
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZn(OTf)2-mediated annulations of N-propargylated tetrahydrocarbolines: divergent synthesis of four distinct alkaloidal scaffolds†
Sadaiwa
Yorimoto
a,
Akira
Tsubouchi
a,
Haruki
Mizoguchi
b,
Hideaki
Oikawa
 c,
Yoshiaki
Tsunekawa
c,
Tomoya
Ichino
c,
Satoshi
Maeda
*cd and
Hiroki
Oguri
*a
c,
Yoshiaki
Tsunekawa
c,
Tomoya
Ichino
c,
Satoshi
Maeda
*cd and
Hiroki
Oguri
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Nakacho, Koganei, Tokyo 184-8588, Japan. E-mail: h_oguri@cc.tuat.ac.jp
bGraduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushimanaka, Kita-ku, Okayama 700-8530, Japan
cDepartment of Chemistry, Faculty of Science, Hokkaido University, Kita-ku Kita 10 Jo Nishi 8 Chome, Sapporo 060-0810, Japan. E-mail: smaeda@eis.hokudai.ac.jp
dInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Sapporo 001-0021, Japan
First published on 1st May 2019
Abstract
Intramolecular hydroarylations of N-propargylated tetrahydrocarbolines were efficiently mediated using a unique combination of Zn(OTf)2 with t-BuOH under neutral conditions. Use of the artificial force induced reaction method in the global reaction route mapping strategy provided insights into the Zn(OTf)2-mediated hydroarylations and the associated intriguing solvent effects of t-BuOH facilitating a protodezincation process without a Brønsted acid activator. We systematically implemented three distinct hydroarylations as well as an unanticipated α-alkenylation of a carbonyl group to obtain the four alkaloidal scaffolds 2–4, and 18. Zn(OTf)2-mediated annulation of 1c proceeded through kinetic formation of the spiroindole 3c followed by an alkenyl shift and concomitant retro-Mannich-type fragmentation to furnish azepino[4,5-b]indole 2 framework. Substituents on substrate 1 in the vicinity of the reaction sites substantially affected the mode of the divergent annulations. Judicious choices of the substituents, solvent and reaction conditions enabled programmable divergent synthesis of the four distinct skeletons.
Introduction
The activation of alkynes with transition metal mediators used as “π-acids” enables efficient carbon–carbon or carbon-heteroatom bond-forming reactions. Whilst various metal catalysts have been reported for the hydroarylation of alkynes,1 synthetic approaches employing zinc(II) reagents for the activation of alkynes currently remain very limited.2 Herein we report Zn(OTf)2-mediated divergent cyclizations of N-propargylated tetrahydrocarbolines 1 towards programmable synthesis of the four distinct skeletons 2–5, reminiscent of naturally occurring indole alkaloids (Fig. 1a and b). This study provides rare examples of the activation of alkynes with zinc(II) reagents in protic solvent (t-BuOH) under almost neutral conditions without Brønsted acid promoters. Exploiting the artificial force induced reaction (AFIR) method in the global reaction route mapping strategy (GRRM),3 we postulated that participation of t-BuOH as a proton donor dramatically facilitates the protodezincation process to promote the zinc(II)-mediated hydroarylations. Furthermore, divergent annulations to access the four scaffolds 2–5 were systematically implemented via appropriate choices of the substituents on 1, as well as the solvent and reaction conditions.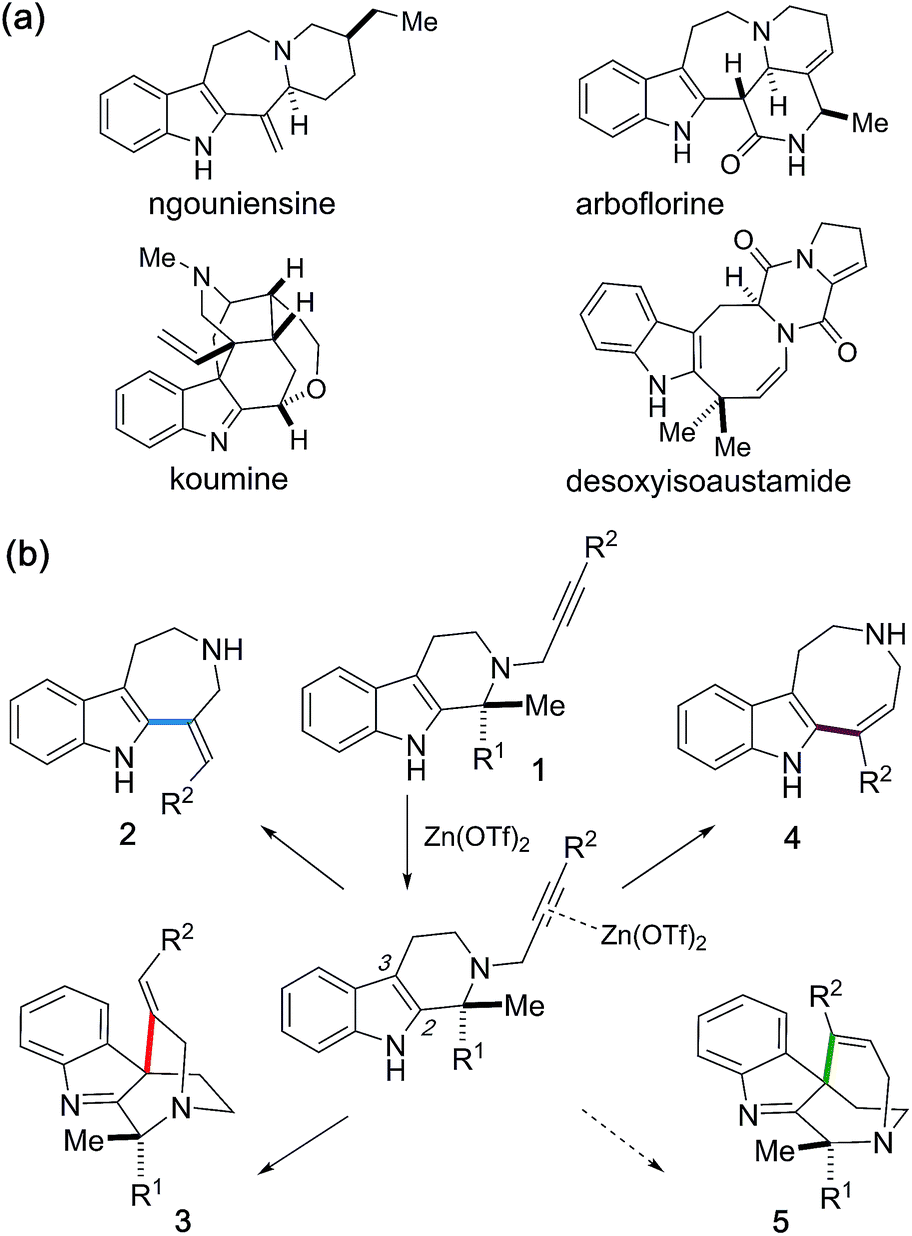 | ||
| Fig. 1 (a) Naturally occurring alkaloids. (b) Zn(OTf)2-mediated divergent cyclizations of 1 for systematic generation of four distinct scaffolds 2–5. | ||
We incidentally discovered the zinc(II)-mediated ring-expansion reactions of N-propargylated tetrahydrocarbolines involving intramolecular hydroarylation of alkynes during the course of our synthetic studies on terpene indole alkaloids.4 Since the hetero-conjugate addition of 6 to methyl propiolate and subsequent Hofmann elimination via the resulting zwitterionic intermediate smoothly proceeded at room temperature to generate 7,4a we originally aimed to conduct essentially the same conversion employing N-propargylated tetrahydrocarboline 1a in place of 6 to afford 7 (Fig. 2a). Contrary to the smooth conversion of 6 (a β-amino acid substructure), the corresponding transformation with the sterically more congested 1a bearing an α, α-disubstituted amino acid moiety resulted in no reaction. To facilitate the initial hetero-conjugate addition of 1a to methyl propiolate, ZnBr2 was added as an oxophilic Lewis acid that could activate methyl propiolate through coordination to the ester moiety. Although we failed to achieve the intended conversion leading to 7, this attempt led to the unexpected discovery of Zn(II)-mediated ring expansion reactions to forge azepino[4,5-b]indole 8 and azocino[5,4-b]indole 9 scaffolds in modest yields (Fig. 2b). We anticipated that the seven and eight-membered rings 2a and 4a could be formed via hydroarylation reactions between the indole C2 position and either the internal or terminal carbon of the alkyne with hydrolytic release of the methyl pyruvate unit. Subsequent hetero-conjugate additions of the resulting secondary amines 2a and 4a to methyl propiolate would yield 8 and 9, respectively.
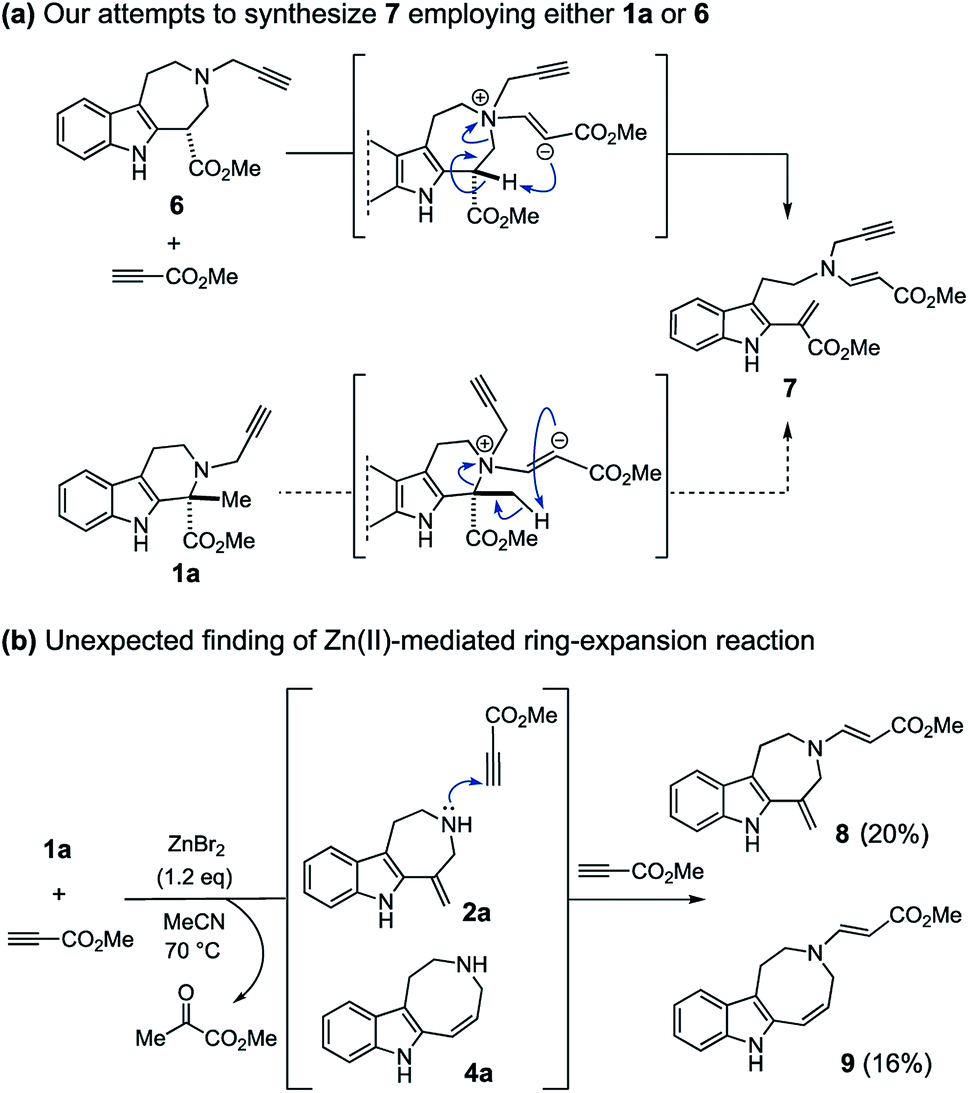 | ||
| Fig. 2 (a) The reaction of 6/1a with methyl propiolate in the absence of metal promoter. (b) Incidental discovery of the Zn(II)-mediated ring-expansion reactions. | ||
As a pioneering example of intramolecular hydroarylation employing an indole group, Echavarren reported both cationic Au(I)-catalyzed 7-exo-dig (10 → 11) and Au(III)-catalyzed 8-endo-dig (10 → 12) cyclizations (Fig. 3).5 Van der Eycken devised a regio-controlled annulation (13 → 14) employing cationic Au(I) catalyst to construct the functionalized azocino[5,4-b]indole scaffold.6 Recently, Wang and co-workers reported Au(I)-catalyzed cyclizations of 15 having either an internal or terminal alkyne to form azocino[5,4-b]indole 16 and spirocyclic scaffold 17, respectively.7 Addition of methanesulfonic acid (MsOH) as the Brønsted acid promoter was reported to be essential for complete conversion of 15. Whilst Au(I)/Au(III)-catalyzed intramolecular hydroarylations of related indole systems have been developed,8 there are very few examples of similar conversions employing zinc(II)9 and other metal promoters.10 Given the advantages of zinc reagents (e.g., their abundance, low cost, low toxicity, biological relevance, distinct catalytic abilities, and functional group tolerance), zinc-mediated reactions could provide a sustainable and environmentally benign alternative to the use of more expensive or toxic transition metals.
