
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical cyanomethylation via vinyl azide cascade-fragmentation†‡
James R.
Donald
 * and
Sophie L.
Berrell
* and
Sophie L.
Berrell
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: james.donald@york.ac.uk
First published on 7th May 2019
Abstract
Herein, a novel methodology for radical cyanomethylation is described. The process is initiated by radical addition to the vinyl azide reagent 3-azido-2-methylbut-3-en-2-ol which triggers a cascade-fragmentation mechanism driven by the loss of dinitrogen and the stabilised 2-hydroxypropyl radical, ultimately effecting cyanomethylation. Cyanomethyl groups can be efficiently introduced into a range of substrates via trapping of α-carbonyl, heterobenzylic, alkyl, sulfonyl and aryl radicals, generated from a variety of functional groups under both photoredox catalysis and non-catalytic conditions. The value of this approach is exemplified by the late-stage cyanomethylation of pharmaceuticals.
Introduction
The recent renaissance of synthetic organic radical chemistry has seen the development of several approaches for the introduction of nitrile functionality into molecules through the trapping of radical intermediates with a variety of closed-shell reagents. These are valuable transformations given the importance of nitriles, which are present within the structures of a number of pharmaceuticals and bioactive natural products.1 Nitriles are also widely used as directing groups in C–H activation chemistry2 and as versatile synthetic intermediates, particularly as precursors to heterocycles3 and functionality at the carboxylic acid oxidation level.4Modern methods to intercept radicals and directly install cyano groups use a range of cyanating reagents and build upon classical studies by Barton using tosyl cyanide and the eponymous Barton esters (Scheme 1A(i)).5a Alkyl examples include photoredox-catalysed deboronative cyanation5b and α-heteroatom C–H cyanation with tosyl cyanide,5c and decarboxylative cyanation with the iodane cyanobenziodoxolone (CBX).5d Enantioselective variants have achieved cyanation at benzylic positions via C–H abstraction under asymmetric copper catalysis5e and decarboxylation of N-hydroxyphthalimido esters under cooperative photoredox-asymmetric Cu catalysis;5f both methods using TMSCN as the cyanide source. The direct C–H cyanation of arenes has also been performed under photoredox catalysis, using cyanide generated from TMSCN to trap an aryl radical cation.5g
 | ||
| Scheme 1 The cyanation, cyanoethylation and cyanomethylation of radicals. | ||
The cyanoethylation of radicals exploits the well-established Giese reaction of radical conjugate addition to acrylonitrile (Scheme 1A(ii)).6a Notable recent examples feature nucleophilic alkyl and acyl radicals generated from enamines6f trifluoroborate salts,6bN-hydroxyphthalimido esters,6b–e and carboxylic acids.6g,h
In contrast to cyanation and cyanoethylation, a method in which radicals can be trapped in a cyanomethylation reaction (i.e. a two-carbon homologation process) is not known. At present, radical cyanomethylation can only be achieved via the converse approach of adding an electrophilic cyanomethyl radical to electron-rich substrates, limiting both substrate scope and the sites at which cyanomethylation is possible.7 Thus, to address this deficiency, we planned to develop a new approach that would enable the facile introduction of useful cyanomethyl groups into a broad range of substrates under mild conditions, such as via the use of visible-light driven photoredox catalysis.
To this end, 3-azido-2-methylbut-3-en-2-ol (1)8 was considered ideally suited to achieve the cyanomethylation of radicals because it encompasses two key design elements: (i) a vinyl azide which can act as a masked cyanomethyl group, and (ii) a dimethylcarbinol as a latent radical leaving group (Scheme 1B). Following radical generation from a substrate e.g. via the oxidative quenching of an excited-state photoredox catalyst (PC* → PC+1), it was anticipated that reagent 1 would intercept open-shell species to initiate a cascade process through radical addition to the olefin,9 affording adduct 2 which would readily expel dinitrogen to produce iminyl radical 3.10 Subsequent fragmentation of iminyl radical 3 through α-C–C bond cleavage and ejection of the stabilised 2-hydroxypropyl radical 4 was envisaged to drive the formation of the nitrile functionality.11 Importantly, the low oxidation potential of radical 4 [EOx1/2 = −0.61 V vs. saturated calomel electrode (SCE)]12 would potentially make reagent 1 amenable to use both under photoredox catalysis, where radical 4 could readily undergo electron transfer to the oxidised form of a photocatalyst (PC+) to close a redox-neutral oxidative quenching cycle, and in other electron transfer processes such as to another molecule of substrate R–X in a chain propagation (see proposed mechanism). Interestingly, azide 1 has previously been utilised in the ionic cyanomethylation of stabilised p-quinone methides, promoted by BF3·OEt2via a distinct mechanism.13 In this paper, we report the successful implementation of vinyl azide 1 as a new reagent for the direct cyanomethylation of a range of radicals generated from a broad variety of precursors under both photoredox-catalysed and non-photocatalysed radical generation.
