
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly diastereoselective preparation of chiral NHC-boranes stereogenic at the boron atom†
Clara
Aupic
a,
Amel
Abdou Mohamed
a,
Carlotta
Figliola
 a,
Paola
Nava
a,
Béatrice
Tuccio
b,
Gaëlle
Chouraqui
a,
Jean-Luc
Parrain
a and
Olivier
Chuzel
*a
a,
Paola
Nava
a,
Béatrice
Tuccio
b,
Gaëlle
Chouraqui
a,
Jean-Luc
Parrain
a and
Olivier
Chuzel
*a
aAix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: olivier.chuzel@univ-amu.fr
bAix Marseille Univ, CNRS, ICR, Marseille, France
First published on 24th May 2019
Abstract
Stereogenic main group elements are clearly generating interest in the enantioselective catalysis field. Surprisingly, while chiral organoboron reagents are very useful in stereoselective transformations, few scaffolds stereogenic at boron and configurationally stable have been reported to date. Herein, we describe an original library of chiral NHC-boranes, stereogenic at the boron atom, that has been prepared in only a few steps and in good yields (up to 93%). Key steps involve a chlorination/arylation sequence in the presence of simple Grignard reagents from bicyclic NHC-boranes. The high and unprecedented diastereoselectivity observed during the second step (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) has been rationalized through a plausible SRN1 mechanism thanks to EPR observations and DFT calculations.
:1 dr) has been rationalized through a plausible SRN1 mechanism thanks to EPR observations and DFT calculations.
Introduction
When it comes to enantioselective catalysis, the literature on the subject is widely dominated by transition metal catalysts. Nevertheless, due to their inherent cost, rarity and toxicity, alternative solutions are needed, and chemists worldwide have focused on developing more environmentally benign systems based on abundant materials and low toxicity metal-, organo- or bio-catalysis.1 In this respect, main-group elements offer great potential and have recently been presented as the fourth pillar of catalysis by Melen2 (after bio-, organo- and metal catalysis as defined by List).3 Particularly, as with transition metal catalysis, p-block elements allow the activation of various chemical bonds and, consequently enable chemical transformations.4Stereogenic main group elements other than carbon have gained interest, and a great deal of work has been done to obtain stereogenic S,5 Si,6 P7 or N atoms.8 Astonishingly, while chiral organoboron reagents are very useful in stereoselective transformations, efforts are still needed regarding the design of scaffolds stereogenic at boron, and only a handful of configurationally stable ones have been reported to date, mainly as racemates (Fig. 1).9 Expanding the chemical space of stereogenic tetrahedral-coordinated boron is of paramount importance and should open interesting new prospects especially in the field of metal-free catalysis where tetravalent boron structures are involved as key intermediates in borane or, more recently, in borenium catalysis.10 This area is still in its infancy and the design and preparation of chiral platforms stereogenic at boron are challenging tasks. This is mostly due to the labile nature of ligands attached to the tetracoordinated boron atom that makes it configurationally unstable. Accordingly, the choice of the nature of boron's ligands is crucial.
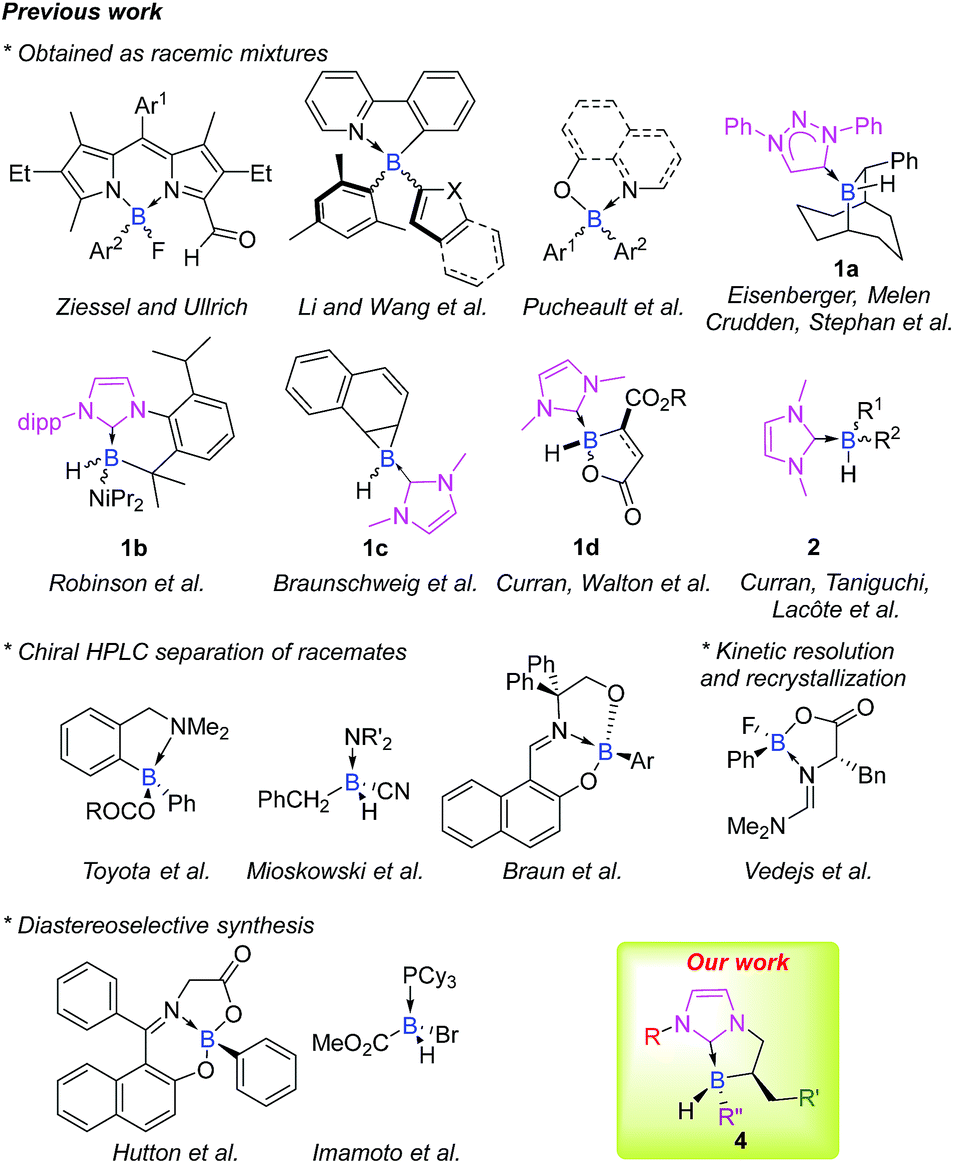 | ||
| Fig. 1 Representative examples of chiral platforms stereogenic at boron and the strategy of preparation. | ||
In this context, N-heterocyclic carbene boranes (NHC-boranes) have been the subject of significant research. Efforts have been devoted to gain better insights into their physicochemical properties and reactivity profiles11 and rapidly, their scope of application in organic synthesis (radical,12 ionic,13 and organometallic reactions14) has showcased their interesting added value. The recent infatuation for NHC ligated boranes also comes from the fact that they are easily available, stable and, more importantly, are quite resistant to dissociation.11 Nevertheless, only a few scaffolds of cyclic 1a–d15 and acyclic 2 (ref. 12d and 16) NHC-boranes stereogenic at the boron atom in the racemic form have previously been reported (Fig. 1).
