:0) |
| 40 °C, 36 h | 6% | |||
| 2 |
|
|
75 °C, 36 h | 64% (31:69) |
| 40 °C, 36 h | 1% | |||
| 3 |

|

|
75 °C, 1 h | 99% (9:91) |
| 40 °C, 36 h | 51% | |||
| 4 |
|

|
75 °C, 0.5 h | 95% (8:92) |
| 40 °C, 24 h | 82% | |||
| 5 |
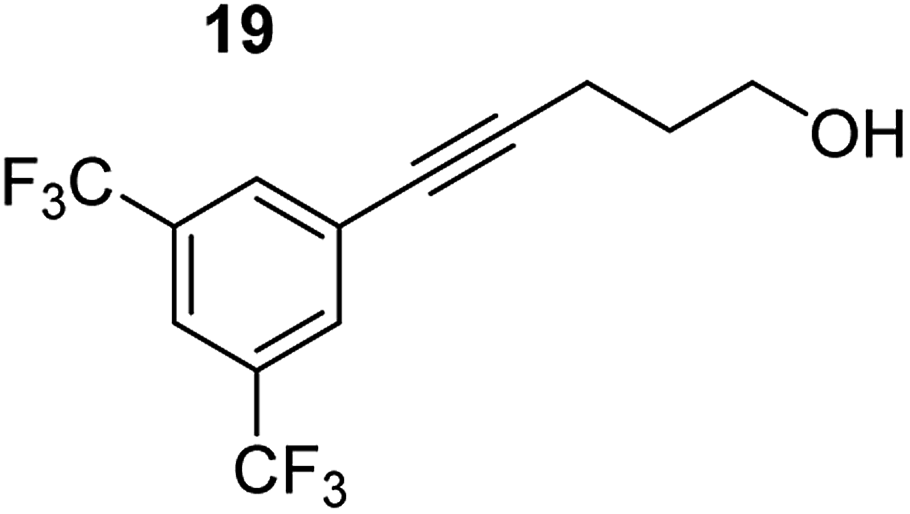
|

|
75 °C, 0.5 h | 97% (4:96) |
| 40 °C, 2 h | 93% | |||
| 6 |
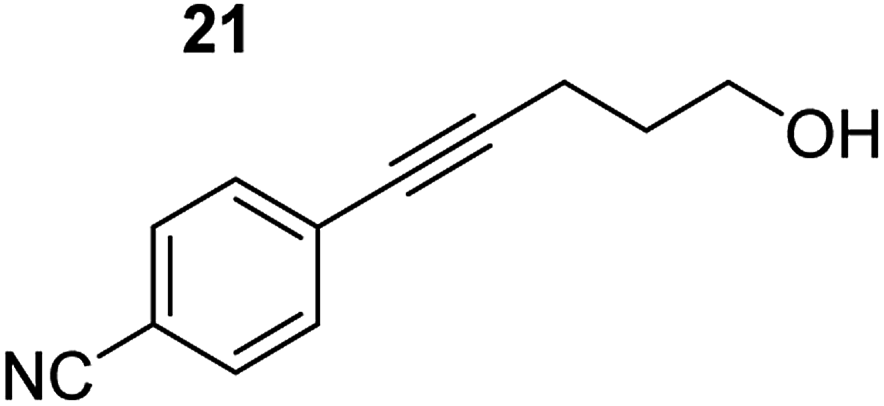
|
|
75 °C, 0.5 h | 84% (6:94) |
| 40 °C, 0.5 h | 76% | |||
| 7 |
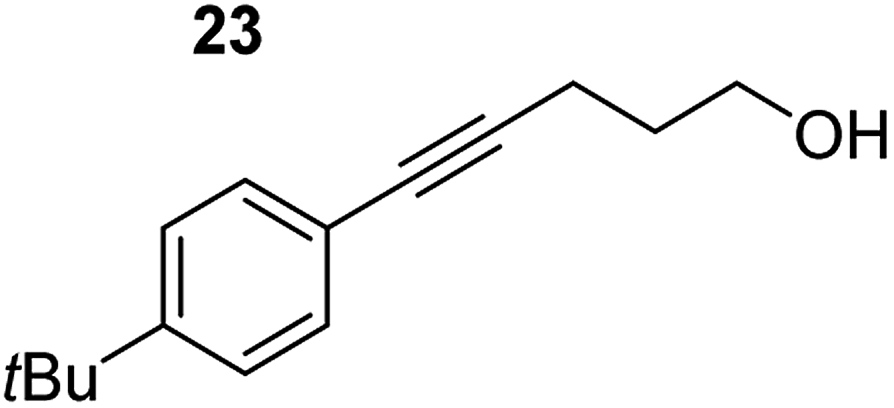
|
|
75 °C, 6 h | 87% (11:89) |
| 40 °C, 36 h | 2% | |||
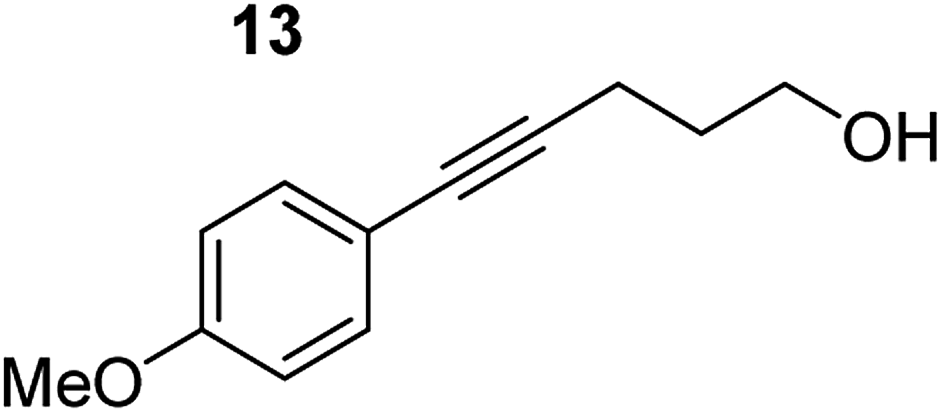
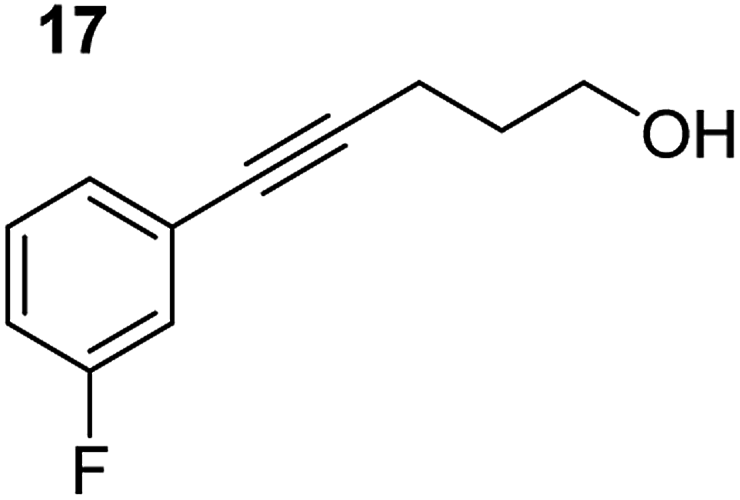
In order to further probe the functional group tolerance of the catalytic magnesiate system, a range of internal alkynes were synthesised with different pendant functional groups, including halogens, and electron-donating and electron-withdrawing groups. It transpired that no side reactions took place when MeO, F, Cl, CF3 or CN functional groups were incorporated into the substrate. Looking at 5-phenylpent-4-yn-1-ol (11) (Table 3, entry 1) the reaction reached completion in 1 h at 75 °C yielding only two product isomers. The major product being the external Z-alkene 12b and the minor external E isomer 12a. In the aforementioned studies by Hill,62 a mixture of three product isomers was obtained, the external alkene isomers (12a, 12b) and an internal alkene (12c) (note these three products are still exocyclic). In this work, it was also noted that the product ratio appeared to have a degree of temperature dependence and required significantly harsher reaction conditions (90–110 °C, 16–40 h).
Despite altering the reaction temperature (reactions performed at both 40 and 75 °C), the same isomer ratios were obtained for all the reactions which tended towards completion (entries 4–6, Table 3). For entries 1–3, Table 3, the low temperature reactions (40 °C) were relatively less efficient which precluded accurate determination of the product ratio at this temperature. Focusing on the specific substrates reacting at 75 °C, when the electron donating methoxy group (entry 2, Table 3) is incorporated onto the 5-phenylpent-4-yn-1-ol scaffold, the reaction rate is dramatically reduced with only a 64% yield achieved in 36 h, compared to 1 h for the non-functionalised substrate (entry 1, Table 3). This reduction in reaction rate is also observed, albeit to a lesser extent, with the weaker donating t-butyl group (entry 7, Table 3). When mildly withdrawing halides are incorporated within the substrate (entries 3 and 4, Table 3) the reaction is complete in less than 