Known vinyl azide 1 and novel diphenyl analogue 7 were prepared from the corresponding alkynes via Bi's Ag(I) catalysed hydroazidation methodology (Scheme 2).8 Careful control of the equivalents of water and modification of the work-up and purification procedures facilitated isolation of product 1 in 80% yield on a 60 mmol scale (6 g obtained,14 see ESI‡ for details). The cyclic voltammogram of azide 1 exhibited a single reduction process with a peak current at −1.68 V vs. SCE. The relatively large magnitude of this value suggests that direct reduction of 1via single-electron transfer is unlikely to be competitive with the proposed reaction mechanism.
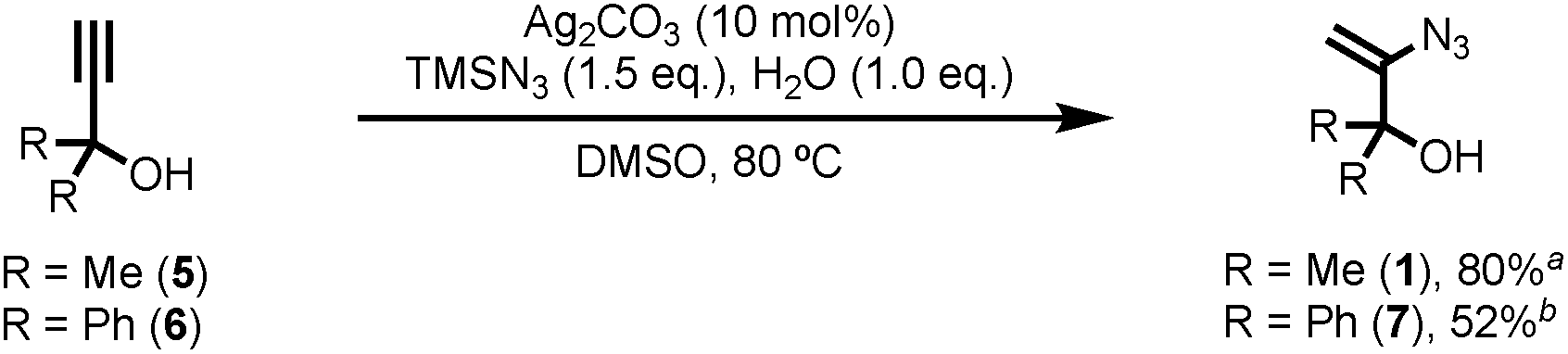 | ||
| Scheme 2 Reagent synthesis. Reaction time a2 h, b6 h. | ||
Reaction development commenced with the evaluation of vinyl azide 1 in the cyanomethylation of 2-bromoacetophenone [Ered1/2 = −1.13 V vs. SCE]15 in the presence of 2,6-lutidine and a range of photocatalysts (1.0 mol%) with strongly reducing photoexcited-states capable of inducing radical formation via spin-centre shift. All of the catalysts tested afforded cyanomethylated product 9 in high efficiency (Table 1, entries 1–3, see ESI‡ for full details). Ru(bpy)3Cl2·6H2O was selected on grounds of cost and commercial availability, providing nitrile 9 in 93% yield by 1H NMR, and 97% isolated yield on a 1.0 mmol scale. Diphenyl bearing vinyl azide 7 performed with similar efficacy in the radical cyanomethylation process (92% yield) suggesting that a family of related structures might be viable reagents for this transformation. Given that the reaction by-products from reagent 1 are simply nitrogen and acetone, it was preferred over azide 7 which liberates benzophenone, for reasons of atom-economy and purification. When run in CH2Cl2 or DMF, the reaction proceeded with efficiency comparable to using MeCN as solvent (entries 5 and 6).