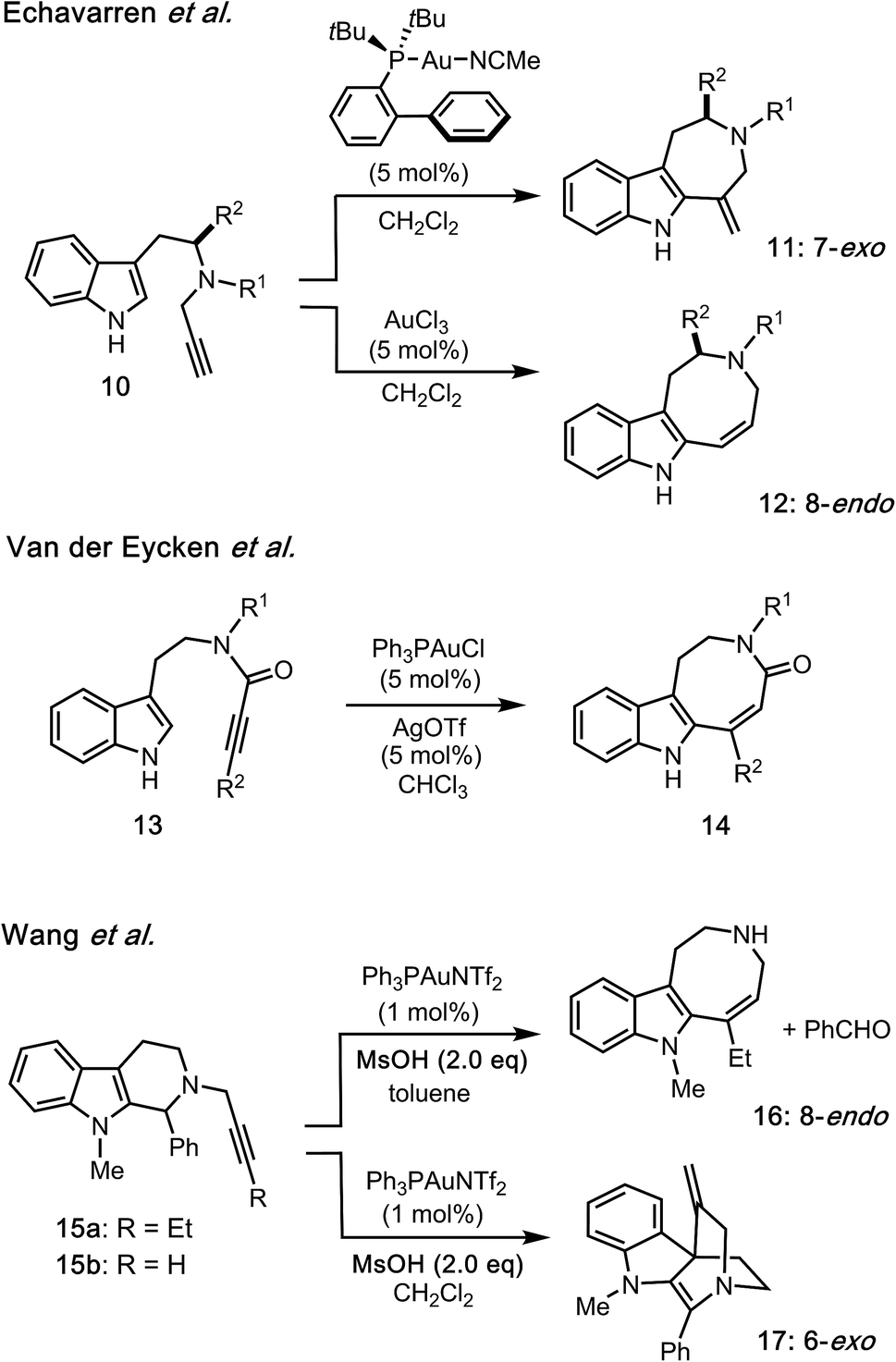 | ||
| Fig. 3 Au(I)/Au(III)-catalyzed intramolecular hydroarylations of alkynes with indoles. | ||
Results and discussion
Screening of Zn(II)-mediated cyclizations
Intrigued by the unexpected finding shown in Fig. 2b, zinc reagents and solvents were screened to improve the ring-expansion reaction of 1a to form 2a (Table 1). The combination of Zn(OTf)2 in t-BuOH appeared to be optimal. The cyclizations mediated by ZnCl2, ZnBr2, ZnI2, and Zn(OAc)2 resulted in poor or moderate conversions (entries 1–4). Other zinc(II) salts composed of counter anions with highly electron-withdrawing properties, such as Zn(NTf2)2 and Zn(NO3)2, were slightly less effective compared to Zn(OTf)2 (entries 5–7).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | ZnX2 | Solvent | Time | Yieldb (%) | Recovery (%) | ||
| 2a | 3a | 4a | |||||
| a All reactions were performed using 1a (0.2 mmol) and Zn(OTf)2 (0.2 mmol) in 1 mL of each solvent at 60 °C. b Determined by 1H-NMR analysis. | |||||||
| 1 | ZnCl2 | t-BuOH | 24 | 5 | 30 | 22 | 23 |
| 2 | ZnBr2 | t-BuOH | 24 | 6 | 35 | 26 | 4 |
| 3 | ZnI2 | t-BuOH | 24 | 8 | 37 | 19 | — |
| 4 | Zn(OAc)2 | t-BuOH | 24 | — | — | — | 99 |
| 5 | Zn(NTf2)2 | t-BuOH | 24 | 33 | — | 6 | — |
| 6 | Zn(NO3)2b | t-BuOH | 24 | 46 | 11 | 20 | 15 |
| 7 | Zn(OTf) 2 | t-BuOH | 1 | 76 | 13 | 6 | — |
| 8 | Zn(OTf)2 | MeCN | 6 | 18 | 5 | 2 | — |
| 9 | Zn(OTf)2 | THF | 24 | 45 | 6 | 8 | — |
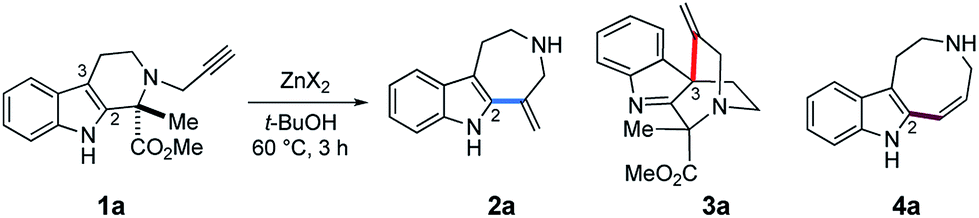
The Zn(OTf)2-mediated hydroarylation of 1a was greatly affected by the reaction solvent. The use of polar aprotic solvents such as acetonitrile and tetrahydrofuran resulted in decreased yields of 2a (entries 8–9). In contrast, use of the sterically demanding alcoholic solvent t-BuOH dramatically increased the reaction rate (entry 7). The substrate 1a was completely consumed within 1 hour to afford 2a in 76% yield as the major product along with formation of byproducts 3a (13%) and 4a (6%) bearing spirocyclic and azocino[5,4-b]indole scaffolds, respectively. The use of methanol, ethanol, DMF, or DMSO as solvent resulted in poor conversions.