1 h. At this juncture, it was deemed necessary to consider reaction times at 40 °C to ascertain whether any difference in relative rates was observed. With a Cl substituent (entry 3, Table 3), the yield was 51% after 36 h compared to 6% for the non-substituted alkynol (entry 1, Table 3). Using a F substituent, this yield can be increased to 82% in 24 h (entry 4, Table 3). Moving to stronger electron-withdrawing groups, incorporating trifluoromethyl (yield, 93%) brings the reaction time down to only 2 h at 40 °C (entry 5, Table 3), and if a cyano substituent (yield, 76%) is utilised this is further reduced to 30 min (entry 6, Table 3).
This set of substrates based on 5-phenylpent-4-yn-1-ol (11) show nicely the benefit of using a bimetallic system for these cyclisation reactions. With the heavier alkaline earth metal amides and lanthanide amides, substrate 11 is more challenging to cyclise, taking 16 h at 90 °C with Ca(HMDS)2 and about 15 h at 120 °C with La(HMDS)2. Marks38 has suggested that the reason for this sluggish reaction time with La(HMDS)3 is sterically driven by the phenyl substituent that prevents interaction between the metal and the internal alkyne. Using a bimetallic system, it appears that this steric clash is somewhat circumvented giving rise to a faster reaction time than the other two systems mentioned.
In addition to reaction scope, kinetic studies have been carried out on the system. Prime motivations were to determine the reaction order of the alkynol substrate and catalyst present in the reaction, with the aim of providing insights into the reaction mechanism. The substrate chosen for these studies was 4-pentynol (1), as this was used for the initial parameterisation studies. A plot of concentration of 1 against time displays a linear fit until half conversion when a rate acceleration is observed [Fig. 1(a)].
 | ||
| Fig. 1 (a) Plot of the consumption of 4-pentynol (1) versus time (0.040 M cat., C6D6, 343 K). (b) Plot of the [4-pentynol (1)]2versus time (0.040 M cat., C6D6, 343 K). | ||
Consistent with substrate inhibition, the plot of [1]2versus time shows a good fit [Fig. 1(b)]. Further evidence was gathered by plotting initial reaction rates at varied alkynol concentrations (Fig. S1†), where greater rates were observed as the concentration of 1 was decreased. This negative first order was observed by Hill62 for the transformation employing heavier group 2 amide catalysts, where its origin is proposed to be the decoordination of a substrate molecule from the active catalyst being required before cyclisation can occur (Scheme 3).