|
|
||||
|---|---|---|---|---|
| Entry | Azide | Photocatalyst | Solvent | Yield of 9 (%) |
| a Reactions performed on a 0.2 mmol scale. Yields were determined by 1H NMR integration against 1,3-benzodioxole as an internal standard. b Isolated yield on a 1.0 mmol scale. c No light. d No 2,6-lutidine. e With TEMPO [(2,2,6,6-tetramethylpiperidin-1-yl)oxyl] (2.0 eq.). | ||||
| 1 | 1 | Ru(bpy)3Cl2·6H2O | MeCN | 93, 97b |
| 2 | 1 | fac-Ir(ppy)3 | MeCN | 97 |
| 3 | 1 | 4CzIPN | MeCN | 95 |
| 4 | 7 | Ru(bpy)3Cl2·6H2O | MeCN | 92 |
| 5 | 1 | Ru(bpy)3Cl2·6H2O | CH2Cl2 | 93 |
| 6 | 1 | Ru(bpy)3Cl2·6H2O | DMF | 93 |
| 7 | 1 | — | MeCN | 0 |
| 8c | 1 | Ru(bpy)3Cl2·6H2O | MeCN | 0 |
| 9d | 1 | Ru(bpy)3Cl2·6H2O | MeCN | 7 |
| 10e | 1 | Ru(bpy)3Cl2·6H2O | MeCN | 0 |
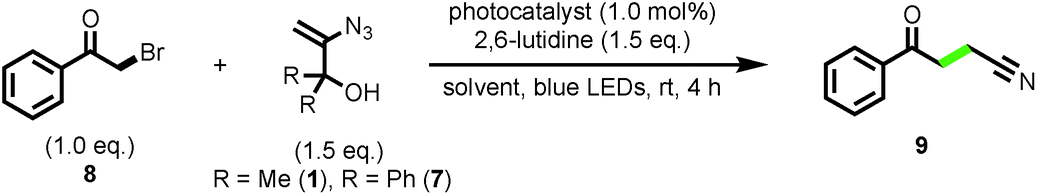
Control experiments confirmed that both photocatalyst and light were necessary for product formation, and that the yield was much lower in the absence of base – presumably due to the acid (HBr) promoted decomposition of vinyl azide 1 (entries 7–9). Performing the reaction in the presence of TEMPO (2.0 eq.) completely suppressed the formation of product 9, and lowered the conversion of bromide 8, with 89% remaining after 4 h; indicative of a radical mechanism (entry 10). Quantum yield measurements for the reactions with azides 1 and 7 (entries 1 and 4) determined values of Φ = 1.8 and Φ = 0.6, respectively; suggesting that mechanistic contributions from radical chain processes cannot be ruled out (see ESI‡ for details).16
The focus turned next to exploration of the nature of the substrates and radical intermediates that could be cyanomethylated with vinyl azide 1. Cyanomethylation of various electrophilic α-carbonyl alkyl radicals prepared from the corresponding bromides was performed in high yield with reagent 1, under photoredox catalysis (products 9–14, Scheme 3). Exchanging Ru(bpy)3Cl2·6H2O [E1/2(RuIII/RuII*) = −0.81 V vs. SCE] for the more strongly reducing photoexcited-state catalyst fac-Ir(ppy)3 [E1/2(IrIV/IrIII*) = −1.73 V vs. SCE] and increasing the reaction time afforded improved yields for the more challenging substrates 10, 11 and 14.17 Particularly pleasing was the formation of β-acetoxy ketone 10 in 88% isolated yield, without any obvious trace of elimination under the reaction conditions, highlighting the advantages of an approach which avoids the strong base mediated functionalization of MeCN.