Zn(OTf)2-mediated cyclization of substrate 1a
Based on the above screening results, we performed Zn(OTf)2-mediated cyclizations under the optimized conditions employing t-BuOH as a solvent in the presence of MS4A (Fig. 4). The use of molecular sieves allowed consistent reproduction of the experimental results, since Zn(OTf)2-mediated conversions in t-BuOH were noticeably affected by trace amounts of water. Due to the practical difficulty of isolating 2a, isolated yields of the products were reported after benzenesulfonylation of the resultant secondary amino group of 2a, in which the major product 2b (67%), along with 4b (6%) and 3a (6%), were obtained via 7-exo, 8-endo ring-expansion reactions and spiro-cyclization, respectively. In this paper, we adopted terms, such as 6-exo, 7-exo, and 8-endo, for indication of the formal cyclization modes regardless of the actual reaction mechanisms.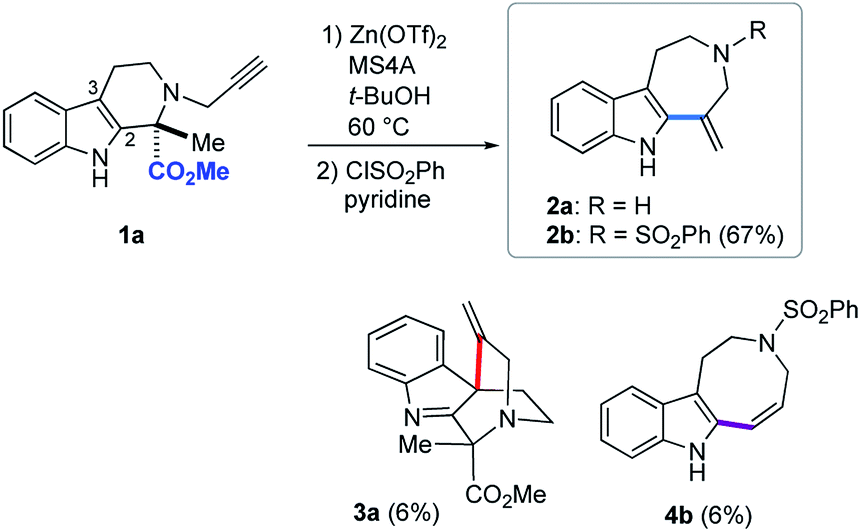 | ||
| Fig. 4 Zn(OTf)2-mediated hydroarylation of 1a. | ||
Mechanistic insights into Zn(OTf)2-mediated hydroarylation
We performed DFT calculations to gain mechanistic insights into the Zn(OTf)2-mediated cyclizations. Stable and transition state (TS) structures were systematically identified using the artificial force induced reaction (AFIR) method implemented in the GRRM program.3,11,12 All structures were optimized at the ωB97X-D/Def2-SVP level of theory and the solvent effects of t-BuOH were evaluated by the solvation model based on density (SMD) method.We will discuss the initial exo-/endo-cyclization of 1a (Fig. 5): the exo-cyclization affords either 2a or 3a, whereas endo-cyclization gives 4a. The propargyl group in 1a coordinates to Zn(OTf)2 to form the pre-reaction complex IM0. A systematic TS structure search identified TS1-i and TS1-ii as the lowest TS structures for the exo- and endo-cyclizations, respectively.
 | ||
| Fig. 5 Two intramolecular cyclization pathways. Gibbs energies (T = 333.15 K) relative to the sum of the Gibbs energies for separately calculated 1a and Zn(OTf)2 are shown in kJ mol−1. | ||
In these TS structures, one of the alkynyl-carbon atoms bridges the indole C2 and C3 atoms. Fig. 5 shows that exo-cyclization viaTS1-i is kinetically preferred over endo-cyclization viaTS1-ii. Our search of the TS structures identified 24 TSs for the exo-cyclization and 34 TSs for the endo-cyclization reactions (see ESI, Fig. S2†). The Boltzmann distribution of these 58 TSs suggests that the ratio between the number of molecules that undergo exo- and endo-cyclization is 80.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19.9. Indeed, 4a was a minor product in our experiment.
:19.9. Indeed, 4a was a minor product in our experiment.
In this study, we focused on the fate of molecules that undergo the kinetically preferred exo-cyclization reaction. As illustrated in Fig. 6, an intrinsic reaction coordinate (IRC) calculation from TS1-i provided IM1 and IM2 as the two end-points. IM2 can isomerize to IM1 through the relatively low barrier TS2. IM3-ii is then generated through a C–C bond scission and subsequent solvent coordination. IM3-ii could be a resting state, since it has the lowest energy of any structure along this path. IM3-ii undergoes protodezincation to yield IM4 and t-BuO−–[ZnII]. IM4 is expected to be hydrolyzed to 2a in the workup procedure.13
 | ||
| Fig. 6 (a) Reaction mechanism for the two intramolecular cyclization pathways. (b) Energy diagrams for the two cyclization pathways. Gibbs energies (T = 333.15 K) relative to the sum of the Gibbs energies for separately calculated 1a and Zn(OTf)2 are shown in kJ mol−1. | ||
The process which converts IM2 to 3a requires t-BuO−–[ZnII] released in the protodezincation processes. The t-BuO− part of t-BuO−–[ZnII] abstracts a proton from the indole N–H moiety. Then, solvent coordination and subsequent protodezincation afford 3a. Notably, all obtained paths not involving t-BuO−–[ZnII] possess much higher barriers than this path, as shown in Fig. S5 (ESI†).
It was previously suggested that dynamical path bifurcation occurs in the exo-/endo-cyclization step of 1a when the process is catalyzed by an Au catalyst,7 and thus we searched for the valley–ridge transition (VRT) point14 along the IRC path from TS1. As shown in Fig. S6 (ESI†), the VRT point was identified, indicating that dynamical path bifurcation indeed occurs in the present system. In other words, the elementary process through TS1 may give not only IM2, but also a certain ratio of IM1, although quantitative determination of this ratio through extensive MD simulations is beyond the scope of this study.
We conducted a kinetic simulation to illustrate the time evolution of the population of each species, with the initial population comprising exclusively IM2. The rate equations shown in ESI† were solved numerically, and the time evolution of the population of each species in 1 hour was computed. Details of this simulation are described in ESI.†Fig. 7 shows the time evolution of the population of intermediates and products. The initial population (IM2) decreases within 1 μs, and simultaneously, the population of IM3-ii increases. As mentioned above, IM3-ii is a resting state and is dominant until around 100 s. Finally, the populations of IM4 and 3a increase gradually until 3600 s, to a final ratio of IM4:3a = 84:8, consistent with the experimental yields of 2a (76%; formed from IM4) and 3a (13%), respectively.
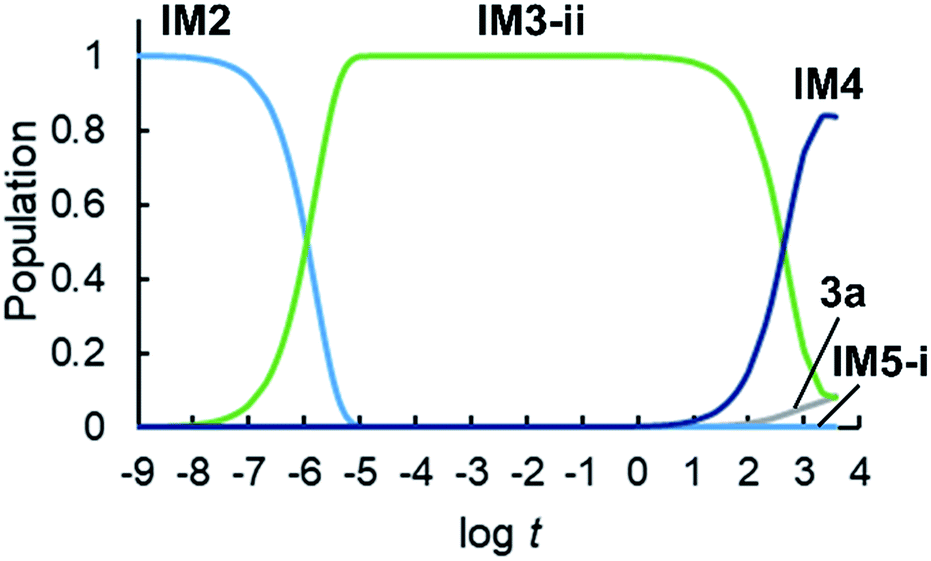 | ||
| Fig. 7 Time evolution of the population of intermediates and products in 3600 s. | ||
Au(I)/Au(III)-catalyzed hydroarylations of alkynes often necessitate a Brønsted acid activator such as methanesulfonic acid (Fig. 3).7 In sharp contrast, reaction conditions employing the unique combination of Zn(OTf)2 and t-BuOH significantly accelerated the intramolecular cyclizations, facilitating the corresponding protodemetallation process under almost neutral conditions. Based on this calculation, the corresponding protodezincation reaction (IM3-ii → IM4) has been demonstrated to have the largest activation energy and thus is the rate-determining step. This computational analysis offers hitherto unmarked mechanistic insights into the participation of t-BuOH in Zn(II)-mediated hydroarylation reactions via transition states (TS4 and TS5) and may provide a rational explanation for our experimental findings of the intriguing solvent effects resulting in substantial rate accelerations.
We verified the involvement of the cationic species IM3-ii, a putative resting state, in the reaction pathway leading to 2a by conducting the Zn(OTf)2-mediated ring-expansion reaction in the presence of Et3SiH (Fig. 8). This attempt resulted in the isolation of the corresponding product 2d (13% yield) via reduction of the iminium cation corresponding to IM3-ii along with 2b (67%). Despite the modest yield of 2d, this experimental result is consistent with the proposed mechanism.