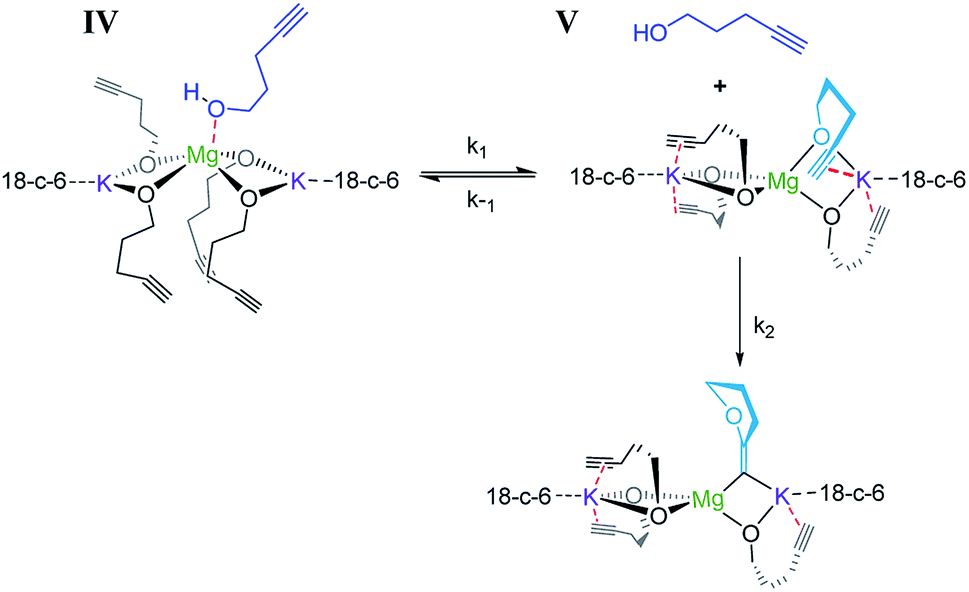 | ||
| Scheme 3 Reaction showing the coordination/decoordination of an additional alkynol molecule. | ||
When coordination occurs (IV), it appears that the cyclisation process is hindered. As the reaction is inverse first order in the substrate (vide infra) it is assumed that the incoming alkynol coordinates to the single Mg centre rather than the K centres, which are already highly coordinated by bonding to the crown ether. In previous studies, the addition of 18-crown-6 has had both beneficial78,81 and detrimental77 impacts on synthetic performance. Here we observe an improvement in catalytic activity, which could perhaps be explained by the 18-crown-6 that sufficiently satisfies the coordination environment of the metal, preventing further excess alcohol coordination from occurring which would cause inhibition.
Steady state approximation
First step slow
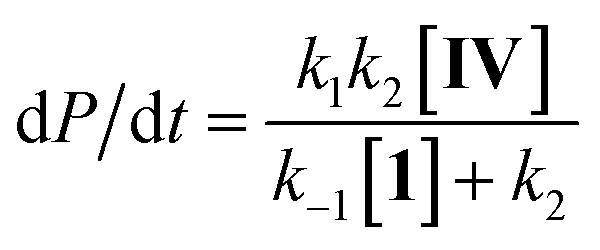 | (1) |
| dP/dt = k1[IV] | (2) |
Pre-equilibrium approximation
First step fast
 | (3) |
Considering this catalytic process as initial dissociation of 1 (k1 and k−1) followed by cyclisation reaction (k2) and using the steady-state approximation eqn (1) can be deduced. This equation can be simplified to eqn (2) at low concentrations of 1 (k2 ≫ k−1[1]), while at high concentrations of 1 (k−1[1] ≫ k2) eqn (3) rules this process. Alternatively, a pre-equilibrium approximation in which dissociation of 1 from magnesium does not influence the reaction rate leads exclusively to eqn (3), and hence to a good inverse first order relationship in 1 at any concentration of this ligand. The plot of the observed initial rates vs. 1/[1] (Fig. 2) reveals a linear fit with a non-zero intercept according to eqn (1), and hence consistent with a slow ligand dissociation step followed by a rapid cyclisation process.