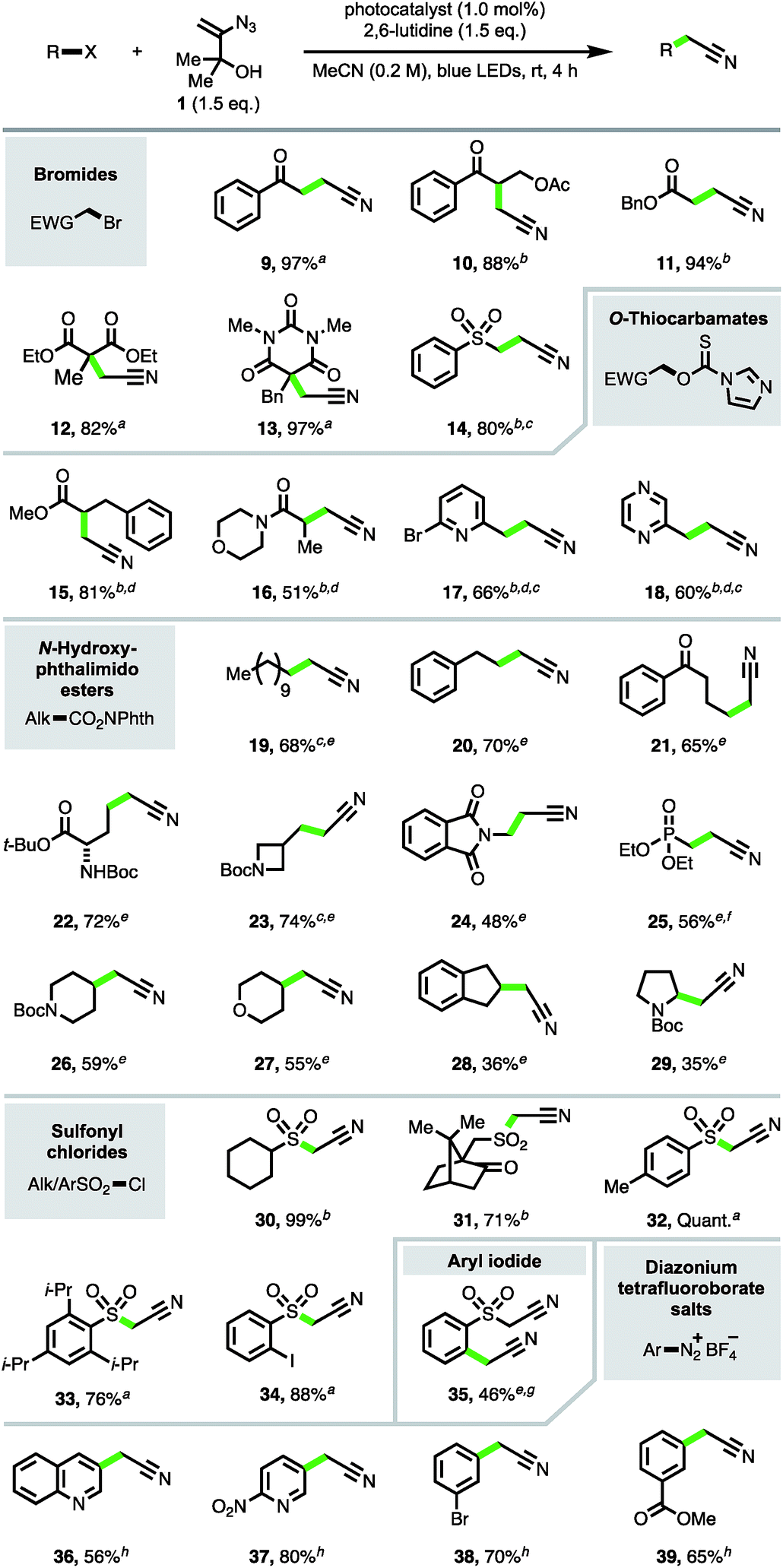 | ||
| Scheme 3 Cyanomethylation of radicals. Reaction conditions: all reactions run on a 1.0 mmol scale; aRu(bpy)3Cl2·6H2O; bfac-Ir(ppy)3; c24 h, dno 2,6-lutidine; efac-Ir(5-Fppy)3, (+)-sodium L-ascorbate (1.5 eq.); fno (+)-sodium L-ascorbate; g8 h; hno photocatalyst, no LEDs. | ||
To expand the substrate scope, we sought to utilise imidazolyl thiocarbamates as radical precursors, which have previously been applied in a Barton–McCombie deoxygenation reaction under photoredox catalysis.18 Lactic acid derivatives 15 and 16 were produced by deoxygenative cyanomethylation via interception of the intermediate α-carbonyl radicals. This approach was also successfully applied to the trapping of heterobenzylic radicals to afford β-heteroarylpropionitriles 17 and 18 in 66% and 60% yields respectively.