Herein, we report the controlled diastereoselective preparation of stable chiral NHC-borane 4 stereogenic at boron (up to 99:1 dr) (Fig. 1) via boron–carbon bond formation starting from commercially available Grignard reagents and our in-house cyclic NHC-boranes 3 synthesized via an enantioselective rhodium-mediated boracyclopentannulation reaction.17 Thanks to this tunable platform a delicate balance between steric and electronic factors could be controlled directly at the boron atom.
Results and discussion
Preliminary results
Based on the literature precedents and starting from our tailor-made cyclic NHC-boranes 3, several methods were at our disposal for the formation of a new boron–carbon bond. However, despite all our efforts and the optimized procedure from the literature, the direct hydroboration of arynes with NHC-boranes16a provided an inseparable mixture of mono-4f and bis-phenylborane 4f′ products (up to a 9:1 ratio) and poor diastereoselectivity for 4f (up to 3:2 dr)‡ (Scheme 1).† On the other hand, insertion of the rhodium carbene 7 into the B–H bond pleasingly delivered the desired product 8a in a good yield (96%),16b albeit with modest diastereoselectivity (up to 4:1 dr)‡ and with a limited substrate scope.† Consequently, a more general approach, starting from easily and commercially available substrates was highly necessary.
 | ||
| Scheme 1 Diastereoselective one-step functionalization of chiral NHC-boranes. | ||
We turned our attention to a two-step diastereoselective chlorination/arylation procedure (Scheme 2).18 The cyclic chiral NHC-borane 3a simply treated with an equivalent of hydrogen chloride yielded the chlorinated NHC-borane 5a in a quantitative yield albeit in a 1:1 ratio of diastereomers.‡ Then, 5a was treated with an equivalent of the mesityl Grignard reagent at 50 °C for 5 hours in benzene. To our delight, the desired adduct 4a was obtained not only in an excellent 93% yield but also as a single diastereomer‡ (Scheme 2). The structure and the absolute stereochemistry of 4a were obtained by X-ray diffraction analysis, thus confirming the anticipated trans relationship between the B-mesityl group and the adjacent methyl substituent.19
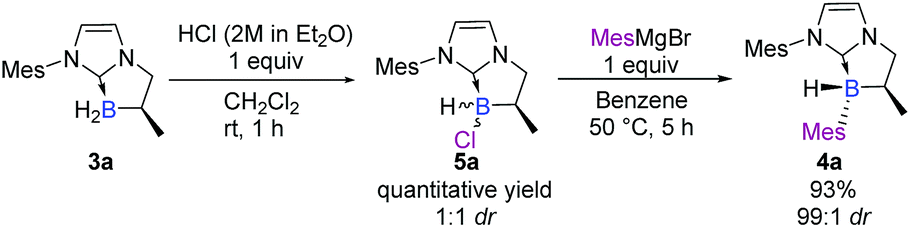 | ||
| Scheme 2 Diastereoselective two-step preparation of chiral NHC-boranes. | ||
Scope and limits of the reaction
Triggered by this impressive and unprecedented result, we next examined the scope and limits of the reaction. Application of these conditions across a variety of Grignard reagents was then undertaken. The reaction proceeded smoothly in most cases with total conversion,20 with yields up to 85% and diastereoselective ratios‡ ranging from 67:33 to 99:1 (Scheme 3). Compounds bearing electron-withdrawing 4c (66% yield, 87:13 dr, trans as the major diastereomer) or electron-donating groups 4d21 (61% yield, 87:13 dr, trans as the major diastereomer) showed similar reactivity, indicating that the electronic nature of the Grignard substrate does not play an essential role in the transformation. The reaction proved to be compatible with an aliphatic Grignard reagent and delivered the desired adduct 4g in a good yield (74%), albeit with lower diastereoselectivity (67:33 dr). As a further demonstration of the generality of the process, 2,6-methylphenyl and 2-methyl-1-naphthyl Grignard reagents were also successfully used in the reaction and yielded the corresponding NHC-boranes 4b22 and 4e in 85 and 35% yields, respectively (99:1 dr, trans as the major diastereomer).