 | ||
| Fig. 2 Observed rate constant at varied substrate concentrations versus inverse substrate concentration (1). | ||
Looking at the catalyst order, a good fit was seen when the deduced rates for the different catalyst loadings (Fig. S2†) were plotted against the concentration of 2 (Fig. 3), indicating that the reaction has a first order dependence on the catalyst. This is similar to that found by Marks,38 but not the second order dependence that was found by Hill;62 however, in previous work by Hill using alkaline earth metal amides, a second order dependence was observed. This is said to be due to the pre-catalysts existing as dimers. The first order dependence on the magnesiate pre-catalyst observed here has also been observed in hydroamination reactions.62
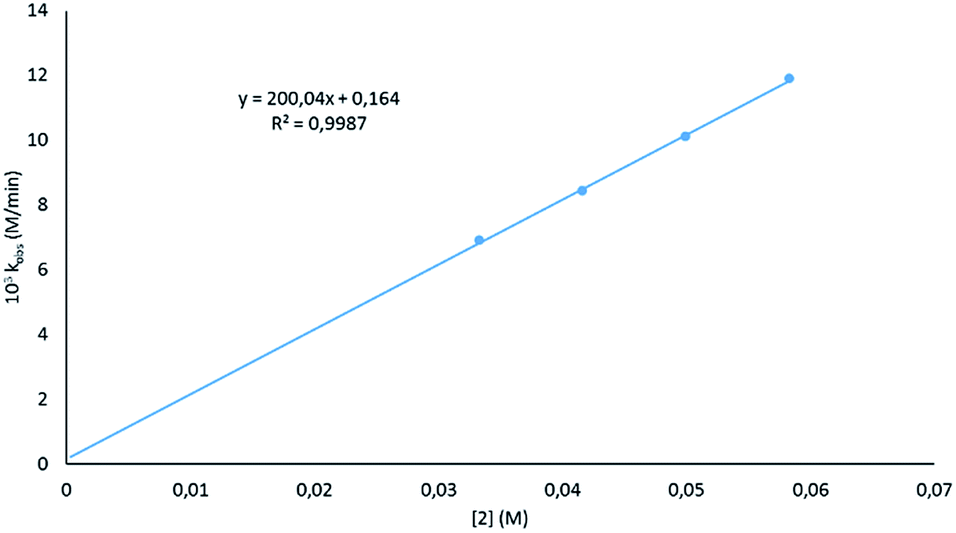 | ||
| Fig. 3 Plot of the observed rate constant at varied catalyst loadings versus catalyst concentration. | ||
In terms of the mechanism, there has been a previous proposition by Hill62 built upon the work of Marks.38 It reasons that there are two catalytic cycles at work simultaneously to allow for the production of the two product isomers observed. One cycle involves the alkyne passing through an allene intermediate, where it was demonstrated that starting from an allenyl alcohol instead of the alkynyl alcohol yielded the same products in a similar yield and ratio. In this work utilizing alkali metal magnesiates in combination with substrate 3, we detected the presence of an allene intermediate (by 1H NMR spectroscopy, at δ 5.19 and 4.63 ppm) during the course of the reaction (Fig. 4). As such we believe that a similar two-cycle reaction mechanism is likely to be at play. The findings from the kinetics were also included with the other results to form a proposed mechanism for this alkali metal magnesiate-mediated catalysis (Scheme 4).
 | ||
| Fig. 4 Evidence for the allene intermediate (via1H NMR spectroscopy, C6D6, 400 MHz) produced during the cyclisation of 5-hexynol (3) (Table 2, entry 1) as proposed by Hill.62 | ||
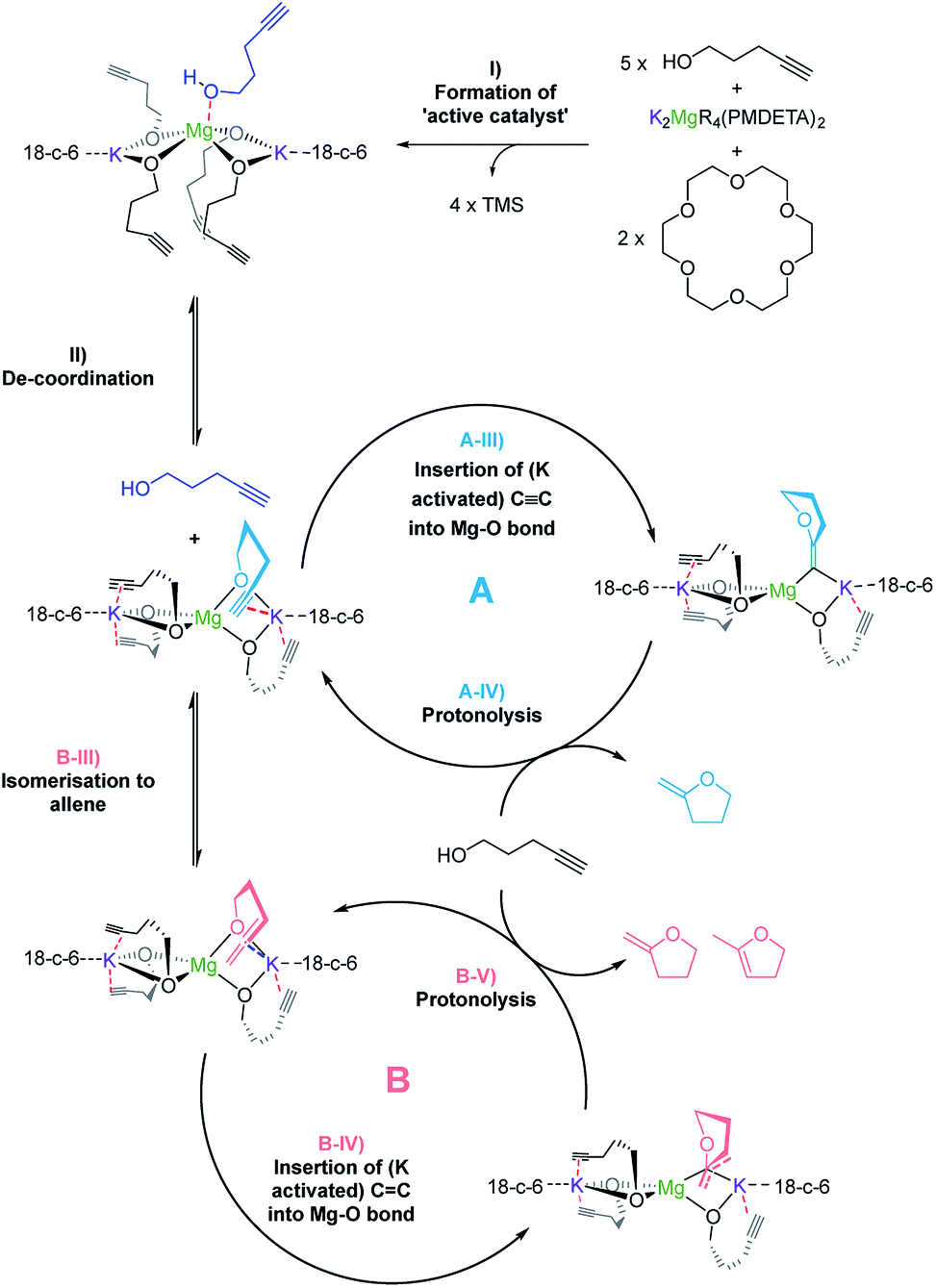 | ||
| Scheme 4 Proposed mechanism for the alkali metal magnesiate + crown ether co-catalyst mediated catalytic cyclisation of alkynols via dual activation. | ||
Overall the deduced mechanism starts with formation of the active catalyst from the magnesiate pre-catalyst. This involves the deprotonation of four alcohol substrate molecules to form a magnesiate alkoxide, liberating tetramethylsilane. This ‘active catalyst’ is then involved in a coordination/decoordination process with an additional substrate molecule as suggested by the kinetic studies. This additional molecule of 1 occupies the coordination sphere of the magnesium inhibiting cyclisation, giving rise to an inverse first order term in the substrate. Cyclisation (insertion of the carbon–carbon multiple bond into the magnesium oxygen bond) only occurs upon its decoordination.