Next, to provide a new one-carbon homologation strategy from carboxylic acids to cyanomethyl groups, the cyanomethylation of electronically unactivated alkyl radicals generated from N-hydroxyphthalimido esters was investigated.6c,d,19 The best results were obtained with the highly reducing photocatalyst fac-Ir(5-Fppy)3 [E1/2(IrIV/IrIII*) = −1.91 V; E1/2(IrIII/IrII) = −2.18 V vs. SCE]20 in conjunction with (+)-sodium L-ascorbate, producing products 19–24 resulting from primary radicals in 48–74% yields. The addition of (+)-sodium L-ascorbate was detrimental to the formation of phosphonate product 25, likely due the lability of the β-phosphonato N-hydroxyphthalimido ester.21 Excitingly, azide 1 was also competent in intercepting secondary alkyl radicals, e.g. to produce cyanomethyl compounds 26–28, and even afforded product 29 derived from trapping of the electron-rich N-Boc pyrrolidinyl radical intermediate, albeit in a modest yield.
Sulfonyl radicals were also efficiently trapped by reagent 1 (products 30–34), providing a direct access to α-sulfonyl acetonitriles from sulfonyl chlorides and obviating the typical synthetic procedure involving reduction to the intermediate sulfinate followed by alkylation with a halo-acetonitrile reagent.22 Resubjection of iodo-α-sulfonyl acetonitrile 34 to the reaction in the presence of fac-Ir(5-Fppy)3 and (+)-sodium L-ascorbate afforded the di-cyanomethylated product 35 in 46% yield. This result highlighted that aryl radicals can participate in the cyanomethylation reaction to afford arylacetonitriles,23 which are valuable synthetic precursors to heterocyclic structures,3e and that sequential radical cyanomethylation is possible, with radical formation gated by the redox potentials of the functional groups involved.24
To further scope the trapping of aryl radicals with vinyl azide 1, aryl diazonium salts were explored as radical precursors.25 Reaction screening of phenyldiazonium tetrafluoroborate with reagent 1 revealed that the addition of 2,6-lutidine alone was sufficient to produce aryl radical intermediates, affording phenylacetonitrile in 52% yield.26 The conditions provided convenient access to substituted arylacetonitriles 36–39 under mild conditions from the corresponding aryldiazonium tetrafluoroborates.
Finally, to demonstrate the cyanomethylation of radicals in more complex settings, the late stage functionalisation of pharmaceutical agents was undertaken. The N-hydroxyphthalimido ester derivative of the nonsteroidal anti-inflammatory (NSAID) oxaprozin (40)27a was subjected to a decarboxylative cyanomethylation, yielding homologated nitrile 41 in 63% isolated yield (47% from oxaprozin, Scheme 4(i)). Secondly, the sulfonyl chloride derivative of the diuretic meticrane (42)27b was readily prepared by heating in chlorosulfonic acid; this isolated intermediate was efficiently cyanomethylated with reagent 1 under photoredox catalysis, affording sulfonylacetonitrile 43 in 80% yield (Scheme 4(ii)). Lastly, the aniline bearing aminoglutethimide (44) was selected for modification, a compound that acts as a steroidogenesis inhibitor used for the treatment of Cushing's syndrome,27c seizures and a number of cancers.27d Following diazotization, the isolated salt was efficiently cyanomethylated under the mild reaction conditions (Scheme 4(iii)). These examples help to highlight the diversity of functional groups widely found within medicinally relevant compounds that after activation, can be employed as substrates for the radical cyanomethylation procedure.