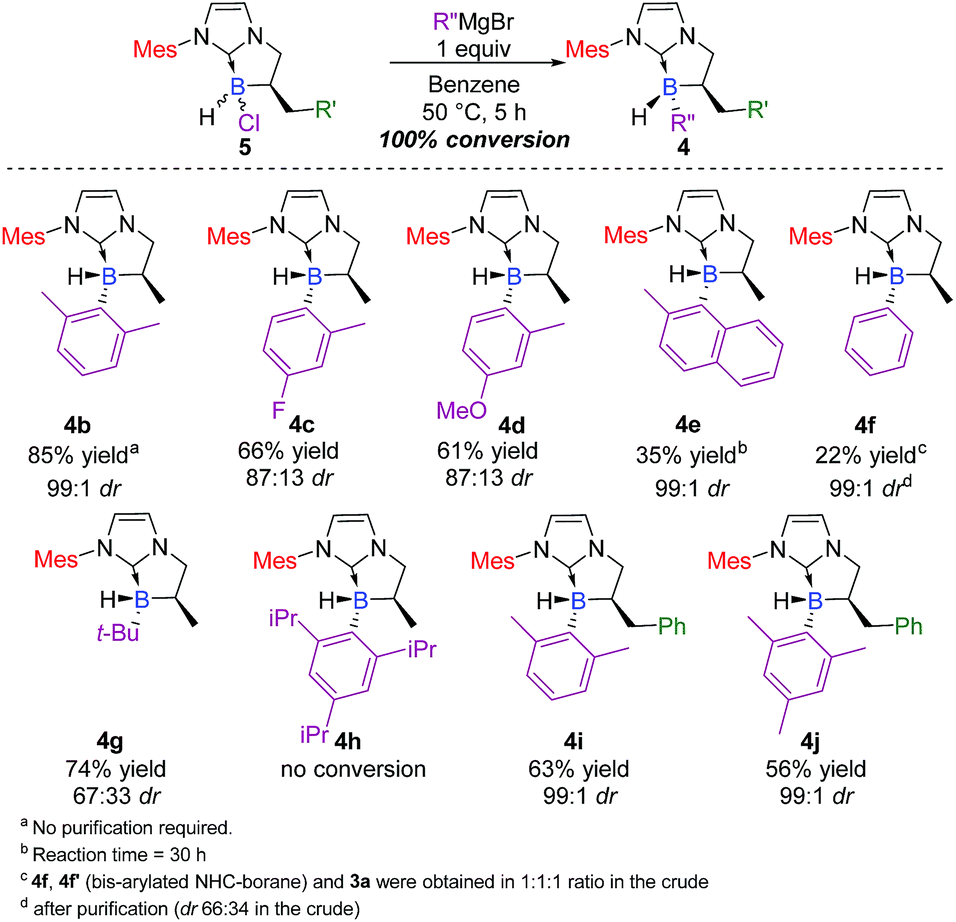 | ||
| Scheme 3 Scope of the diastereoselective functionalization of NHC-borane 5 with various Grignard reagents (ESI†). | ||
Limits were reached with simple phenyl magnesium bromide which gave a mixture of mono-(4f) and bis-phenyl-functionalized (4f′) NHC-boranes, and surprisingly, with the concomitant formation of 3a (1:1:1 ratio in the crude, see below in the Mechanism section for a plausible explanation and in the ESI for complementary experiments, pages S75 and S76†). On the other hand, no reaction could take place with a much hindered organomagnesium reagent (see 4 h). Finally, the wide scope of the process was evaluated with respect to the α-benzyl-substituted boron atom. The reaction yielded the corresponding trisubstituted NHC-boranes 4i–j in good yields (63 and 56% respectively) and excellent diastereoselectivities (99:1 dr).
Attention was then turned to the N-substituted NHC-backbone. As shown in Scheme 4, the reaction was compatible with Dipp (2,6-diisopropylphenyl) (4k–o) or Ph (4r–t) N-substituents providing a library of novel chiral NHC-boranes. Unfortunately, NHC-borane 5 did not react with the perfluorinated phenyl Grignard reagent, and a ratio of only 94:6 of the starting material to 4q was observed in the crude by 1H NMR. On the other hand, total conversion of the starting material and good diastereoselectivity (81:19 dr) was observed in the case of the vinyl derivative 4p; however, the latter decomposed during purification, but the yield was estimated to be 45% in the crude by 1H NMR after filtration on a short pad of Celite©.
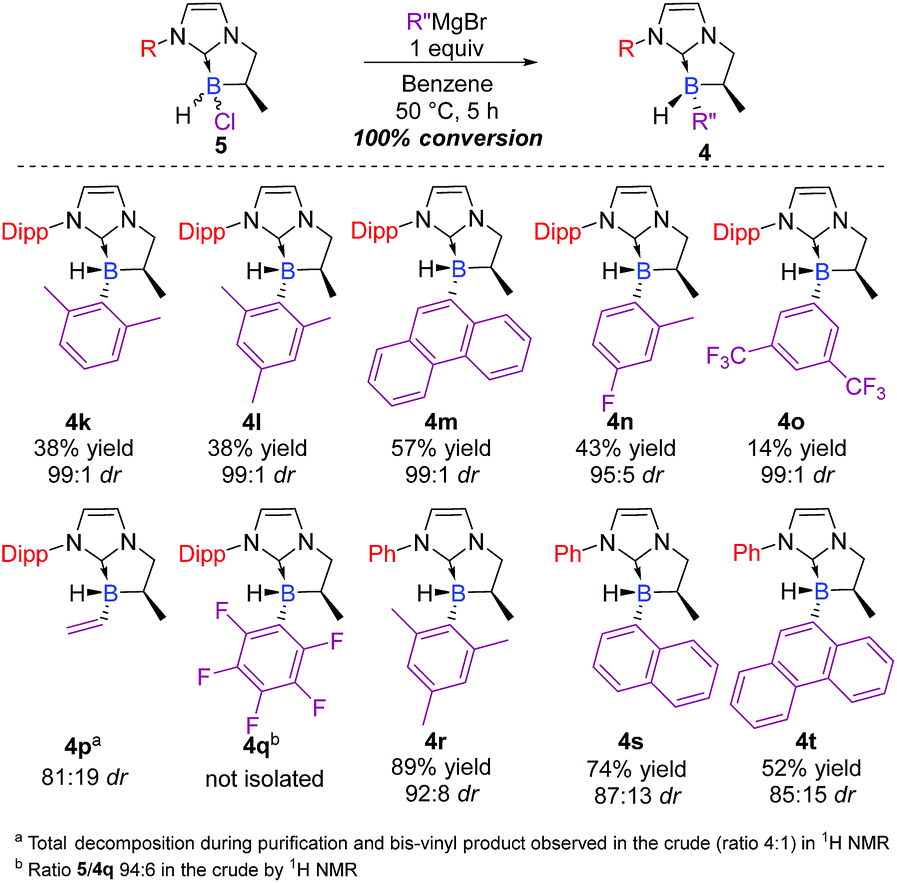 | ||
| Scheme 4 Scope of the diastereoselective functionalization of NHC-boranes with various N-substituents on the starting NHC-borane scaffold 5 (ESI†). | ||
To summarize, this two-step sequence led to the highly diastereoselective formation of novel NHC-boranes 4, stereogenic at boron, in good yields and with high diastereoselectivities, driven notably by bulky substituents on the nitrogen atom of the NHC-core (i.e. Mes or Dipp). Moreover, the excellent diastereoselectivity observed in the last step drew our attention and naturally led us to an understanding of the reaction mechanism. Obviously, the SN2 pathway was immediately ruled out. Therefore, two plausible mechanisms via a trivalent boron species remained: an ionic (SN1) pathway or a radical (SRN1) one (Scheme 5).