In cycle A the carbon–carbon triple bond of the alkyne is directly inserted into the metal–oxygen bond, leading to the formation of only one product upon protonolysis. Cycle B on the other hand involves an equilibrium isomerisation from alkyne to allene which upon insertion into the metal–oxygen bond, and subsequent protonolysis, can yield two product isomers. Protonolysis releases the cyclised products and reforms the active catalyst completing the cycle.
Disclosing a unique cooperative behaviour under catalytic conditions, each of the metals plays a key role in this process, by simultaneously activating the O–H and CC bonds of the substrate. Thus, coordination of the CC bond to the larger more π-philic potassium centre enables further activation of this unsaturated group and brings it into close proximity to the di-anionic (magnesiate) {Mg(OR)4}2− component, which is significantly more nucleophilic than a charge-neutral magnesium compound, facilitating intermolecular ring-closure to furnish the relevant oxygen-heterocycle.
Experimental
General considerations
All reactions were carried out under a protective atmosphere of argon using standard Schlenk techniques. Non-deuterated solvents were dried by heating to reflux over sodium ketyl radicals under nitrogen and deuterated solvents were degassed and stored over molecular sieves. Pre-catalysts [Mg(CH2SiMe3)2, K(CH2SiMe3), LiMg(CH2SiMe3)3, NaMg(CH2SiMe3)3, KMg(CH2SiMe3)3, Li2Mg(CH2SiMe3)4(TMEDA)2, Na2Mg(CH2SiMe3)4(TMEDA)2, K2Mg(CH2SiMe3)4(TMEDA)2 and K2Mg(CH2SiMe3)4(PMDETA)2] were prepared following literature procedures2 and handled in a glovebox. Li(CH2SiMe3), 1 and 5 were obtained from Sigma-Aldrich; 1 and 3 from Alfa-Aesar; and 7 from Fluorochem. Substrates 11, 13, 15, 17, 19 and 21 were synthesised based on literature procedures82 (characterisation data provided for new substrates and products 13–22 can be found in the ESI†). NMR spectra were recorded on a Bruker AV III 400 MHz spectrometer operating at 400.1 MHz for 1H, 100.6 MHz for 13C or 128.4 MHz for 19F.Procedure for pre-catalyst synthesis
Pre-catalysts were prepared and isolated prior to being employed in reactions. Na(CH2SiMe3),83,84 K(CH2SiMe3)84,85 and Mg(CH2SiMe3)2 (ref. 69) were prepared from literature procedures. TMEDA and PMDETA were distilled over CaH2 before use. K2Mg(CH2SiMe3)4(PMDETA)2 was prepared by suspending K(CH2SiMe3) (0.25 g, 2 mmol) and Mg(CH2SiMe3)2 (0.20 g, 1 mmol) in hexane (10 mL). The mixture was stirred at ambient temperature for 1 h. PMDETA (0.43 mL, 2 mmol) was added to this white suspension and the mixture was gently heated until homogeneity was achieved. After storage at −26 °C overnight, the mixture was filtered to yield a light yellow solid (typical yield; 0.62 g, 78%).Li2Mg(CH2SiMe3)4(TMEDA)2, Na2Mg(CH2SiMe3)4(TMEDA)2, and K2Mg(CH2SiMe3)4(TMEDA)2 were prepared using similar methods to K2Mg(CH2SiMe3)4(PMDETA)269 with the following modifications: substitution of TMEDA (0.30 mL, 2 mmol); Li(CH2SiMe3) (2 mL, 2 M in pentanes); and/or Na(CH2SiMe3) (0.22 g, 2 mmol) where appropriate.
LiMg(CH2SiMe3)3,70 NaMg(CH2SiMe3)3,32 and KMg(CH2SiMe3)3 were prepared using half the molar quantity of M(CH2SiMe3)3 (1 mL/0.11 g/0.13 g respectively), and toluene (5 mL) was added in place of the multidentate amine in order to achieve dissolution upon heating.