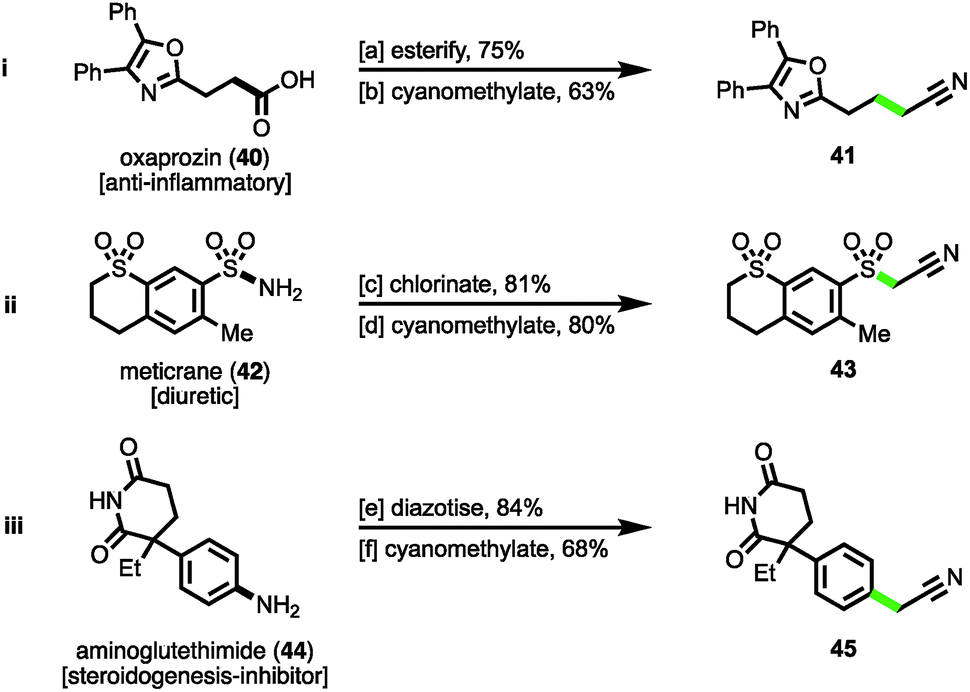 | ||
| Scheme 4 Late-stage cyanomethylation of pharmaceuticals. Reaction conditions: aoxaprozin (40), PhthNOH, DCC, DMAP, CH2Cl2, rt, 16 h; bN-hydroxyphthalimido ester (1.0 mmol), 1 (1.5 mmol), fac-Ir(5-Fppy)3 (0.01 mmol), DMSO (0.2 M), blue LEDs, rt, 24 h; cmeticrane (42), ClSO3H, 100 °C, 2 h; dsulfonyl chloride (1.0 mmol), 1 (1.5 mmol), 2,6-lutidine (1.5 mmol), Ru(bpy)3Cl2·6H2O (0.01 mmol), MeCN (0.2 M), blue LEDs, rt, 4 h; eaminoglutethimide (44), NaNO2, aq. HBF4, H2O, 0 °C, 30 min; fdiazonium salt (1.0 mmol), 1 (1.5 mmol), 2,6-lutidine (1.5 mmol), MeCN (0.2 M), rt, 4 h. | ||
Conclusions
In conclusion, by exploiting the radical decomposition of functionalised vinyl azide 1via loss of dinitrogen and fragmentation of the resultant iminyl radical, a cascade-fragmentation approach towards the cyanomethylation of radicals has been developed. Reagent 1 is readily prepared on scale and can be used to intercept α-carbonyl, heterobenzylic, alkyl, sulfonyl and aryl radicals prepared from a range of precursors under both photoredox catalysis and more classical radical generation. This methodology facilitates access to synthetically versatile cyanomethyl groups without the need for cyanide or strong base, under mild conditions, making it amenable to the derivatisation of more complex substrates as demonstrated in the late-stage cyanomethylation of pharmaceutical agents. Further exploration of this reactivity pattern in the design of reagents with which to trap radicals is on-going and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Elsevier (J. R. D.) and the EPSRC Dial-a-Molecule Network (EP/P007589/1) for financial support, and Prof. Richard J. K. Taylor (University of York) for insightful discussions.Notes and references
- For reviews, see: (a) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 RSC; (b) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
- For a review, see: Y. Ping, L. Wang, Q. Ding and Y. Peng, Adv. Synth. Catal., 2017, 359, 3274–3291 CrossRef CAS.
- For selected reviews, see: (a) Z. Rappoport, Chemistry of the Cyano Group, Wiley, London, 1970 Search PubMed; (b) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085–1094 RSC; (c) M. D. Hill, Chem.–Eur. J., 2010, 16, 12052–12062 CrossRef CAS PubMed; tetrazole synthesis: (d) J. Roh, K. Vávrová and A. Hrabálek, Eur. J. Org. Chem., 2012, 6101–6118 CrossRef CAS; (e) P. J. Lindsay-Scott and P. T. Gallagher, Tetrahedron Lett., 2017, 58, 2629–2635 CrossRef CAS.
- For selected reviews see: (a) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, VCH, New York, 1989 Search PubMed; (b) A. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef.
- (a) D. H. R. Barton, J. C. Jaszberenyi and E. A. Theodorakis, Tetrahedron, 1992, 48, 2613–2626 CrossRef CAS; (b) J.-J. Dai, W.-M. Zhang, Y.-J. Shu, Y.-Y. Sun, J. Xu, Y. S. Feng and H. J. Xu, Chem. Commun., 2016, 52, 6793–6796 RSC; (c) T. Wakaki, K. Sakai, T. Enomoto, M. Kondo, S. Masaoka, K. Oisaki and M. Kanai, Chem.–Eur. J., 2018, 24, 8051–8055 CrossRef CAS PubMed; (d) F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790–1800 RSC; (e) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed; (f) D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632–15635 CrossRef CAS PubMed; (g) J. B. McManus and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 2880–2883 CrossRef CAS PubMed.