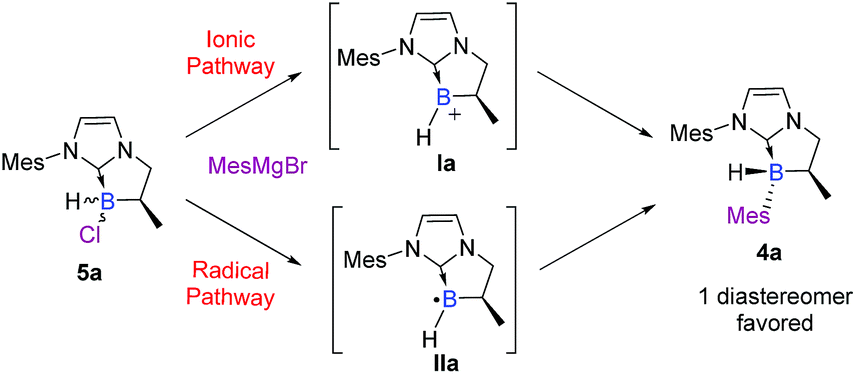 | ||
| Scheme 5 Suggested pathways for the diastereoselective functionalization of NHC-boranes. | ||
Insights into the mechanism
To further elucidate the mechanism, we decided to first examine the ionic route. We hypothesized that an intermediate borenium trivalent species Ia could result from chloride abstraction by a magnesium Lewis acid. Grignard reagents in solution are particularly complex since multiple species are present at thermodynamic equilibrium. More precisely, via the Schlenk equilibrium, magnesium bromide (MgBr2),23 a weak halophile, could promote the formation of the borenium derivative at thermodynamic equilibrium. However, when borane 5a was treated with MgBr2 (1 equiv) in benzene-d6, the intermediate borenium Ia was not observed by 11B NMR (Scheme 6, eqn (1)). Needless to say, the non-observation of Ia by 11B NMR is not evidence to rule out the formation of the intermediate borenium and excludes a plausible ionic pathway. Accordingly, the experiment with MgBr2 was next repeated in the presence of a borenium trap [P(O)Et3] to displace the potential thermodynamic equilibrium (Scheme 6, eqn (2)). However, the sole product observed was the MgBr2·P(O)Et3 complex (31P NMR; δ = 64.4 ppm in benzene-d6) and no trace of the expected boronium phosphine oxide complex was found. Based on these new results, the ionic pathway seems to be ineffective and additional studies were then undertaken towards the radical pathway.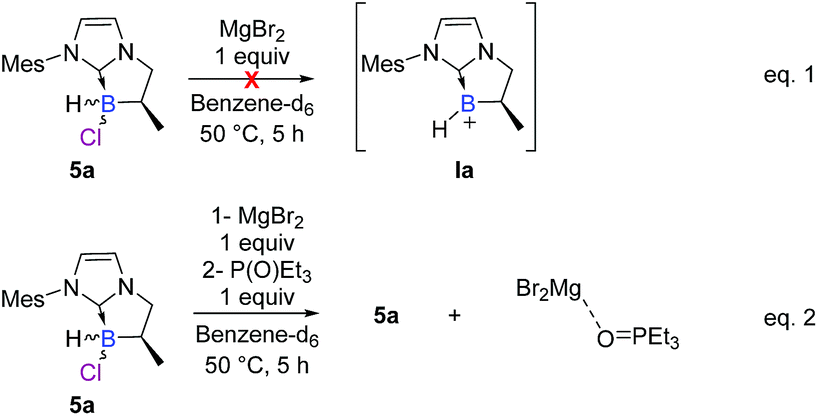 | ||
| Scheme 6 Control experiments of the hypothetical SN1 mechanism via the borenium cation Ia. | ||
A control EPR experiment was performed. A solution of NHC-borane 5a and the mesityl Grignard reagent in deoxygenated toluene was placed in the cavity of a CW-X-band EPR spectrometer. Before heating the medium, a spectrum showing three main lines of equal intensity, broadened by several weak hyperfine couplings, was systematically recorded (Scheme 7d).
 | ||
| Scheme 7 EPR spectra and DFT analysis (uM06-2X/6-311G+(d,p) level of theory) of 5a, 5a˙− and IIa. (a) Optimized structure of trans-5a. Selected bond distances (Å): B–CNHC 1.62; B–Cl 1.91. Contour plots (b) of the SOMO (isovalue = 0.05) and (c) of the electron density (isovalue = 0.004) of 5a˙−. Selected bond distances (Å): B–CNHC 1.59; B–Cl 2.01. (d) Experimental (black) X-band EPR spectrum of 5a˙− recorded at 25 °C in toluene and its superimposed simulation (red), aN1 = 4.7 G, aN2 = 0.8 G, and aB = 0.5 G. Contour plots (e) of the SOMO (isovalue = 0.05, NBO charges: B(0.219); CNHC(0.114)) and (f) of the electron density (isovalue = 0.004) of IIa. Selected bond distance (Å): B–CNHC 1.50. (g) Experimental (black) X-band EPR spectrum of IIa recorded at 50 °C in toluene and its superimposed simulation (red), aB = 4.5 G, aH = 0.7 G and aH = 1.6 G. | ||
Considering that Grignard reagents are subject to one-electron oxidation through a SET mechanism,24,25 this spectrum could reasonably be assigned to the radical anion species of 5a (5a˙−), as routinely described for the initiation step in the chain mechanism of the SRN1 reaction.26 Its computer simulation yielded the following hyperfine coupling constant (hfcc) values: aN1 = 4.7 G, aN2 = 0.8 G, and aB = 0.5 G, which are consistent with major localization of the unpaired electron on the carbenic atom of the NHC.