Procedure for NMR scale reactions
In the catalytic cyclisation of 4-pentynol (1) with K2Mg(CH2SiMe3)4(PMDETA)2 + 18-c-6, 4-pentynol (56 μL, 0.6 mmol, 1.2 eq.) (0.2 eq./20 mol% excess required to form the ‘active catalyst’) was added to a Young's tap NMR tube alongside C6D6 (0.54 mL), 1,2,3,4-tetraphenylnaphthalene (0.022 g, 0.05 mmol, 0.1 eq.) and 18-crown-6 (0.013 g, 0.05 mmol, 0.1 eq.). To this K2Mg(CH2SiMe3)4(PMDETA)2 (0.020 g, 0.025 mmol, 0.05 eq.) was added and the tube was placed in an oil bath at 75 °C. The reaction was periodically monitored by 1H NMR spectroscopy and the yields obtained were calculated using NMR spectroscopic integrals and were relative to the internal standard. Optimisation and substrate scope reactions were carried out using similar procedures, employing 10 mol% of an appropriate internal standard [1,2,3,4-tetraphenylnaphthalene (0.022 g) or ferrocene (0.009 g)] and with the oil bath temperature being changed where necessary based on observed reaction times. 0.5 mmol alkynol substrate was used with the monometallic species and lower-order magnesiates, and 0.6 mmol with higher-order magnesiate pre-catalysts. 0.025 mmol pre-catalyst was employed in all cases, quantities shown in Table 3. In general, the reactions remain homogeneous until nearing completion. At this point an unidentified solid/gel forms, which is presumably a species akin to the ‘K2Mg(OAlk)4(18-c-6)2 resting-state’.Procedure for kinetic studies
In the kinetic studies to determine the rate dependence of alkynol in the cyclisation of 4-pentynol (1) with K2Mg(CH2SiMe3)4(PMDETA)2 + 18-c-6, 4-pentynol (56 μL, 0.6 mmol) was placed in a Young's tap NMR tube with C6D6 (0.6 mL), 1,2,3,4-tetraphenylnaphthalene (0.022 g, 0.05 mmol) and 18-crown-6 (0.010 g, 0.04 mmol). To this K2Mg(CH2SiMe3)4(PMDETA)2 (0.016 g, 0.02 mmol) was added. The reaction was maintained at 70 °C in the NMR spectrometer and was monitored by 1H NMR spectroscopy. Yields were calculated using NMR spectroscopic integrals characteristic of 2a relative to the internal standard, 1,2,3,4-tetraphenylnaphthalene. This procedure was repeated with 0.90, and 1.20 mmol alkynol (1) to provide the data presented in Fig. S1† which was subsequently used in Fig. 2.The same procedure was applied to the investigation of the dependence of the initial rates of the reaction on the catalyst. The same concentration of 1 was used while varying the concentration of the catalyst. In terms of the pre-catalyst, 0.025, 0.030 and 0.035 mmol quantities of K2Mg(CH2SiMe3)4(PMDETA)2 were employed, necessitating 0.05, 0.06 and 0.07 mmol 18-crown-6 co-catalyst. The data from these experiments with varying catalyst concentration are presented in Fig. S2† and are subsequently used in Fig. 3.
Conclusions
In summary, alkali metal magnesiates have been shown to successfully promote the catalytic intramolecular hydroalkoxylation of alkynols through cooperative bimetallic catalysis. The roles of both magnesium and potassium components are crucial for the success of the process, affording a unique type of substrate activation that is not possible in conventional single-metal systems. Through cooperativity, the utilisation of an alkali metal magnesiate has overcome the inherent problems associated with homometallic magnesium systems. The optimised catalyst system, (PMDETA)2K2Mg(CH2SiMe3)4 paired with 18-crown-6 has been shown to function well with both terminal and internal alkyne substrates having a range of functional groups. Kinetic studies have revealed an inhibition effect of substrate on the catalyst under standard conditions (high concentrations of alkynol) by the formation of a coordination adduct which requires dissociation prior to the cyclisation step. Initial reactivity studies suggest that coordination of the 18-crown-6 to K finely tunes the reactivity of the bimetallic system, probably minimising the coordination of additional substrate molecules to the catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All high-resolution mass spectrometry (HRMS) was performed at the EPSRC UK National Mass Spectrometry Facility at Swansea University. We would like to thank the European Research Council (ERC-STG grant to EH), EPSRC UK (EP/N011384/1 grant to EH) and Leverhulme Trust (research grant RPG-2016-281 to CTOH) for the generous sponsorship of this research. We are also grateful to Professor Robert Mulvey for insightful discussions and Cristian Vidal for synthesising substrates 7 and 9. J. G.-A. is indebted to the MINECO of Spain (CTQ2016-81797-REDC and CTQ2016-75986-P) and the Gobierno del Principado de Asturias (project GRUPIN14-006) for financial support. J. G.-A. also thanks: (i) the Fundación BBVA for the award of “Beca Leonardo a Investigadores y Creadores Culturales 2017”; and (ii) PhosAgro/UNESCO/IUPAC for the award of “Green Chemistry for Life Grant”. The Fundación BBVA accepts no responsibility for the opinions, statements and contents included in the project and/or the results thereof, which are entirely the responsibility of the author. Data used within this publication can be accessed at https://doi.org/10.15129/5d0b6ec6-569c-4c8b-ae12-69d703afbe3d.Notes and references
- G. Wittig, F. J. Meyer and G. Lange, Justus Liebigs Ann. Chem., 1951, 571, 167–201 CrossRef CAS.
- T. Klatt, J. T. Markiewicz, C. Sämann and P. Knochel, J. Org. Chem., 2014, 79, 4253–4269 CrossRef CAS PubMed.
- A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958–2961 CrossRef CAS PubMed.