- (a) B. Giese, J. A. González-Gómez and T. Witzel, Angew. Chem., Int. Ed. Engl., 1984, 23, 69–70 CrossRef; (b) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef CAS; (c) G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef CAS PubMed; (d) Y. Slutskyy and L. E. Overman, Org. Lett., 2016, 18, 2564–2567 CrossRef CAS PubMed; (e) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS PubMed; (f) J. A. Terrett, M. D. Clift and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 6858–6861 CrossRef CAS PubMed; (g) A. Millett, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464–13468 CrossRef PubMed; (h) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830–4833 CrossRef CAS PubMed.
- For examples of generation of the electrophilic cyanomethyl radical from bromoacetronitrile and addition to electron-rich closed-shell substrates see: (a) E. R. Welin, A. A. Warkentin, J. C. Conrad and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 9668–9672 CrossRef CAS PubMed; (b) Q. Chang, Z. Liu, P. Liu, L. Yu and P. Sun, J. Org. Chem., 2017, 82, 5391–5397 CrossRef CAS PubMed; (c) C. J. O'Brien, D. G. Droege, A. Y. Jiu, S. S. Ghandi, N. A. Paras, S. H. Olson and J. Conrad, J. Org. Chem., 2018, 83, 8926–8935 CrossRef PubMed; for a review of radical cyanomethylation initiated by C–H abstraction from acetonitrile, see: (d) X.-Q. Chu, D. Ge, Z.-L. Xhen and T.-P. Loh, ACS Catal., 2018, 8, 258 CrossRef CAS.
- Z. Liu, J. Liu, L. Zhang, P. Liao, J. Song and X. Bi, Angew. Chem., Int. Ed., 2014, 53, 5305–5309 CrossRef CAS PubMed.
- For examples of our own work featuring radical cascades, see: (a) W. F. Petersen, R. J. K. Taylor and J. R. Donald, Org. Lett., 2017, 19, 874–877 CrossRef CAS PubMed; (b) W. F. Petersen, R. J. K. Taylor and J. R. Donald, Org. Biomol. Chem., 2017, 15, 5831–5845 RSC.
- For the original example of this reactivity, see: (a) A. Suzuki, M. Tabata and M. Ueda, Tetrahedron Lett., 1975, 2195–2198 CrossRef CAS; for elucidation of the mechanism, see: (b) A. F. Bamford, M. D. Cook and B. P. Roberts, Tetrahedron Lett., 1983, 3779–3782 CrossRef CAS; for reviews, see: (c) B. Hu and S. G. DiMagno, Org. Biomol. Chem., 2015, 13, 3844–3855 RSC; (d) J. Fu, G. Zanoni, E. A. Anderson and X. Bi, Chem. Soc. Rev., 2017, 46, 7208–7228 RSC.
- For examples of iminyl radical α-C–C bond cleavage under photoredox catalysis, see: (a) L. Li, H. Chen, M. Mei and L. Zhou, Chem. Commun., 2017, 53, 11544–11547 RSC; (b) X.-Y. Yu, J.-R. Chen, P.-Z. Wang, M.-N. Yang, D. Liang and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738–743 CrossRef CAS PubMed; (c) E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS PubMed; (d) C. Zhu, F. Chen, C. Liu, H. Zeng, Z. Yang, W. Wu and H. Jiang, J. Org. Chem., 2018, 83, 14713–14722 CrossRef CAS PubMed; for a review, see: (e) J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem.–Eur. J., 2018, 24, 12154–12163 CrossRef CAS PubMed.
- T. Lund, D. D. M. Wayner, M. Jonsson, A. G. Larsen and K. Daasbjerg, J. Am. Chem. Soc., 2001, 123, 12590–12595 CrossRef CAS.
- C. Lin, Y. Shen, B. Huang, Y. Liu and S. Cui, J. Org. Chem., 2017, 82, 3950–3956 CrossRef CAS PubMed.
- Whilst low molecular weight organic azides can potentially be explosive, we have observed no issues of instability in either the handling or storage of reagent 1. This compound fulfills the ‘rule of six’: six carbons (or heavier atoms) per energetic functional group, which has been empirically observed to provide sufficient dilution to render such compounds relatively inert. See: H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed . To probe the stability of compound 1, a 20 mg sample was heated on an aluminium block. After melting at 33 °C, the material appeared stable in the liquid state with no visible decomposition up to ca. 100 °C, whereupon it had evaporated.