To rationalize these EPR observations, calculations were performed on the radical anion 5a˙− in the gas phase by means of DFT methods at the uM06-2X/6-311G+(d,p) level of theory (see the ESI† for details). The shape of the calculated singly occupied molecular orbital (SOMO) shows considerable localization of the unpaired electron on the carbenic atom of the NHC (Scheme 7b and c), as revealed in the EPR study. Moreover, the optimized geometry of the radical anion 5a˙− exhibits an elongated B–Cl bond length (201.1 pm) compared to that of the chlorinated NHC borane 5a (191.4 pm), which helps to predict the easy fragmentation of the B–Cl bond at 50 °C to generate the boryl radical intermediate IIa.
The medium was then heated to 50 °C directly in the spectrometer cavity and the EPR signal shown in Scheme 7g was recorded. Its analysis revealed the presence of a second radical species showing a hfcc with the boron isotope 11B in mixture with the one observed at room temperature. After simulation, the following hyperfine coupling constants were determined for this second radical: aB = 4.5 G, aH = 0.7 G and aH = 1.6 G. These results suggest considerable localization of the unpaired electron on the boron atom of IIa and are consistent with the literature precedent.27 The resulting half-life of the putative radical IIa is ca. 2.5 min (at 50 °C), revealing its relative persistence under our experimental conditions. Calculations were also performed on the boryl radical IIa (DFT methods at the uM06-2X/6-311G+(d,p) level of theory, see the ESI† for details). Firstly, the optimized geometry of the NHC boryl radical IIa exhibits a shorter B–CNHC bond length (149.6 pm) compared to that of the NHC-borane 5a (162.0 pm). The boryl radical is planar at the boron atom and the radical is partially delocalized into the π-system of the NHC. The boryl radical character of IIa is further supported by the shape of the calculated singly occupied molecular orbital (SOMO) and the respective contributions of the carbenic atom of NHC and the boron atom (B/CNHC ratio of the spin density in IIa is about 8.1:1.9 by NBO analysis),† fitting perfectly with the EPR observations (Scheme 7e and f).
Unfortunately, classical attempts to experimentally trap the putative radical IIa, in the presence of the Grignard reagent, did not reach our expectations (spin traps used: PBN, MNP, DMPO, and TEMPO). On the other hand, the boryl radical IIa was not EPR-observed after treatment of 5a in toluene at 25 °C or 50 °C with lithium or magnesium metals or a sodium/mercury metal amalgam. Based on some observations of the reactivity of our NHC-boranes with disulfide reagents, we managed to indirectly trap IIa and ascertain its reactivity (Scheme 8). In the presence of diphenyl disulfide, NHC-borane 3a gave the corresponding phenylsulfide NHC-borane adduct 6a in a 67% yield (88:12 dr),‡ together with the disubstituted adduct 6a′ (88:12 ratio)‡ (Scheme 8, eqn (1)) through a postulated radical mechanism.28 Under the same conditions, NHC-borane 5a was found to be inert (Scheme 8, eqn (2)).
 | ||
| Scheme 8 Reactivity of disulfide reagents with NHC-boranes 3a and 5a, and cross-reactivity with the Grignard reagent. | ||
A control spin trapping experiment was then performed with 3a in the presence of phenyldisulfide and α-phenyl-N-tertiary-butyl nitrone (PBN) as the spin trap (Scheme 9); an intense spin adduct signal was recorded. Its computer simulation allowed us to assign this signal to the boron-centered radical adduct of PBN (aN = 14.7 G, aB = 2.9 G and aH = 3.9 G), i.e. the nitroxide radical 9. This undoubtedly confirms the radical pathway for the formation of 6a. Interestingly, a mixture of NHC-borane 5a (1 equiv), phenyldisulfide (0.4 equiv) and MesMgBr (0.6 equiv) in benzene at 50 °C led to the NHC-boranes 4a and 6a and in a 3:2 ratio as the sole products in the crude by 1H and 11B NMR,† thus demonstrating that the boron centered radical IIa putatively arises from the reduction of the chlorinated NHC borane 5a by the Grignard reagent and then reacts with phenyldisulfide (Scheme 8, eqn (3)).
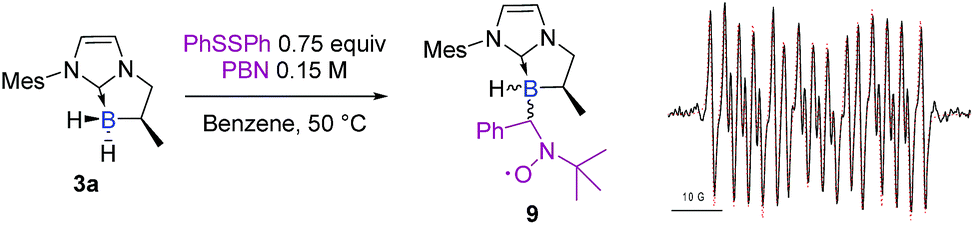 | ||
| Scheme 9 Experimental (black) X-band EPR spectrum of 9 recorded at 50 °C in toluene and its superimposed simulation (red), aN = 14.7 G, aH = 3.9 G, and aB = 2.9 G. | ||
Finally, and still from the viewpoint of rationalizing the formation of the by-products (3 and 4′) via a radical pathway, the stability of the final product 4a was analyzed under the reaction conditions. It turned out that a radical species formed from MesMgBr was able to abstract the remaining hydrogen atom (B–H). Accordingly, a medium containing NHC-borane 4a (1.0 equiv) and MesMgBr (0.3 equiv) was prepared in toluene and submitted to EPR analysis. If no signal was observed before heating, raising the temperature to 50 °C yielded an intense and wide signal (Scheme 10, eqn (1)). Its simulation (superimposed red dotted lines) was achieved by considering a single paramagnetic species with hyperfine couplings with a boron, a hydrogen and two nitrogen nuclei, and led to the following hyperfine splitting constants: aH = 4.2 G, aN = 2.0 G (2N), and aB = 6.4 G (11B–80%) or aB = 2.1 G (10B–20%) (Scheme 10b). The value obtained for aH is consistent with a hydrogen nucleus in the α-position towards the radical center. It should also be noted that the values obtained for aB11 and aB10 perfectly fit with the gyromagnetic ratio of these two isotopic nuclei: γB11/γB10 = 2.99 and aB11/aB10 = 3.05. According to this experiment, the spectrum was assigned to a boron-centered radical IIIa formed after the reaction with MesMgBr as a reductant (Scheme 10a). Calculations were also performed on the boryl radical IIIa (DFT methods at the uM06-2X/6-311G+(d,p) level of theory).† As in the boryl radical IIa, IIIa presents a shorter B–CNHC bond length (150.9 pm) compared to the NHC-borane 5a (162.0 pm). IIIa is planar at the boron atom and the radical is partially delocalized into the π-system of the NHC (B/CNHC ratio of the spin density in IIIa is about 8.0:2.0 by NBO analysis in the SOMO).† These results clearly show that Grignard reagents can abstract hydrogen atoms by homolytic cleavage, therefore explaining the formation of the subsequent disubstituted NHC-boranes with less hindered Grignard reagents (as observed with phenyl and vinyl Grignard, see 4f and 4p).