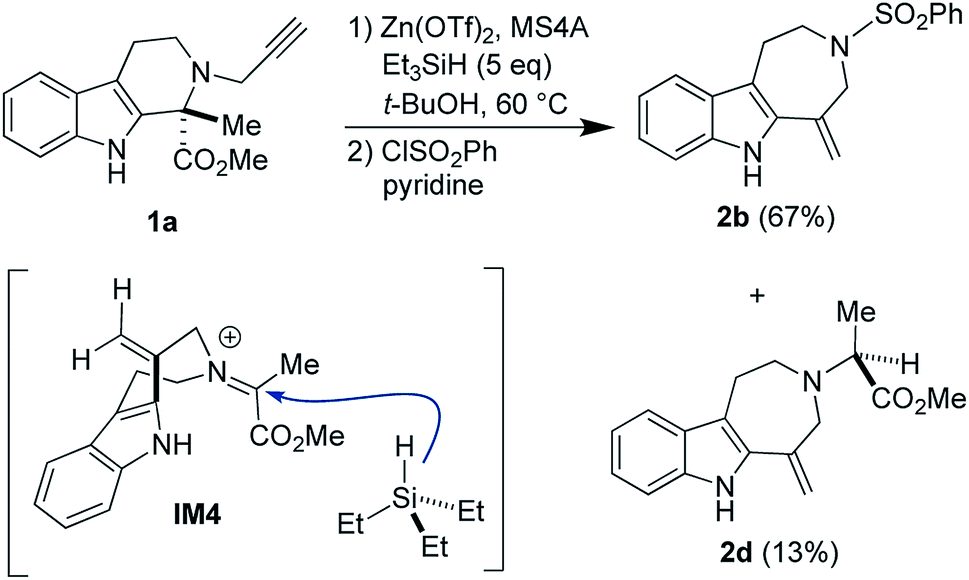 | ||
| Fig. 8 An attempt to trap the intermediate IM4 to form 2d. | ||
Zn(OTf)2-mediated annulations with distinct substrates
Next, the substrates were modified (Fig. 9) not only to explore the scope of potential substrates but also to change the annulation modes for gaining divergent access to distinct skeletons. To this end, we designed and synthesized substrate 1b, which bears a benzyl amide group in place of the methyl ester substituent of 1a. Zn(OTf)2-mediated cyclization of 1b at 60 °C in t-BuOH furnished 3b with a quaternary carbon center via 6-exo dearomatizing spirocyclization in 60% yield as the major product. This reaction is in sharp contrast to the conversion of substrate 1a to 2bvia 7-exo cyclization shown in Fig. 4. Annulations of 1c possessing a gem-dimethyl moiety resulted in the formation of approximately equimolar amounts of 2b (44%) and 3c (44%). The relative stereochemistries of the spirocyclic products 3a–c were determined based on either NOE or X-ray analysis (Fig. 9).15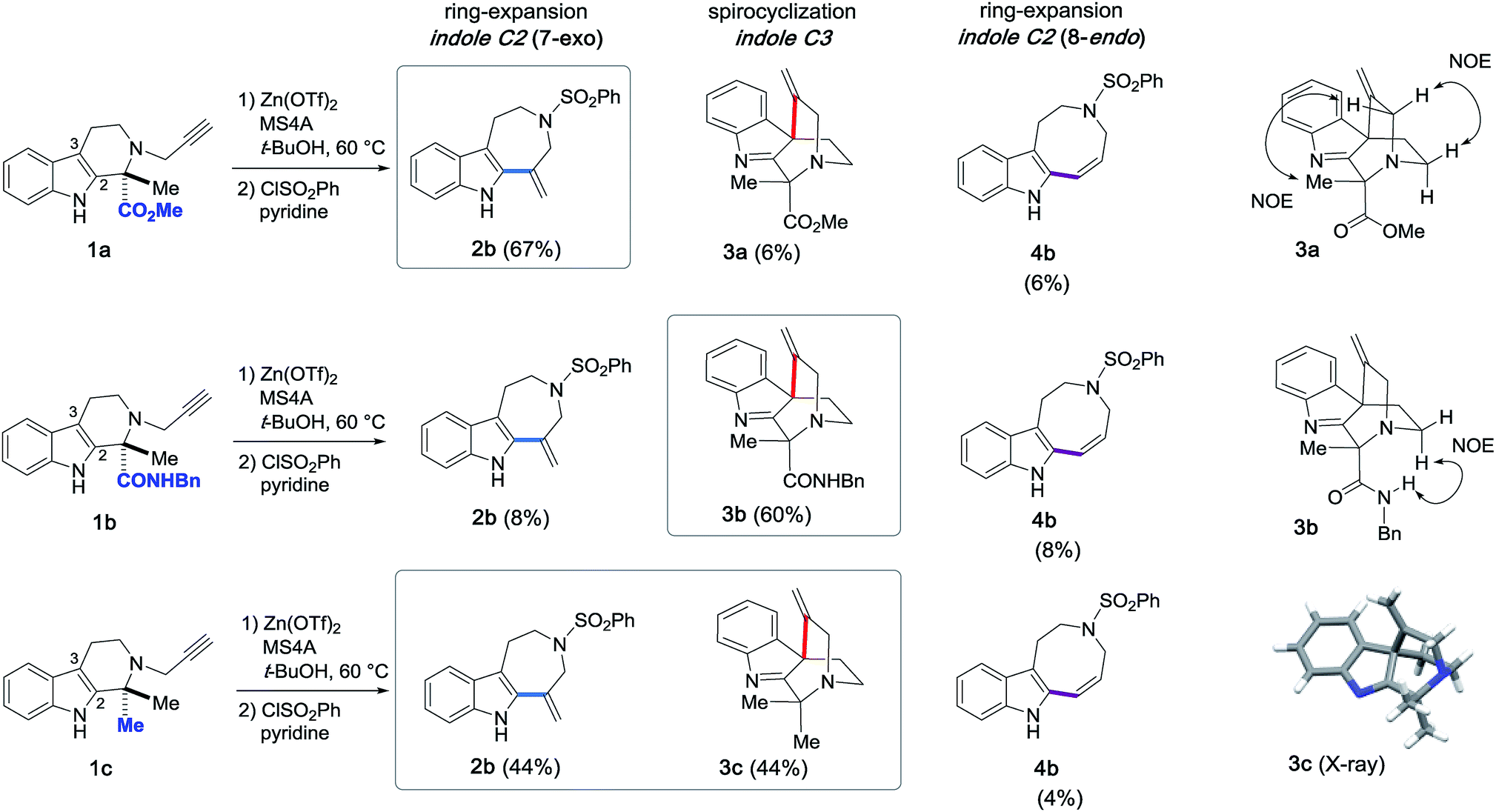 | ||
| Fig. 9 Zn(OTf)2-mediated intramolecular hydroarylations of alkynes with indoles and structural elucidation of the spirocyclized products 3a–c. | ||
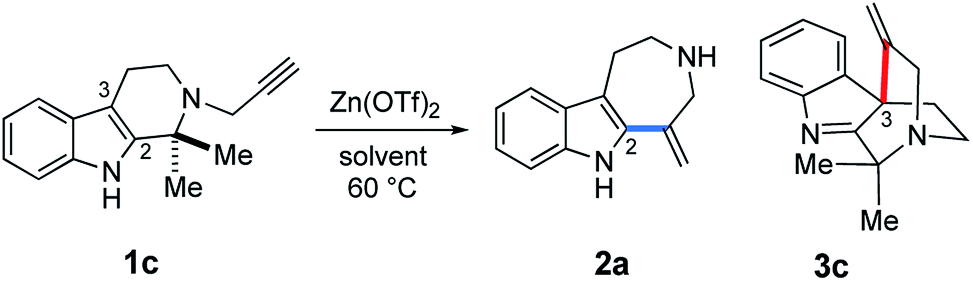
The Zn(OTf)2-mediated annulation of 1c provided a 3c:2a product ratio that reversed over time (Fig. S1, see, ESI†). The spirocyclic product 3c was the major product at the beginning of the reaction but decreased with time as the yield of 2a gradually increased and eventually exceeded that of 3c after approximately 80 h.
Zn(OTf)2-mediated conversion of spiroindole 3 to the azepino[4,5-b]indole scaffold 2
Given these experimental results, we hypothesized that the Zn(OTf)2-mediated hydroarylations of 1a–c likely involve the kinetically-controlled formation of spirocyclic products (3a–c) and subsequent rearrangement of the alkenyl group concomitant with fragmentation leading to azepino[4,5-b]indole 2a as a thermodynamic product. In fact, treatment of 3c with Zn(OTf)2 at elevated temperature (100 °C) in 2-BuOH resulted in the expected efficient conversion of 3c into 2a, and subsequent benzenesulfonylation of the resulting 2a afforded 2b in high yield (93%) (Fig. 10). This experimental result clearly indicated the conversion of 3c to 2a under high temperature conditions.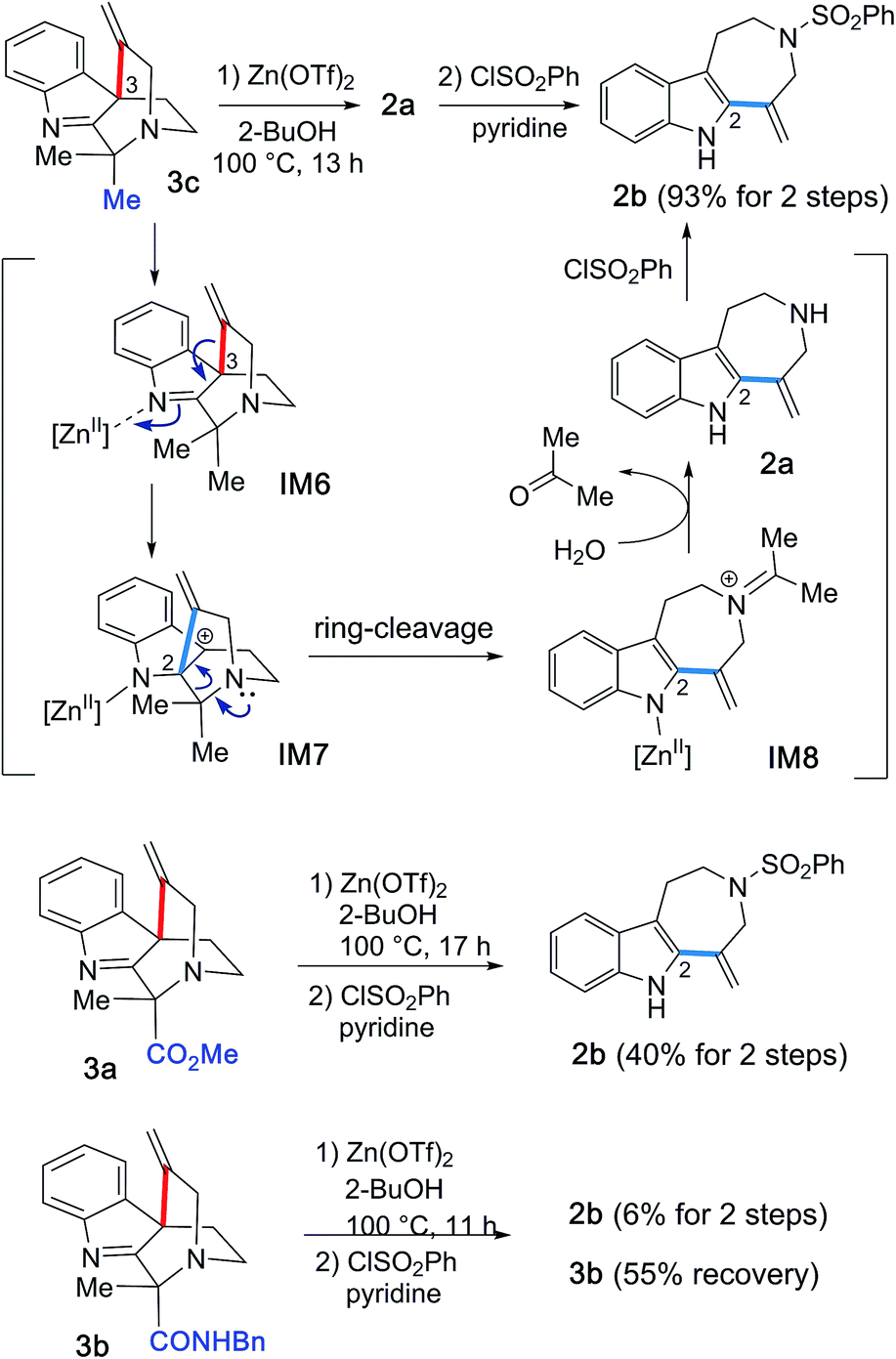 | ||
| Fig. 10 Conversions of 3a–c to 2b and a mechanistic rationale. | ||
Based on these experimental findings, we propose the reaction mechanism shown in Fig. 10. Activation of an imine group by Zn(OTf)2 could facilitate an alkenyl shift from the C3 to the C2 position to generate a tertiary cation at the benzylic C3 position (IM6 → IM7). Retro-Mannich-type reaction of the tertiary cation IM7 would entail ring-cleavage and formation of iminium cation intermediate IM8 and regeneration of the indole system. Hydrolysis of the resulting iminium cation IM8 would liberate secondary amine 2a and acetone, followed by sulfonylation of 2a to provide 2b. It is worth noting that to our knowledge, there is no previous report of the unexpected and highly efficient conversion of spiroindole 3 to the azepino[4,5-b]indole scaffold 2 upon the activation with Lewis acid promoters, whilst the Au(I)/Au(III)-catalyzed related intramolecular hydroarylations of indole and alkyne linear substrates have been reported.7
A similar conversion of the spirocycle 3a bearing a methylester group proceeded, presumably via an alkenyl shift followed by ring-cleavage, to afford 2b in 45% isolated yield after sulfonylation of the resultant 2a. In contrast, the reaction of 3b, which bears a benzyl amide group, resulted in poor conversion to form 2b in 6% yield over two steps and recovery of the substrate 3b (55%). These experimental results imply that the substituent (R1: Me, CO2Me, and CONHBn) in the vicinity of the bridgehead aliphatic nitrogen can greatly influence the migration ability of the alkenyl group for the spirocyclic substrates 3a–c, thereby changing the product distribution between the azepino[4,5-b]indole 2 and spirocyclic 3 scaffolds.