- A. J. Martínez-Martínez, S. Justice, B. J. Fleming, A. R. Kennedy, I. D. H. Oswald and C. T. O'Hara, Sci. Adv., 2017, 3, e1700832 CrossRef PubMed.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- R. L.-Y. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 6884–6900 RSC.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.1029b00047.
- R. E. Mulvey and S. D. Robertson, in Organo-di-Metallic Compounds (or Reagents): Synergistic Effects and Synthetic Applications, ed. Z. Xi, Springer International Publishing, Cham, 2014, pp. 129–158 Search PubMed.
- F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563–7727 CrossRef CAS PubMed.
- D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2014, 114, 1207–1257 CrossRef CAS PubMed.
- M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042–2043 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS PubMed.
- M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009–10011 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 206–216 CrossRef CAS.
- M. Arrowsmith, W. M. S. Shepherd, M. S. Hill and G. Kociok-Kohn, Chem. Commun., 2014, 50, 12676–12679 RSC.
- H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nature Catalysis, 2018, 1, 40–47 CrossRef.
- M. J. Butler, A. J. P. White and M. R. Crimmin, Organometallics, 2018, 37, 949–956 CrossRef CAS.
- H. Elsen, C. Färber, G. Ballmann and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 7156–7160 CrossRef CAS PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- S. Kobayashi and Y. Yamashita, Acc. Chem. Res., 2011, 44, 58–71 CrossRef CAS PubMed.
- R. J. Less, R. L. Melen and D. S. Wright, RSC Adv., 2012, 2, 2191–2199 RSC.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362–13365 CrossRef CAS PubMed.
- B. Liu, T. Roisnel, J. F. Carpentier and Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943–4946 CrossRef CAS PubMed.
- D. Mukherjee, H. Osseili, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2016, 138, 10790–10793 CrossRef CAS PubMed.
- J. Penafiel, L. Maron and S. Harder, Angew. Chem., Int. Ed., 2015, 54, 201–206 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed.
- C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC.
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. Commun., 2015, 51, 14477–14480 RSC.
- F. M. Younis, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2015, 34, 3577–3585 CrossRef CAS.
- S. E. Baillie, W. Clegg, P. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and L. Russo, Chem. Commun., 2011, 47, 388–390 RSC.
- M. De Tullio, A. Hernán-Gómez, Z. Livingstone, W. Clegg, A. R. Kennedy, R. W. Harrington, A. Antiñolo, A. Martínez, F. Carrillo-Hermosilla and E. Hevia, Chem.–Eur. J., 2016, 22, 17646–17656 CrossRef CAS PubMed.
- A. Hernán-Gómez, T. D. Bradley, A. R. Kennedy, Z. Livingstone, S. D. Robertson and E. Hevia, Chem. Commun., 2013, 49, 8659–8661 RSC.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- D. J. Faulkner, Nat. Prod. Rep., 1984, 1, 251–280 RSC.
- T. L. B. Boivin, Tetrahedron, 1987, 43, 3309–3362 CrossRef CAS.
- S. Y. Seo, X. H. Yu and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 263–276 CrossRef CAS PubMed.
- B. Seiller, C. Bruneau and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1994, 493–494 RSC.
- P. Pale and J. Chuche, Eur. J. Org. Chem., 2000, 1019–1025 CrossRef CAS.
- A. Varela-Fernández, C. García-Yebra, J. A. Varela, M. A. Esteruelas and C. Saá, Angew. Chem., Int. Ed., 2010, 49, 4278–4281 CrossRef PubMed.
- E. Benedetti, A. Simonneau, A. Hours, H. Amouri, A. Penoni, G. Palmisano, M. Malacria, J.-P. Goddard and L. Fensterbank, Adv. Synth. Catal., 2011, 353, 1908–1912 CrossRef CAS.
- V. Diachenko, M. J. Page, M. R. D. Gatus, M. Bhadbhade and B. A. Messerle, Organometallics, 2015, 34, 4543–4552 CrossRef CAS.
- C. Vidal, L. Merz and J. García-Álvarez, Green Chem., 2015, 17, 3870–3878 RSC.
- J. L. Arbour, H. S. Rzepa, A. J. P. White and K. K. Hii, Chem. Commun., 2009, 46, 7125–7127 RSC.
- A. G. Nair, R. T. McBurney, M. R. D. Gatus, S. C. Binding and B. A. Messerle, Inorg. Chem., 2017, 56, 12067–12075 CrossRef CAS PubMed.
- R. J. Harris, R. G. Carden, A. N. Duncan and R. A. Widenhoefer, ACS Catal., 2018, 8, 8941–8952 CrossRef CAS.
- V. Cadierno, J. Díez, J. García-Álvarez, J. Gimeno, N. Nebra and J. Rubio-García, Dalton Trans., 2006, 5593–5604 RSC.
- M. J. Pouy, S. A. Delp, J. Uddin, V. M. Ramdeen, N. A. Cochrane, G. C. Fortman, T. B. Gunnoe, T. R. Cundari, M. Sabat and W. H. Myers, ACS Catal., 2012, 2, 2182–2193 CrossRef CAS.
- X. Xu, J. Liu, L. Liang, H. Li and Y. Li, Adv. Synth. Catal., 2009, 351, 2599–2604 CrossRef CAS.
- Y. Sheng, D. G. Musaev, K. S. Reddy, F. E. McDonald and K. Morokuma, J. Am. Chem. Soc., 2002, 124, 4149–4157 CrossRef CAS PubMed.
- S. B. Moilanen and D. S. Tan, Org. Biomol. Chem., 2005, 3, 798–803 RSC.