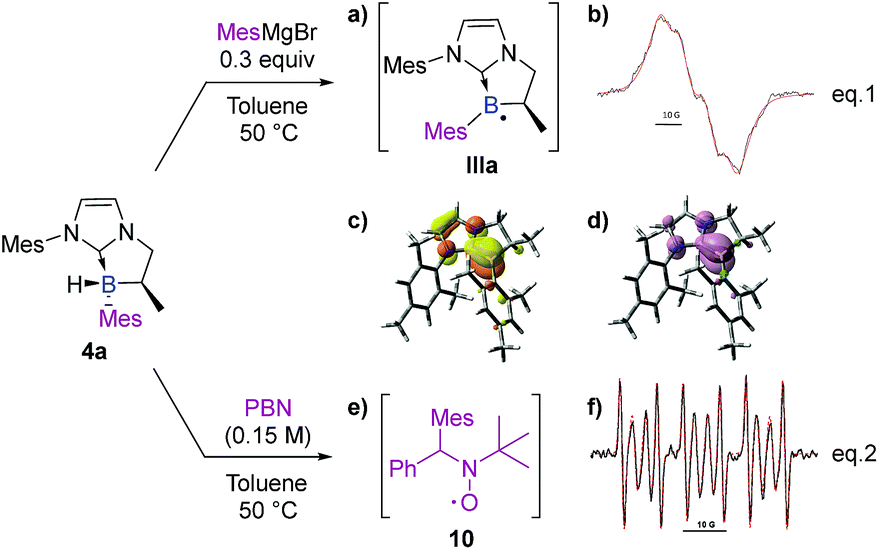 | ||
| Scheme 10 EPR spectra and DFT analysis (uM06-2X/6-311G+(d,p) level of theory) of IIIa and 10. (a) NHC-boryl radical IIIa. (b) Experimental (black) X-band EPR spectrum of IIIa recorded at 50 °C in toluene and its superimposed simulation (red), aH = 4.2 G, aN = 2.0 G (2N), and aB = 6.4 G (11B–80%) and aB = 2.1 G (10B–20%). Contour plots (c) of the SOMO (isovalue = 0.05, NBO charges: B(0.521); CNHC(0.093)) and (d) of the electron density (isovalue = 0.004) of IIIa. Selected bond distances (Å): B–CNHC 150.9; B–CMes 157.2. (e) Mesityl/PBN radical adduct 10. (f) Experimental (black) X-band EPR spectrum obtained by heating 4a in benzene at 50 °C in the presence of PBN, and its superimposed simulation (red); the major signal corresponds to the Mes/PBN adduct 10 (aN = 14.6 G, aH = 8.5 G, 60%). | ||
Interestingly, despite the fact that borane 4a in toluene was found to be EPR silent, additional spin-trapping experiments performed at 50 °C on 4a in the presence of the spin trap α-phenyl-N-tertiary-butylnitrone (PBN) clearly gave a 6 line EPR spectrum as shown in Scheme 10f (aN = 14.6 G and aH = 8.5 G), which could be assigned to the mesityl/PBN radical adduct 10 (Scheme 10e, eqn (2)), as previously described by Yoshida et al.,29 mixed with a minor unidentified carbon-centered radical (aN = 14.5 G, aH = 3.2 G, 40%, signal also observed before heating, see the ESI†). This last experiment clearly demonstrates the possible existence of an equilibrium between the radical species IIa and the formation of 4a through homolytic cleavage of the B–CAr bond. Consequently, hydrogen atom transfer may arise from any species able to produce hydrogen radicals. To acquire thermodynamic information, DFT calculations [M06-2X/6-311 g(d,p)] were performed on trans and cis isomers of 4a and 4d. Energy differences of 2.99 and 1.63 kcal mol−1 were, respectively, found in favor of the trans-diastereomer which is in full agreement with the experimental diastereomeric ratios obtained.†
Accordingly, based on these experimental observations and theoretical investigations, all these data suggest a probable SRN1-type mechanism initiated by a SET from the Grignard reagent to chlorinated NHC-borane 5a. The radical anion 5a˙− thus generated reacts upon fragmentation at 50 °C and provides the corresponding boryl radical IIa along with the chloride ion. Then, IIa reacts with the Grignard reagent in the chain mechanism as classically described in the SRN1 mechanism.26
Perspective
In the view point of using chiral platform 4 as a chiral hydride source in reduction reactions, the diastereoselective regeneration of the NHC-borane from the corresponding planar NHC-borenium is the key step in a hypothetical catalytic cycle, by analogy with the concept developed by Seebach on carbon stereocenters30 and transposed here to the boron atom. Accordingly, after the smooth and quantitative formation of the intermediate trivalent borenium IVa (11B NMR δ = 71.7 ppm, CD2Cl2), from 4a in the presence of trityl tetrakis(pentafluorophenyl) borate in deuterated dichloromethane, the borocation was treated with lithium borohydride as a quantitative hydride donor.§ To our delight, the hydride addition to borenium IVa fully regenerated the original diastereomer of NHC-borane 4a with a total retention of the configuration at the boron atom as indicated by 1H NMR for the crude. Moreover, starting from a less diastereo-enriched NHC-borane 4d (87:13 dr), the reaction of the borenium intermediate IVd (11B NMR δ = 64.1 ppm, CD2Cl2) with LiBH4 regenerates 4d with improved diastereoselectivity (93:7 dr) compared to that of the starting material (Scheme 11), showing that hydride addition on this borenium platform is highly diastereoselective. These experiments nicely illustrate the high potential of the designed species 4. This original chiral environment imparted by the stereogenic boron atom and assisted by the α-carbon stereocenter could be groundbreaking in the field of borenium-catalyzed reduction reactions, for example, where the stereo defining step is the hydride delivery one. To give more substance to the concept, the synthetic utility of these B-stereogenic NHC-boranes was checked in the borenium-catalyzed hydrosilylation of (E)-N,1-diphenylethan-1-imine with phenylsilane. The borenium was generated in situ from NHC-borane 4j with trityl tetrakis(pentafluorophenyl)borate. The catalytic process gave the corresponding amine in a 90% yield and a 60:40 enantiomeric ratio (Scheme 12). Despite the modest enantioselectivity, this is the best example of enantioselective hydrosilylation of an imine catalyzed by a chiral NHC-borenium.