Attempts to control product distribution by the choice of reaction solvent
We changed the reaction solvent in an attempt to improve the selectivities of the Zn(OTf)2-mediated cyclizations of 1c leading to 2a and 3c (Table 2). Use of either a secondary alcohol (2-BuOH/i-PrOH) or ethanol instead of t-BuOH provided the spirocyclic product 3c as the major product (entries 1–4), whereas the use of methanol substantially impeded the conversion (entry 5). In contrast, the use of polar aprotic solvents, such as acetonitrile or THF, enabled preferential formation of 2a as the major product in good yield (entries 6 and 7). Thus, appropriate choice of the reaction solvent allowed the divergent synthesis of azepino[4,5-b]indole 2a and spirocycle 3c in practical yields. The Zn(OTf)2-mediated conversion of substrates 1a–c bearing a terminal alkyne was accompanied by the competing formation of azocino[5,4-b]indole scaffold 4a, giving the corresponding 4a and 4b in less than 10% yield.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Time | Yieldb (%) | Recovery (%) |
2a:3c |
||
| 2a | 3c | 4a | |||||
| a All reactions were performed using 1c (0.2 mmol) and Zn(OTf)2 (0.2 mmol) in 1 mL of each solvent at 60 °C. b Determined by 1H-NMR analysis. | |||||||
| 1 | t-BuOH | 24 | 45 | 55 | 5 | — | 45:55 |
| 2 | 2-BuOH | 8 | 25 | 70 | 5 | — | 26:74 |
| 3 | i-PrOH | 8 | 23 | 68 | 5 | — | 25:75 |
| 4 | EtOH | 24 | 31 | 63 | 5 | — | 33:67 |
| 5 | MeOH | 24 | 13 | 40 | 3 | 37 | 25:75 |
| 6 | CH3CN | 8 | 84 | 7 | 7 | 2 |
92:8 |
| 7 | THF | 24 | 80 | 6 | 7 | 6 |
93:7 |
Regio-controlled hydroarylation to form the azocino[5,4-b]indole scaffold
Whilst formation of the azocino[5,4-b]indole scaffold 4a was a minor pathway for substrates 1a–c bearing a terminal alkyne, changing the terminal alkyne into an internal alkyne (1a → 1d) reversed the cyclization mode to afford 4c as the major product via 8-endo-dig mode with loss of a methyl pyruvate unit (Fig. 11). Subsequent acetylation afforded crystalline 4d in 56% yield (2 steps), and the structure of 4d was unambiguously confirmed by X-ray analysis. Similarly, Zn(OTf)2-mediated 8-endo cyclization of 1e, followed by acetylation, provided 4d in good yield (91% over 2 steps).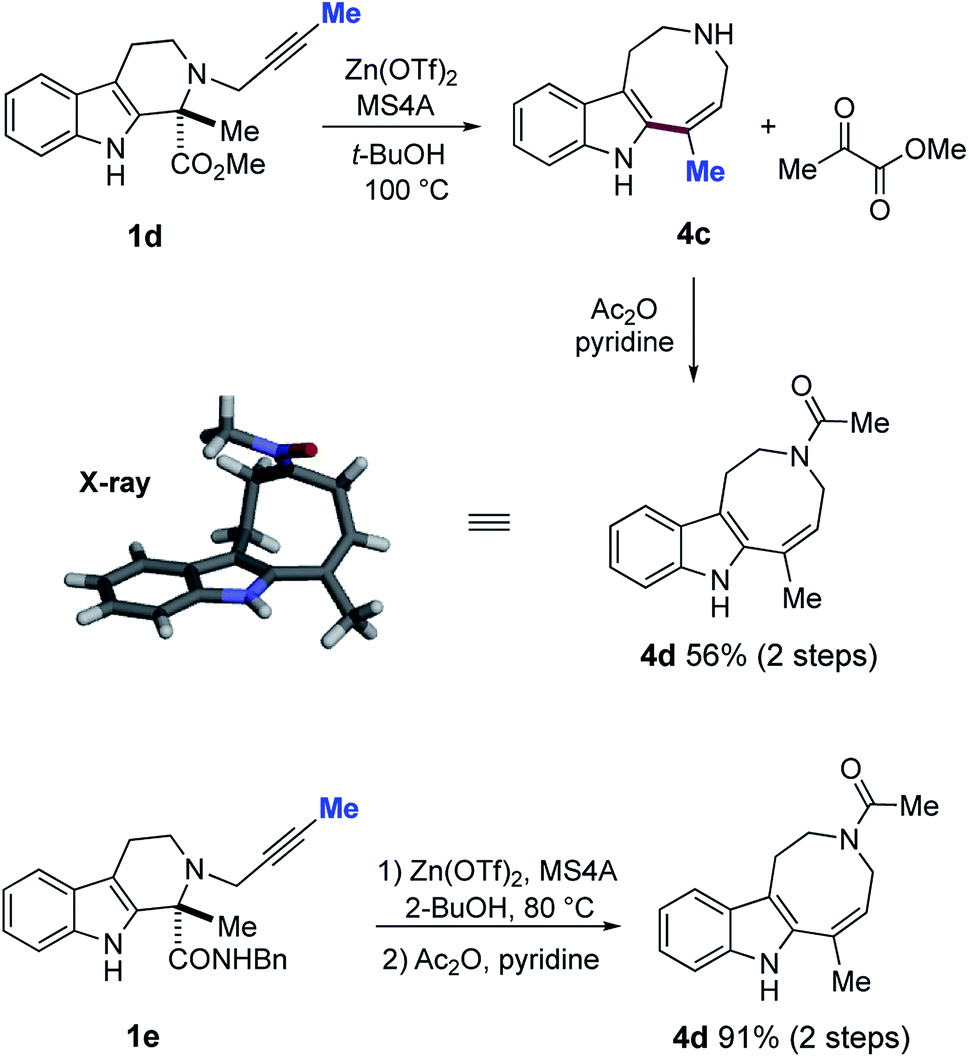 | ||
| Fig. 11 Zn(OTf)2-mediated ring expansion reaction of 1d–e and subsequent acetylation to form 4d and X-ray analysis of 4d. | ||
Substrate design for 7-endo cyclization and the unexpected α-alkenylation of a carbonyl group
We integrated the computational and experimental insights obtained throughout this study to investigate the remaining 7-endo cyclization between the indole C3 position and the distal alkynyl carbon to construct the spirocyclic scaffold 5 (Fig. 1b). To this end, we designed the new substrate 1f possessing both an amide moiety and an α-proton at the carbonyl group (Fig. 12a). We anticipated that kinetically-favored spirocyclization at the indole C3 position would form the iminium cation IM9 stabilized by hydrogen bonding between the bridgehead nitrogen and the amide N–H proton.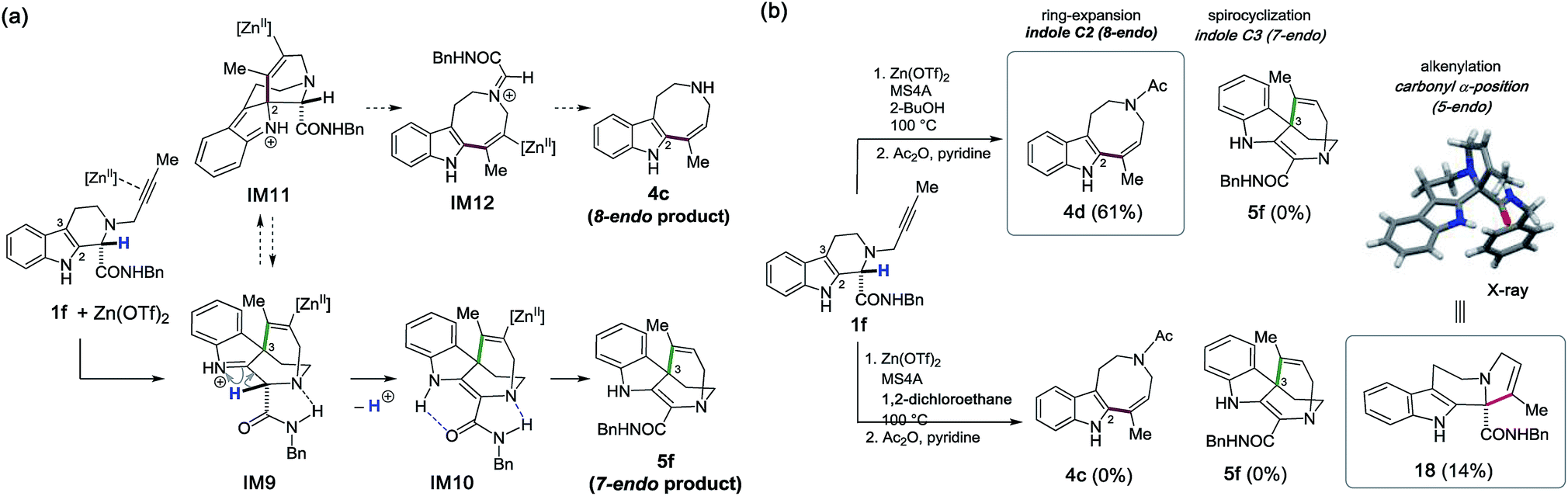 | ||
| Fig. 12 (a) A strategy for the 7-endo cyclization reaction to form 5f. (b) Zn(OTf)2-mediated cyclizations with substrate 1f. | ||
More importantly, installation of an α-proton would facilitate conversion of the iminium cation into the relatively stable enamine (IM9 → IM10) by preventing the competitive alkenyl shift to form intermediate IM11 for the 8-endo cyclization, which leads to undesired product 4cviaIM12.
Contrary to expectation, Zn(OTf)2-mediated cyclization of 1f in 2-BuOH did not afford the desired 7-endo product 5f; rather, the 8-endo product 4d was obtained in 61% yield after acetylation (Fig. 12b). Nonetheless, changing the solvent from 2-BuOH to 1,2-dichloroethane led to unanticipated results. Treatment of 1f in 1,2-dichloroethane at 100 °C enabled the intriguing Zn(OTf)2-mediated α-alkenylation of the carbonyl group to afford the tetracyclic scaffold 18 containing a quaternary carbon center and a trisubstituted alkene, albeit in 14% yield.16 Presumably, replacement of the protic solvent (2-BuOH) with the hydrophobic solvent (1,2-dichloroethane) played a crucial role in facilitating Zn(OTf)2-mediated enolization and α-alkenylation reactions which led to 18. Thus, three distinct modes of intramolecular hydroarylations between the indole C2/C3 positions and the internal/distal alkenyl carbons, as well as the unexpected α-alkenylation of the carbonyl group, were systematically realized by employing Zn(OTf)2 as a versatile “π-acid” mediator. A divergent synthetic process was developed in a programmable manner by the rational choice of substituents in the vicinity of the reaction sites, as well as of the solvents and reaction conditions.