- E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322–3330 CrossRef CAS PubMed.
- Φ values are the average of three experiments. See: M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- (a) Data for Ru(bpy)3·6H2O: C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef CAS; (b) Data for fac-Ir(ppy)3: L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- L. Chenneberg, A. Baralle, M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2014, 356, 2756–2762 CrossRef CAS.
- (a) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS; (b) G. Pratsch, G. L. Lackner and L. E. Overman, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS PubMed.
- Data for fac-Ir(5-Fppy)3: K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- 31% yield with (+)-sodium L-ascorbate (1.5 eq.), 61% yield without, as determined by 1H NMR analysis against 1,3-benzodioxole as an internal standard.
- For recent examples, see: (a) J. Kim, J. Kwon, D. Lee, S. Jo, D.-S. Park, J. Choi, E. Park, J. Y. Hwang, Y. Ko, I. Choi, M. K. Ju, J. Ahn, J. Kim, S.-J. Han, T.-H. Kim, J. Cechetto, J. Nam, S. Ahn, P. Sommer, M. Liuzzi, Z. No and J. Lee, Bioorg. Med. Chem. Lett., 2013, 23, 153–157 CrossRef CAS PubMed; (b) G. Barker, R. Davenport, R. Downham, W. Farnaby, A. Goldby, D. Hannah, D. Harrison and H. Willems, WO 198045 A1, (Takeda) 2015.
- For selected recent approaches to arylacetonitriles, see: (a) J. Velcicky, A. Soicke, R. Steiner and H.-G. Schmalz, J. Am. Chem. Soc., 2011, 133, 6948–6951 CrossRef CAS; (b) P. J. Lindsay-Scott, A. Clarke and J. Richardson, Org. Lett., 2015, 17, 476–479 CrossRef CAS PubMed; (c) Y. Satoh and Y. Obora, RSC Adv., 2014, 4, 15736–15739 RSC.
- As determined by cyclic voltammetry, 2-Iodobenzenesulfonyl chloride is reduced at −0.66 V vs. SCE, this reactivity is attributed to the sulfonyl chloride and lies within the ‘redox window’ of Ru(bpy)3Cl2·6H2O [E1/2(RuIII/RuII*) = −0.81 V vs. SCE]. 2-Iodo-α-sulfonyl acetonitrile 34 exhibits reductions at −1.19 V and −1.62 V vs. SCE, within the ‘redox-window’ of fac-Ir(5-Fppy)3 [E1/2(IrIV/IrIII*) = −1.91 V; E1/2(IrIII/IrII) = −2.18 V vs. SCE] in the presence of (+)-sodium L-ascorbate. Potentials were measured vs. Ag/AgCl (3.0 M KCl) and converted to vs. SCE (see ESI‡ for details).
- For a review of photoredox-catalysed arylation, see: I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- (a) R. A. Abramovitch and J. G. Saha, Tetrahedron, 1965, 21, 3297–3303 CrossRef CAS; (b) M. Gurczynski and P. Tomasik, Org. Prep. Proced. Int., 1991, 23, 438–441 CrossRef CAS; (c) T. Sakakura, M. Hara and M. Tanaka, J. Chem. Soc., Perkin Trans. 1, 1994, 283–288 RSC; (d) G. Iakobson, J. Du, A. M. Z. Slawin and P. Beier, Beilstein J. Org. Chem., 2015, 11, 1494–1502 CrossRef CAS PubMed.
- (a) K. McCormack and K. Brune, Drugs, 1991, 41, 533–547 CrossRef CAS PubMed; (b) I. V. Ukrainets and N. L. Bereznyakova, Chem. Heterocycl. Compd., 2012, 48, 155–165 CrossRef CAS; (c) D. E. Schteingart, Expert Opin. Emerging Drugs, 2009, 14, 661–671 CrossRef CAS PubMed; (d) R. J. Santen, H. Brodie, E. R. Simpson, P. K. Siiteri and A. Brodie, Endocr. Rev., 2009, 30, 343–375 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Richard J. K. Taylor on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental protocols, cyclic voltammetry, quantum yield measurements and spectral data. See DOI: 10.1039/c9sc01370a |
| This journal is © The Royal Society of Chemistry 2019 |