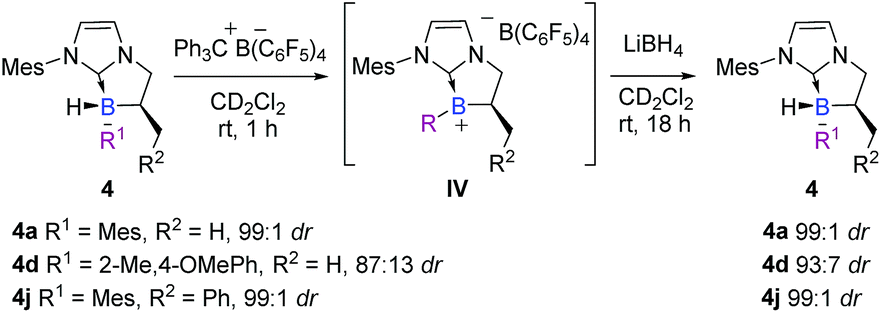 | ||
| Scheme 11 Highly diastereoselective addition of hydride on the borenium IV intermediate to recover chiral NHC-borane 4. | ||
 | ||
| Scheme 12 Imine reduction with silane catalyzed by chiral NHC-borenium. | ||
Conclusions
In conclusion, the diastereoselective synthesis of a series of new stable and enantioenriched NHC-borane complexes stereogenic at boron has been reported. Our results demonstrate that single diastereomers can be obtained in a high yield starting with a 1:1 diastereomeric mixture of chlorinated NHC-borane 5, representing a rare example of stereoselective synthesis of a chiral scaffold stereogenic at the boron atom.31 Preliminary results regarding the mechanistic studies indicate that a radical pathway (SRN1) is more likely to occur. Another important finding from this study is the stereoselective regeneration of the boron stereogenic center, from a borenium intermediate with a hydride, with excellent diastereoselectivity, therefore providing a promising strategy for metal-free enantioselective reduction catalysis.15a Future studies should explore the catalytic activity of this unprecedented scaffold, and more specifically, the scope and the selectivity of the borenium-catalyzed reactions, particularly FLP-type reduction reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the ANR (ANR-16-CE07-0002) for their generous support of our program. We gratefully acknowledge Dr Michel Giorgi (Spectropole, Aix Marseille Université) for X-Ray structural analysis, the CNRS research infrastructure RENARD for access to the EPR facilities, and Novachim, Aix Marseille Université, the CNRS, and the “Provence-Alpes-Côte d’Azur” Council for financial support.Notes and references
- R. A. Sheldon, I. Arends and U. Hanefeld, Green chemistry and catalysis, Wiley-VCH, 2007 Search PubMed.
- L. C. Wilkins and R. L. Melen, Coord. Chem. Rev., 2016, 324, 123 CrossRef CAS.
- B. List, Chem. Rev., 2007, 107, 5413 CrossRef CAS.
- (a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed; (b) F. G. Fontaine and E. Rochette, Acc. Chem. Res., 2018, 51, 454 CrossRef CAS PubMed; (c) R. L. Melen, Science, 2019, 363, 479 CrossRef CAS PubMed.
- S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147 CrossRef CAS PubMed.
- M. Oestreich, Synlett, 2007, 11, 1629 CrossRef.
- (a) M. J. Johansson and N. C. Kann, Mini-Rev. Org. Chem., 2004, 1, 233 CrossRef CAS; (b) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS.
- (a) D. Qian and J. Sun, Chem.–Eur. J., 2019, 25, 3740 CrossRef CAS PubMed; (b) F. A. Davis and R. H. Jenkins Jr., Synthesis and utilization of compounds with chiral nitrogen centers, in Asymmetric synthesis, ed. J. D. Morrison, J. W. Scott, Academic Press, San Diego, 1984, ch. 4, vol. 4 Search PubMed.