Experimental section
All reactions were performed under a nitrogen atmosphere unless otherwise noted. Microwave reactions were performed using a Biotage® Initiator. Reactions were monitored by thin layer chromatography using Merck Millipore TLC Silica gel F254 plates (0.25 mm), which were visualized using UV light, phosphomolybdic acid (PMA) stain, PMS stain and p-anisaldehyde stain. Flash column chromatography was performed using Kanto Silica Gel 60N and Wako Alumina, Activated. The medium pressure liquid chromatography (MPLC) purifications were performed on a YAMAZEN YFLC-AI-580 and Biotage® Isolera. NMR spectra were recorded on JEOL ECA 500 (1H/500 MHz, 13C/125 MHz) and JEOL ECA 400 (1H/400 MHz, 13C/100 MHz) spectrometers. Chemical shifts (δ) were quoted in parts per million (ppm) from chloroform, acetonitrile, and dimethyl sulfoxide as an internal standard of 7.26, 1.94, 2.50 and 77.16, 1.32, 39.52 for 1H and 13C-NMR, respectively. Data for 1H NMR were reported as follows: chemical shift (number of hydrogen, multiplicity, coupling constant). Multiplicity was abbreviated as follows: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), m (multiplet), br (broad). ESI-Mass spectra were recorded on Bruker Daltonics microTOF-QII.Materials
Solvents used for the Zn(OTf)2-promoted reaction were dried and stored over activated molecular sieves 4A. Zn(OTf)2 was purchased from Tokyo Chemical Industry Co., Ltd. (TCI) and used as received. The substrates 1a–f were prepared from commercially available tryptamine hydrochloride (see ESI†).General procedure for Zn(OTf)2-promoted reaction of N-propargylated tetrahydrocarbolines 1
All reactions were carried out using 1.00 mmol of 1. A solution (0.2 M) of Zn(OTf)2 in t-BuOH was prepared prior to use as follows. Zn(OTf)2 (436 mg, 1.20 mmol) and t-BuOH (6 mL) were mixed in a Biotage microwave vial. The vial was sealed with a Teflon septum lined cap, and the mixture was then heated for 5 min at 100 °C under microwave irradiation to give a homogenous clear solution.Finely powdered molecular sieves 4A (200 mg) were placed in a microwave vial and dried by heating with a heat gun in vacuo. The t-BuOH solution of Zn(OTf)2 (0.2 M, 5 mL, 1.00 mmol) prepared as mentioned above and compound 1 (1.00 mmol) were successively added. The vial was then sealed with a Teflon septum lined cap. The mixture was stirred for 1 h at room temperature and then heated at 60 °C for appropriate time. After being cooled to room temperature, EtOAc (1 mL) and a saturated aqueous solution (1 mL) of the Rochelle salt were added at 0 °C. The mixture was stirred at 0 °C for 1 h and insoluble materials were filtered off and washed with EtOAc (50 mL) and H2O (5 mL). The organic layer was separated and washed with a saturated aqueous solution (10 mL) of NaHCO3. The aqueous layer was extracted with EtOAc (50 mL × 2). Combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure to give a crude mixture of products 2, 3, and 4.
The products 2 and 4 were isolated after sulfonylation as follows. The crude mixture was dissolved in pyridine (5 mL) and then treated with benzenesulfonyl chloride (0.640 mL, 5.00 mmol) at 0 °C. After being stirred for 10 min at 0 °C and then 30 min at room temperature, the reaction was quenched by addition of a saturated aqueous solution of NaHCO3 at 0 °C. The resulting mixture was extracted with EtOAc (100 mL) and washed with a saturated aqueous solution of NaHCO3 (10 mL) and H2O (10 mL). Combined aqueous layers were extracted with EtOAc (100 mL). Combined organic extracts were washed with brine and dried over Na2SO4. After filtration and concentration in vacuo, the products were isolated by column chromatography.
Reaction of 1a bearing a methylester group
According to the general procedure, Zn(OTf)2-promoted reaction of 1a (283 mg, 1.00 mmol) was carried out. The resulting mixture was purified by silica gel column chromatography (CH2Cl2/MeCN) to give a fraction containing 2b and 4b (248 mg, 0.733 mmol, 73%, 2b:4b = 92:8) as well as a fraction containing 3a. The fraction mainly composed of 3a was further purified by alumina column chromatography (CH2Cl2) to give 3a (17 mg, 0.0602 mmol, 6%). Whilst the mixture composed of 2b and 4b was separable via repetitive column chromatography, it caused significant decrease in isolated yields. 2b: Rf = 0.30 (CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.82 (1H, br s), 7.78–7.76 (2H, m), 7.47–7.44 (2H, m), 7.38–7.35 (2H, m), 7.27–7.25 (1H, m), 7.20–7.11 (1H, m), 7.10–7.07 (1H, m), 5.24 (1H, s), 5.22 (1H, s), 4.34 (2H, s), 3.74 (2H, t, J = 5.7 Hz), 3.06 (2H, t, J = 5.7 Hz); 13C NMR (125 MHz, CDCl3) δ 139.9, 137.5, 136.0, 133.1, 132.4, 128.9, 128.4, 127.1, 123.2, 119.7, 118.5, 113.1, 111.8, 110.8, 52.7, 49.1, 24.2. HRMS (ESI, m/z): calcd for C19H18KN2O2S [M + K]+ 377.0721, found 377.0721. 3a: Rf = 0.30 (CH2Cl2–MeCN = 1:2); 1H NMR (500 MHz, CDCl3): δ 7.74 (1H, d, J = 8.6 Hz), 7.42–7.38 (2H, m), 7.21 (1H, t, J = 7.5 Hz), 4.81–4.80 (1H, m), 4.72–4.71 (1H, m), 3.94 (1H, br d, J = 18.1 Hz), 3.84 (3H, s), 3.74 (1H, ddd, J = 18.1, 2.3, 2.3 Hz), 3.22 (1H, ddd, J = 14.8, 10.2, 5.4 Hz), 3.07–3.00 (1H, m), 2.31 (1H, ddd, J = 12.3, 10.6, 5.2 Hz), 1.77 (3H, s), 1.63 (1H, ddd, J = 12.3, 10.6, 5.2 Hz); 13C NMR (125 MHz, CDCl3): δ 185.4, 171.4, 157.1, 143.9, 137.4, 128.5, 125.5, 124.3, 121.8, 106.4, 67.8, 59.2, 53.4, 51.1, 46.8, 27.0, 23.7; HRMS (ESI, m/z): calcd for C17H19N2O2 [M + H]+ 283.1441, found 283.1466. 4b: Rf = 0.28 (CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.65–7.64 (2H, m), 7.62 (1H, br s), 7.47–7.42 (2H, m), 7.35–7.32 (2H, m), 7.25 (1H, d, J = 8.6 Hz), 7.19–7.15 (1H, m), 7.09 (1H, t, J = 7.4 Hz), 6.49 (1H, d, J = 11.5 Hz), 5.85 (1H, dt, J = 11.5, 6.6 Hz), 4.00 (2H, d, J = 6.9 Hz), 3.60–3.58 (2H, m), 3.00–2.97 (2H, m); 13C NMR (125 MHz, CDCl3): δ 140.1, 136.1, 132.2, 131.6, 128.9, 128.1, 127.4, 127.0, 123.2, 122.8, 119.8, 118.3, 112.4, 110.9, 46.3, 46.1, 24.4; HRMS (ESI, m/z): calcd for C19H18N2O2S [M + H]+ 339.1162, found 339.1166.
Reaction of 1b bearing a benzylamide group
According to the general procedure, Zn(OTf)2-promoted reaction of 1b (357 mg, 1.00 mmol) was carried out. The resulting crude mixture was purified by silica gel column chromatography (CH2Cl2/MeCN) to give a mixture of 2b and 4b (50.4 mg, 0.145 mmol, 15%; 2b:4b = 1:1) as well as 3b (213 mg, 0.596 mmol, 60%). 3b: Rf = 0.31 (EtOAc–MeOH = 10:1); 1H NMR (500 MHz, CD3CN): δ 8.19 (1H, t, J = 5.5 Hz), 7.68 (1H, d, J = 8.0 Hz), 7.46 (1H, d, J = 7.5 Hz), 7.34–7.22 (7H, m), 4.78–4.75 (2H, m), 4.48–4.38 (2H, m), 3.90 (1H, br d, J = 17.8 Hz), 3.70 (1H, br d, J = 17.8 Hz), 3.16 (1H, ddd, J = 14.3, 10.3, 4.0 Hz), 2.90–2.82 (1H, m), 2.37–2.30 (1H, m), 1.61 (3H, s), 1.31 (1H, ddd, J = 12.3, 10.6, 4.0 Hz); 13C NMR (125 MHz, CD3CN): δ 189.5, 172.4, 157.3, 144.5, 139.7, 138.6, 129.6, 129.2, 128.21, 128.17, 126.7, 125.5, 122.2, 106.5, 68.4, 60.8, 51.1, 47.3, 43.8, 29.4, 26.9; HRMS (ESI, m/z): calcd for C23H24N3O [M + H]+ 358.1914, found 358.1943.
Reaction of 1c bearing a gem-dimethyl group
According to the general procedure, Zn(OTf)2-promoted reaction of 1c (238 mg, 0.999 mmol) was carried out. The resulting crude mixture was purified by silica gel column chromatography (CH2Cl2/MeCN) to give a fraction containing 2b and 4b (163 mg, 0.482 mmol, 48%; 2b:4b = 91:9) as well as a fraction containing 3c. The fraction mainly composed of 3c was further purified by alumina column chromatography (CH2Cl2) to afford 3c (104 mg, 0.436 mmol, 44%). 3c: Rf = 0.11 (CH2Cl2–MeCN = 1:1); 1H NMR (500 MHz, CDCl3): δ 7.61 (1H, d, J = 8.0 Hz), 7.36 (1H, d, J = 7.5 Hz), 7.33 (1H, dd, J = 8.0, 7.5 Hz), 7.20 (1H, dd, J = 8.0, 7.5 Hz), 4.81–4.80 (1H, m), 4.70–4.67 (1H, m), 3.92 (1H, br d, J = 18.1 Hz), 3.63 (1H, br d, J = 18.1 Hz), 3.37–3.31 (1H, m), 3.05–2.98 (1H, m), 2.32–2.27 (1H, m), 1.57 (3H, s), 1.53–1.46 (1H, m), 1.49 (3H, s); 13C NMR (125 MHz, CDCl3): δ 192.9, 157.1, 143.7, 137.8, 128.2, 124.9, 124.2, 121.0, 105.8, 59.4, 58.6, 52.4, 44.4, 29.6, 26.4, 26.1; HRMS (ESI, m/z): calcd for C16H19N2 [M + H]+ 239.1543, found 239.1548.
Reaction of 1d and 1e bearing an internal alkyne
:40 mixture of rotamers): Rf = 0.29 (CH2Cl2–EtOAc = 1:1); 1H NMR (500 MHz, DMSO-d6, 90 °C): δ 10.7 (1H, br s), 7.52 (1H, br d, J = 7.5 Hz), 7.34 (1H, br d, J = 8.0 Hz), 7.11–7.08 (1H, m), 7.04–6.97 (1H, br m), 5.86–5.80 (0.40H, m), 5.80–5.74 (0.60H, m), 3.83 (0.80H, m), 3.80 (1.20H, m), 3.55 (0.80H, m), 3.46 (1.20H, m), 2.91 (1.20H, m), 2.82 (0.80H, m), 2.16 (1.20H, br s), 2.11 (1.80H, br s), 2.01 (1.20H, br s), 1.67 (1.80H, br s); 13C NMR (125 MHz, DMSO-d6, 90 °C): δ 169.5, 169.0, 136.9, 136.8, 136.0, 135.1, 131.1, 129.4, 129.1, 128.6, 128.0, 125.8, 125.1, 123.8, 121.9, 119.1, 118.5, 118.2, 111.9, 111.5, 110.4, 55.2, 47.7, 47.6, 45.7, 42.7, 25.3, 24.7, 23.3, 21.7; HRMS (ESI, m/z): calcd for C16H19N2O [M + H]+ 255.1492, found 255.1494.