- F. E. McDonald and C. C. Schultz, J. Am. Chem. Soc., 1994, 116, 9363–9364 CrossRef CAS.
- F. E. McDonald, K. S. Reddy and Y. Díaz, J. Am. Chem. Soc., 2000, 122, 4304–4309 CrossRef CAS.
- J. A. Varela and C. Saá, Synthesis, 2016, 48, 3470–3478 CrossRef CAS.
- T. Cai, W.-W. Li, W.-B. Qin and T.-B. Wen, Chem.–Eur. J., 2017, 24, 1606–1618 CrossRef PubMed.
- M. Mellor, A. Santos, E. G. Scovell and J. K. Sutherland, J. Chem. Soc., Chem. Commun., 1978, 528–529 RSC.
- M. D. Weingarten and A. Padwa, Tetrahedron Lett., 1995, 36, 4717–4720 CrossRef CAS.
- S. D. Wobser and T. J. Marks, Organometallics, 2013, 32, 2517–2528 CrossRef CAS.
- X. H. Yu, S. Seo and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 7244–7245 CrossRef CAS PubMed.
- J. Martínez, A. Otero, A. Lara-Sánchez, J. A. Castro-Osma, J. Fernández-Baeza, L. Sánchez-Barba and A. M. Rodríguez, Organometallics, 2016, 35, 1802–1812 CrossRef.
- C. Brinkmann, A. G. M. Barrett, M. S. Hill, P. A. Procopiou and S. Reidt, Organometallics, 2012, 31, 7287–7297 CrossRef CAS.
- D. Y. Li, K. J. Shi, X. F. Mao, Z. L. Zhao, X. Y. Wu and P. N. Liu, Tetrahedron, 2014, 70, 7022–7031 CrossRef CAS.
- J. K. Vandavasi, W.-P. Hu, G. C. Senadi, S. S. K. Boominathan, H.-Y. Chen and J.-J. Wang, Eur. J. Org. Chem., 2014, 6219–6226 CrossRef CAS.
- S. Gandhi and B. Baire, ChemistrySelect, 2018, 3, 4490–4494 CrossRef CAS.
- L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684–12693 RSC.
- A. Motta, I. L. Fragalà and T. J. Marks, Organometallics, 2010, 29, 2004–2012 CrossRef CAS.
- N. T. Coles, M. F. Mahon and R. L. Webster, Chem. Commun., 2018, 54, 10443–10446 RSC.
- S. E. Baillie, T. D. Bluemke, W. Clegg, A. R. Kennedy, J. Klett, L. Russo, M. de Tullio and E. Hevia, Chem. Commun., 2014, 50, 12859–12862 RSC.
- S. E. Baillie, W. Clegg, P. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and L. Russo, Organometallics, 2012, 31, 5131–5142 CrossRef CAS.
- C. Lichtenberg, T. P. Spaniol, L. Perrin, L. Maron and J. Okuda, Chem.–Eur. J., 2012, 18, 6448–6452 CrossRef CAS PubMed.
- P. B. Hitchcock, M. F. Lappert and A. V. Protchenko, J. Am. Chem. Soc., 2001, 123, 189–190 CrossRef CAS PubMed.
- I. Koehne, S. Bachmann, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2017, 56, 15141–15145 CrossRef CAS PubMed.
- J. Kretsch, A. Kreyenschmidt, R. Herbst-Irmer and D. Stalke, Dalton Trans., 2018, 47, 12606–12612 RSC.
- S. Neander, F. E. Tio, R. Buschmann, U. Behrens and F. Olbrich, J. Organomet. Chem., 1999, 582, 58–65 CrossRef CAS.
- A. Imberdis, G. Lefèvre, P. Thuéry and T. Cantat, Angew. Chem., Int. Ed., 2018, 57, 3084–3088 CrossRef CAS PubMed.
- S. M. Härling, S. Krieck, H. Görls and M. Westerhausen, Inorg. Chem., 2017, 56, 9255–9263 CrossRef PubMed.
- H. Abas, J. Mas-Roselló, M. M. Amer, D. J. Durand, R. R. Groleau, N. Fey and J. Clayden, Angew. Chem., Int. Ed., 2019, 58, 2418–2422 CrossRef CAS PubMed.
- R. Rochat, K. Yamamoto, M. J. Lopez, H. Nagae, H. Tsurugi and K. Mashima, Chem.–Eur. J., 2015, 21, 8112–8120 CrossRef CAS PubMed.
- S. Seo and T. J. Marks, Chem.–Eur. J., 2010, 16, 5148–5162 CrossRef CAS PubMed.
- M. Shigeno, K. Hanasaka, K. Sasaki, K. Nozawa-Kumada and Y. Kondo, Chem.–Eur. J., 2019, 25, 6077–6081 CrossRef CAS PubMed.
- G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2007, 129, 6328–6335 CrossRef CAS PubMed.
- P. B. Hitchcock, M. F. Lappert, W.-P. Leung, L. Diansheng and T. Shun, J. Chem. Soc., Chem. Commun., 1993, 1386–1387 RSC.
- W. Clegg, B. Conway, A. R. Kennedy, J. Klett, R. E. Mulvey and L. Russo, Eur. J. Inorg. Chem., 2011, 721–726 CrossRef CAS.
- P. B. Hitchcock, A. V. Khvostov and M. F. Lappert, J. Organomet. Chem., 2002, 663, 263–268 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, additional kinetic plots, NMR spectra of catalytic reactions, and characterization data for new compounds (PDF). See DOI: 10.1039/c9sc01598a |
| This journal is © The Royal Society of Chemistry 2019 |