:1); 1H NMR (400 MHz, CDCl3): δ 9.09 (1H, s), 8.43 (1H, t, J = 5.7 Hz), 7.50 (1H, d, J = 7.3 Hz), 7.38 (1H, d, J = 8.2 Hz), 7.35–7.22 (5H, m), 7.16 (1H, ddd, J = 8.1, 6.9, 0.9 Hz), 7.10–7.06 (1H, m), 5.50–5.48 (1H, m), 4.50 (1H, dd, J = 15.1, 6.4 Hz), 4.41 (1H, dd, J = 15.1, 6.0 Hz), 3.81–3.78 (2H, m), 3.33–3.25 (1H, m), 3.08–2.98 (2H, m), 2.60–2.50 (1H, m), 2.01–1.99 (3H, m); 13C NMR (100 MHz, CDCl3): δ 172.3, 140.0, 138.6, 136.5, 133.7, 128.8, 127.5, 126.8, 123.9, 121.9, 119.3, 118.4, 111.6, 109.1, 74.4, 55.4, 45.3, 43.1, 16.8, 14.0; HRMS (ESI, m/z): calcd for C23H24N3O [M + H]+ 358.1914, found 358.1912.
According to the above procedure except for changing the solvent from 1,2-dichloroethane to 2-BuOH, Zn(OTf)2-promoted reaction of 1f (72.0 mg, 0.201 mmol) was carried out at 100 °C for 24 h. The resulting crude reaction mixture containing 4c was treated with acetic anhydride (0.473 mL, 5.00 mmol) in pyridine (5 mL) at 0 °C. After being stirred at room temperature, essentially identical workup protocol as described above was applied to give 4d (31.0 mg, 0.112 mmol, 61%).
Trapping of iminium cation IM4via reduction with Et3SiH
Finely powdered molecular sieves 4A (400 mg) were placed in a microwave vial and dried by heating with a heat gun in vacuo. The t-BuOH solution of Zn(OTf)2 (0.2 M, 10 mL, 2.00 mmol) prepared as described above, the compound 1a (565 mg, 2.00 mmol), and triethylsilane (1.59 mL, 10.0 mmol) were successively added. The vial was sealed with a Teflon septum lined cap and the mixture was stirred at 30 °C for 1 h. The resulting mixture was further heated at 60 °C for 3 h. After being cooled to rt, the reaction mixture was diluted with toluene (10 mL) and concentrated to one fifth of its initial volume under reduced pressure. The residue was diluted with EtOAc (20 mL) and then treated with a saturated aqueous solution (10 mL) of the Rochelle salt at 0 °C. The mixture was vigorously stirred at 0 °C for 1 h, and insoluble materials were filtered off and washed with EtOAc (80 mL) and H2O (5 mL). The organic layer was separated and washed with a saturated aqueous solution of NaHCO3 (10 mL × 2). The aqueous layer was extracted with EtOAc (100 mL × 2). Combined organic extracts were washed with brine (10 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a crude mixture of 2a and 2d. The resultant mixture was dissolved in pyridine (10 mL) and treated with benzenesulfonyl chloride (1.28 mL, 10.0 mmol) at 0 °C. After being stirred at room temperature, the mixture was treated with a saturated aqueous solution of NaHCO3 (10 mL). The mixture was extracted with EtOAc (100 mL × 3), washed with brine (15 mL), and dried over Na2SO4. After filtration and concentration, the residue was purified by silica gel column chromatography (CH2Cl2/EtOAc/MeOH) to give 2d (72.2 mg, 0.253 mmol, 13%). The fraction mainly containing 2b was further purified by passing through a short alumina column (CH2Cl2) to give 2b (456 mg, 1.35 mmol, 67%). 2d: Rf = 0.21 (Hex–EtOAc = 2:1); 1H NMR (500 MHz, CDCl3): δ 7.97 (1H, s), 7.49 (1H, d, J = 8.0 Hz), 7.28 (1H, d, J = 8.0 Hz), 7.20–7.16 (1H, m), 7.11–7.07 (1H, m), 5.26 (1H, s), 5.07 (1H, s), 3.84 (1H, d, J = 14.9 Hz), 3.74–3.72 (4H, m), 3.67 (1H, q, J = 6.9 Hz), 3.32 (1H, ddd, J = 13.1, 7.9, 4.3 Hz), 3.21 (1H, ddd, J = 13.1, 7.9, 4.3 Hz), 3.05 (1H, ddd, J = 16.6, 7.6, 3.9 Hz), 2.97 (1H, ddd, J = 16.6, 7.6, 3.9 Hz),1.43 (3H, d, J = 7.5 Hz); 13C NMR (125 MHz, CDCl3): δ 174.8, 140.0, 136.0, 134.0, 129.1, 123.0, 119.5, 118.8, 114.6, 111.2, 110.6, 76.9, 60.3, 58.5, 52.5, 51.8, 24.3, 16.7; HRMS (ESI, m/z): calcd for C17H21N2O2 [M + H]+ 285.1598, found 285.1614.
General procedure for conversion of 3 into 2b
Zn(OTf)2 (72.7 mg, 0.200 mmol) and 2-BuOH (2 mL) was mixed in a Biotage microwave vial. The vial was sealed with a Teflon septum lined cap. The mixture was heated at 100 °C for 5 min to give a homogenous clear solution. After being cooled to rt, compound 3 (0.200 mmol) were added, and the vial was sealed again with a Teflon septum lined cap. The mixture was then heated at 100 °C for appropriate time. After being cooled to room temperature, the reaction mixture was poured into a mixture of EtOAc and a saturated aqueous solution of the Rochelle salt at 0 °C. The resulting mixture was vigorously stirred at 0 °C for 1 h. The organic layer was separated and washed with a saturated aqueous solution of NaHCO3. The aqueous layer was extracted with EtOAc. Combined organic extracts were washed with brine, dried over Na2SO4 and concentrated under reduced pressure. After azeotropic distillation with toluene, the residue was dissolved in pyridine (2 mL) and then treated with benzenesulfonyl chloride (0.128 mL, 1.00 mmol) at 0 °C. After being stirred at room temperature, the resulting mixture was quenched with saturated aqueous solution of NaHCO3 and extracted with EtOAc. Combined organic extracts were washed with brine and dried over Na2SO4. After filtration and concentration under reduced pressure, the residue was purified by silica gel column chromatography (CH2Cl2/MeCN) to give 2b and 3.Computational session
We have performed an initial reaction path search adopting a simplified model to reduce computational costs. In the model, the benzene ring in the indole moiety was substituted by methyl groups at the C4 and C5 positions. In the initial reaction stage, we focused only on bond reorganizations among four carbon atoms, i.e., two alkynyl carbon atoms in the propargyl moiety and indole C2 and C3 atoms, based on the reaction mechanism proposed by Wang and coworkers.7 In this stage, the model collision energy parameter γ of the AFIR method was set to 200 kJ mol−1. The subsequent C–C bond fission steps were studied by manually applying the artificial force to specific atom pairs with γ = 100 kJ mol−1. TSs for the protodezincation steps were searched by shooting t-BuOH to IM3-i or IM5-i from various random mutual positions and directions by the AFIR method with γ = 200 kJ mol−1, where the OH groups in t-BuOH and Zn, and the Zn–bound alkenyl carbon atom in IM3-i or IM5-i were set as two fragments in the AFIR function. In these search jobs, DFT calculations were carried out at the RI-PBE17/def2-SV(P)18 level implemented in Turbomole v.7.0.1 program.19The AFIR paths obtained in the above calculations were optimized by the locally updated plane (LUP) method20 to obtain initial structures for TS optimizations. From all optimized TS structures, IRC calculations were performed to see path connections. Harmonic vibrational analysis was carried out at all optimized TS and stable structures. In these jobs, DFT calculations were performed at the B3LYP21/Def2SVP level combined with the SMD (t-BuOH) method22 implemented in Gaussian 09 program.23
The reaction pathways of 1a leading to 2a/3a were obtained based on corresponding paths obtained in the simplified model system. Intermediate and TS structures were reoptimized with the ωB97X-D24/Def2SVP level with the SMD (t-BuOH) method. To identify the lowest conformation at TS, TS conformation sampling was made for intramolecular cyclization and protodezincation steps. The conformation at these TSs were systematically searched by the AFIR method with fixing positions of atoms involved in each bond reorganization, at the PBEPBE/Def2-SV(P) level with the SMD (t-BuOH) method. These TSs with various conformations were re-optimized at the ωB97X-D/Def2SVP level with the SMD (t-BuOH) method. All these DFT calculations were carried out with Gaussian 09 program.
Conclusions
Triggered by the incidental finding of zinc(II)-promoted activation of alkynes, we explored intramolecular hydroarylations of N-propargylated tetrahydrocarbolines to develop a versatile synthetic process for generating scaffold and functional group variations. Protic and sterically demanding alcoholic solvents such as t-BuOH dramatically accelerated the Zn(OTf)2-promoted annulations. Computational investigations employing the AFIR method in the GRRM strategy provided a convincing rationale: participation of t-BuOH as a proton donor plays a pivotal role in facilitating the protodezincation process. The unique combination of Zn(OTf)2 and t-BuOH allows the protodemetallation of alkenyl zinc species under mild and almost neutral conditions without the addition of Brønsted acid promoters.We demonstrated that the modes of the Zn(OTf)2-mediated cyclizations are controlled by appropriate choices of the reaction solvents and the substituents (R1 and R2) on the substrates 1a–f. Divergent annulations involving two kinds of ring-expansion reactions and two dearomatizing spirocyclizations were systematically employed to gain programmable access to four distinct scaffolds: azepino[4,5-b]indole 2, spirocycle 3, azocino[5,4-b]indole 4, and spirocyclic 5. Zn(OTf)2-mediated annulations of substrates 1a–c bearing a terminal alkyne group allow divergent cyclizations between an internal alkyne carbon with either the indole C2 or C3 position to effect 7-exo cyclizations or 6-exo spirocyclizations, respectively. Notably, this study revealed that 6-exo dearomatizing spirocyclizations of 1c resulting in formation of a quaternary center is a kinetically-favored pathway. Furthermore, treatment of the spirocyclic products 3a–c with Zn(OTf)2 under thermodynamic conditions can cause cascade reactions via migration of the alkenyl group and concomitant retro-Mannich-type fragmentation. Subsequent hydrolysis of the resulting iminium cation leads to irreversible formation of the azepino[4,5-b]indole scaffold 2a. The R1 substituents (Me, CO2Me, CONHBn) of substrates 1a–c were shown to greatly influence the susceptibilities of substrates for the irreversible reaction leading to 2a at elevated temperature. In this cascade process, Zn(OTf)2 could play key roles not only in the hydroarylation reaction (by activating the alkyne as a “π-acid”) but also on alkenyl migration as a Lewis acid promoter for the imine group. In contrast to substrates 1a–c which have a terminal alkyne and were converted to 2a and 3a, Zn(OTf)2-mediated annulations of substrates 1d–f, which have an internal alkyne, preferentially proceeded through the 8-endo ring expansion reaction to afford azocino[5,4-b]indole 4a in good yield. Furthermore, substrate 1f was demonstrated to affect the intriguing α-alkenylation of the carbonyl group to forge the tetracyclic scaffold 18 containing a quaternary carbon center and a trisubstituted olefin.
These experimental findings underscore that Zn(OTf)2-mediated activation of alkynes provides relatively unexplored but versatile synthetic methodologies for the direct and flexible synthesis of skeletally diverse and densely-functionalized alkaloidal scaffolds reminiscent of natural products. The artificial force induced reaction method allows comprehensive discussion of transition states and could offer both rational and unanticipated guidelines for designing divergent reactions. Integration of synthetic strategies for generating skeletal variations with a systematic computational approach for identifying unforeseen reaction pathways could provide a new route for advancing the combinatorial chemical synthesis of functional molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial supports from the Japan Science and Technology Agency (JST) (JPMJPR13K3) “Precursory Research for Embryonic Science and Technology” (PRESTO to H. O.) and “Core Research for Evolutional Science and Technology (CREST to S. M.)” for projects of “Molecular technology and creation of new functions”, JSPS KAKENHI (Grant No. 15H03117 to H. O., Grant No. JPMJCR14L5 to S. M.), the Asahi Grass Foundation (H. O.), and Astellas Foundation for Research on Metabolic Disorders (H. O.). This work was also inspired by the international and interdisciplinary environments of the JSPS Asian CORE Program, “Asian Chemical Biology Initiative” as well as the JSPS A3 Foresight Program “Asian Chemical Probe Research Hub”. Institute for Chemical Reaction Design and Discovery (ICReDD) was established by World Premier International Research Initiative (WPI), MEXT, Japan.Notes and references
- (a) Y. Nakao, Chem. Rec., 2011, 11, 242–251 CrossRef CAS PubMed; (b) A. Fürstner, Acc. Chem. Res., 2014, 47, 925–938 CrossRef PubMed; (c) Y. Yamamoto, Chem. Soc. Rev., 2014, 43, 1575–1600 RSC; (d) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (e) P. Gandeepan and C. H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (f) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190–222 CrossRef CAS; (g) R. Manikandan and M. Jeganmohan, Org. Biomol. Chem., 2015, 13, 10420–10436 RSC; (h) E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503–1514 CrossRef CAS PubMed.
- (a) X.-F. Wu and H. Neumann, Adv. Synth. Catal., 2012, 354, 3141–3160 CrossRef CAS; (b) S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS; (c) M. J. González, L. A. Lopez and R. Vicente, Tetrahedron Lett., 2015, 56, 1600–1608 CrossRef.
- (a) S. Maeda, Y. Harabuchi, M. Takagi, T. Taketsugu and K. Morokuma, Chem. Rec., 2016, 16, 2232–2248 CrossRef CAS PubMed; (b) W. M. Sameera, S. Maeda and K. Morokuma, Acc. Chem. Res., 2016, 49, 763–773 CrossRef CAS PubMed.
- (a) H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef CAS PubMed; (b) H. Mizoguchi, R. Watanabe, S. Minami, H. Oikawa and H. Oguri, Org. Biomol. Chem., 2015, 13, 5955–5963 RSC; (c) R. Watanabe, H. Mizoguchi, H. Oikawa, H. Ohashi, K. Watashi and H. Oguri, Bioorg. Med. Chem., 2017, 25, 2851–2855 CrossRef CAS PubMed.
- (a) C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105–1109 CrossRef CAS PubMed; (b) C. Ferrer, C. H. M. Amijs and A. M. Echavarren, Chem.–Eur. J., 2007, 13, 1358–1373 CrossRef CAS PubMed.
- (a) S. G. Modha, D. D. Vachhani, J. Jacobs, L. Van Meervelt and E. V. Van der Eycken, Chem. Commun., 2012, 48, 6550–6552 RSC; (b) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Adv. Synth. Catal., 2012, 354, 2841–2848 CrossRef CAS.
- (a) L. Zhang, L. Chang, H. Hu, H. Wang, Z. J. Yao and S. Wang, Chem.–Eur. J., 2014, 20, 2925–2932 CrossRef CAS PubMed; (b) L. Zhang, Y. Wang, Z. J. Yao, S. Wang and Z. X. Yu, J. Am. Chem. Soc., 2015, 137, 13290–13300 CrossRef CAS PubMed.
- (a) Y. Liu, W. Xu and X. Wang, Org. Lett., 2010, 12, 1448–1451 CrossRef CAS PubMed; (b) G. Li and Y. Liu, J. Org. Chem., 2010, 75, 3526–3528 CrossRef CAS PubMed; (c) A. S. K. Hashmi, W. Yang and F. Rominger, Adv. Synth. Catal., 2012, 354, 1273–1279 CrossRef CAS; (d) J. D. Podoll, Y. Liu, L. Chang, S. Walls, W. Wang and X. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15573–15578 CrossRef CAS PubMed; (e) P. Michael Barbour, J. D. Podoll, L. J. Marholz and X. Wang, Bioorg. Med. Chem. Lett., 2014, 24, 5602–5605 CrossRef CAS PubMed; (f) W. Xu, W. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 9546–9549 CrossRef CAS PubMed; (g) Y. Li, S. Zhu, J. Li and A. Li, J. Am. Chem. Soc., 2016, 138, 3982–3985 CrossRef CAS PubMed; (h) D. Nishiyama, A. Ohara, H. Chiba, H. Kumagai, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2016, 18, 1670–1673 CrossRef CAS PubMed; (i) P. M. Barbour, W. Wang, L. Chang, K. L. Pickard, R. Rais, B. S. Slusher and X. Wang, Adv. Synth. Catal., 2016, 358, 1482–1490 CrossRef CAS PubMed; (j) Y. Zhu, W. He, W. Wang, C. E. Pitsch, X. Wang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 12206–12209 CrossRef CAS PubMed; (k) Y. Zhu, L. Cleaver, W. Wang, J. D. Podoll, S. Walls, A. Jolly and X. Wang, Eur. J. Med. Chem., 2017, 125, 130–142 CrossRef CAS PubMed; (l) N. Glinsky-Olivier, P. Retailleau and X. Guinchard, Eur. J. Org. Chem., 2018, 5823–5829 CrossRef CAS; (m) X. Zhang, B. N. Nakde, R. Guo, S. Yadav, Y. Gu and A. Li, Angew. Chem., Int. Ed., 2019, 58, 6053–6058 CrossRef CAS PubMed.
- (a) M. Nakamura, C. Liang and E. Nakamura, Org. Lett., 2004, 6, 2015–2017 CrossRef CAS PubMed; (b) S. Morikawa, S. Yamazaki, Y. Furusaki, N. Amano, K. Zenke and K. Kakiuchi, J. Org. Chem., 2006, 71, 3540–3544 CrossRef CAS PubMed; (c) C.-L. Deng, R.-J. Song, Y.-L. Liu and J.-H. Li, Adv. Synth. Catal., 2009, 351, 3096–3100 CrossRef CAS; (d) X.-L. Fang, R.-Y. Tang, X.-G. Zhang, P. Zhong, C.-L. Deng and J.-H. Li, J. Organomet. Chem., 2010, 696, 352–356 CrossRef; (e) V. Srinivas, K. V. Sajna and K. C. Kumara Swamy, Tetrahedron Lett., 2011, 52, 5323–5326 CrossRef CAS; (f) W. Hess and J. W. Burton, Adv. Synth. Catal., 2011, 353, 2966–2970 CrossRef CAS; (g) G. Li and H. Nakamura, Angew. Chem., Int. Ed., 2016, 55, 6758–6761 CrossRef CAS PubMed.
- (a) P. A. Donets, K. Van Hecke, L. Van Meervelt and E. V. Van der Eycken, Org. Lett., 2009, 11, 3618–3621 CrossRef CAS PubMed; (b) H. Mizoguchi, H. Oikawa and H. Oguri, Org. Biomol. Chem., 2012, 10, 4236–4242 RSC.
- (a) S. Maeda and K. Morokuma, J. Chem. Phys., 2010, 132, 241102/1–241102/4 CrossRef CAS PubMed; (b) S. Maeda and K. Morokuma, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS PubMed; (c) S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC; (d) S. Maeda, T. Taketsugu and K. Morokuma, J. Comput. Chem., 2014, 35, 166–173 CrossRef CAS PubMed.
- (a) R. Uematsu, S. Maeda and T. Taketsugu, Chem.–Asian J., 2014, 9, 305–312 CrossRef CAS PubMed; (b) R. Uematsu, E. Yamamoto, S. Maeda, H. Ito and T. Taketsugu, J. Am. Chem. Soc., 2015, 137, 4090–4099 CrossRef CAS PubMed; (c) K. K. Vong, S. Maeda and K. Tanaka, Chem.–Eur. J., 2016, 22, 18865–18872 CrossRef CAS PubMed; (d) E. Yamamoto, S. Maeda, T. Taketsugu and H. Ito, Synlett, 2017, 28, 1258–1267 CrossRef CAS; (e) T. Yoshimura, S. Mori, S. Maeda, T. Taketsugu, M. Sawamura and K. Morokuma, Chem. Sci., 2017, 8, 4475–4488 RSC.
- The corresponding cyclization pathways to form the minor diastereomers such as syn-product 3a′ were also calculated to provide both a schematic and an energy profile for the reaction paths from 1a′ to 2a/3a′ (see ESI, Fig. S3 and S4†), although the minor syn-product 3a′ was not obtained experimentally.
- S. Maeda, Y. Harabuchi, Y. Ono, T. Taketsugu and K. Morokuma, Int. J. Quantum Chem., 2015, 115, 258–269 CrossRef CAS.
- The crystallographic data (3c: CCDC-1905916, 4d: CCDC-1905889, and 18: CCDC-1905890).
- (a) F. Dénès, A. Pérez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366–2447 CrossRef PubMed; (b) Y. Liu, R.-J. Song and J.-H. Li, Synthesis, 2010, 3663–3669 CrossRef CAS; (c) W. Hessa and J. W. Burton, Adv. Synth. Catal., 2011, 353, 2966–2970 CrossRef; (d) D. Hack, M. Blümel, P. Chauhan, A. Philipps and D. Enders, Chem. Soc. Rev., 2015, 44, 6059–6093 RSC; (e) M. Cao, A. Yesilcimen and M. Wasa, J. Am. Chem. Soc., 2019, 141, 4199–4203 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- TURBOMOLE V7.0.1 2015, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, Since 2007 Search PubMed.
- C. Choi and R. Elber, J. Chem. Phys., 1991, 94, 751–760 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1905889, 1905890 and 1905916. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01507h |
| This journal is © The Royal Society of Chemistry 2019 |