- (a) A. Haefele, C. Zedde, P. Retailleau, G. Ulrich and R. Ziessel, Org. Lett., 2010, 12, 1672 CrossRef CAS PubMed; (b) S. Toyota, T. Hakamata, N. Nitta and F. Ito, Chem. Lett., 2004, 33, 206 CrossRef CAS; (c) S. Toyota, F. Ito, N. Nitta and T. Hakamata, Bull. Chem. Soc. Jpn., 2004, 77, 2081 CrossRef CAS; (d) S. Toyota, F. Ito, T. Yamamoto, H. Akashi and T. Iwanaga, Bull. Chem. Soc. Jpn., 2006, 79, 796 CrossRef CAS; (e) L. Charoy, A. Valleix, L. Toupet, P. Le Gall, P. Pham van Chuong and C. Mioskowski, Chem. Commun., 2000, 2275 RSC; (f) T. Imamoto and H. Morishita, J. Am. Chem. Soc., 2000, 122, 6329 CrossRef CAS; (g) M. Braun, S. Schlecht, M. Engelmann, W. Frank and S. Grimme, Eur. J. Org. Chem., 2008, 5221 CrossRef CAS; (h) S. Schlecht, W. Frank and M. Braun, Beilstein J. Org. Chem., 2011, 7, 615 CrossRef CAS PubMed; (i) E. Vedejs, S. C. Fields, R. Hayashi, S. R. Hitchcock, D. R. Powell and M. R. Schrimpf, J. Am. Chem. Soc., 1999, 121, 2460 CrossRef CAS; (j) E. Vedejs, S. C. Fields, S. Lin and M. R. Schrimpf, J. Org. Chem., 1995, 60, 3028 CrossRef CAS; (k) P. F. Kaiser, J. M. White and C. A. Hutton, J. Am. Chem. Soc., 2008, 130, 16450 CrossRef CAS PubMed; (l) S. K. Mellerup, C. Li, J. Radtke, X. Wang, Q.-S. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 9634 CrossRef CAS PubMed; (m) L. Marciasini, B. Cacciuttolo, M. Vaultier and M. Pucheault, Org. Lett., 2015, 17, 3532 CrossRef CAS PubMed.
- (a) P. Eisenberger and C. M. Crudden, Dalton Trans., 2017, 46, 4874 RSC and see references therein; (b) T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246 CrossRef CAS PubMed; (c) M. J. Ingleson, in Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, Springer-Verlag, Berlin, 2015, vol. 49, pp. 39–71 Search PubMed; (d) B. Rao and R. Kinjo, Chem. - Asian J., 2018, 13, 1279 CrossRef CAS PubMed.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294 CrossRef CAS PubMed.
- Selected examples: (a) S. Ueng, M. M. Brahmi, E. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082 CrossRef CAS PubMed; (b) J. C. Walton, M. M. Brahmi, L. Fensterbank, E. Lacôte, M. Malacria, Q. Chu, S. Ueng, A. Solovyev and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 2350 CrossRef CAS PubMed; (c) X. Pan, E. Lacôte, J. Lalevée and D. P. Curran, J. Am. Chem. Soc., 2012, 134, 5669 CrossRef CAS PubMed; (d) T. Kawamoto, S. Geib and D. P. Curran, J. Am. Chem. Soc., 2015, 137, 8617 CrossRef CAS PubMed; (e) T. Watanabe, D. Hirose, D. P. Curran and T. Taniguchi, Chem.–Eur. J., 2017, 23, 5404 CrossRef CAS PubMed; (f) S. Ren, F. Zhang, J. Qi, Y. Huang, A. Xu, H. Yan and Y. Wang, J. Am. Chem. Soc., 2017, 139, 6050 CrossRef CAS PubMed; (g) Y. Yu, F. Zhang, J. Cheng, J. Hei, W. Deng and Y. Wang, Org. Lett., 2018, 20, 24 CrossRef CAS PubMed.
- Selected Examples: (a) M. Horn, H. Mayr, E. Lacôte, E. Merling, J. Deaner, S. Wells, T. McFadden and D. P. Curran, Org. Lett., 2012, 14, 82 CrossRef CAS PubMed; (b) Q. Chu, M. M. Brahmi, A. Solovyev, S.-H. Ueng, D. P. Curran, D. M. Malacria, L. Fensterbank and E. Lacôte, Chem.–Eur. J., 2009, 15, 12937 CrossRef CAS PubMed; (c) T. Taniguchi and D. P. Curran, Org. Lett., 2012, 14, 4540 CrossRef CAS PubMed; (d) V. Lamm, X. Pan, T. Taniguchi and D. P. Curran, Beilstein J. Org. Chem., 2013, 9, 675 CrossRef CAS PubMed; (e) M.-H. Wang and L.-Y. Chen, Tetrahedron Lett., 2017, 58, 732 CrossRef CAS; (f) T. Liu, L. Chen and Z. Sun, J. Org. Chem., 2015, 80, 11441 CrossRef CAS PubMed.
- Selected Examples: (a) J. Monot, M. M. Brahmi, S.-H. Ueng, C. Robert, M. D.-E. Murr, D. P. Curran, M. Malacria, L. Fensterbank and E. Lacôte, Org. Lett., 2009, 11, 4914 CrossRef CAS PubMed; (b) S. Nerkar and D. P. Curran, Org. Lett., 2015, 17, 3394 CrossRef CAS PubMed.
- (a) J. Lam, B. A. R. Günther, J. M. Farrell, P. Eisenberger, B. P. Bestvater, P. D. Newman, R. L. Melen, C. M. Crudden and D. W. Stephan, Dalton Trans., 2016, 45, 15303Y RSC; (b) Z. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326 CrossRef PubMed; (c) P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704 CrossRef CAS PubMed; (d) W. Dai, T. R. McFadden, D. P. Curran, H. A. Früchtl and J. C. Walton, J. Am. Chem. Soc., 2018, 140, 15868 CrossRef CAS PubMed; (e) W. Dai, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2019, 141, 3623 CrossRef CAS PubMed.
- (a) T. Taniguchi and D. P. Curran, Angew. Chem., Int. Ed., 2014, 53, 13150 CrossRef CAS PubMed; (b) X. Li and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 12076 CrossRef CAS PubMed; (c) A.-L. Vallet and E. Lacôte, Org. Biomol. Chem., 2019, 17, 4234 RSC.
- M. Toure, O. Chuzel and J.-L. Parrain, J. Am. Chem. Soc., 2012, 134, 17892 CrossRef CAS PubMed.
- For an example of arylation of borane chloride with Grignard reagent reaction, see: D. L. Crossley, R. J. Kahan, S. Endres, A. J. Warner, R. A. Smith, J. Cid, J. J. Dunsford, J. E. Jones, I. Vitorica-Yrezabal and M. J. Ingleson, Chem. Sci., 2017, 8, 7969 RSC.
- 4a, CCDC 1856403..
- In some cases, partial decomposition of NHC-boranes 4 was observed during the purification step, accounting for the observed decrease in yield (compared to conversion)..
- 4d, CCDC 1857909..
- 4b, CCDC 1857908..
- (a) W. Schlenck and W. Schlenck, Ber. Dtsch. Chem. Ges. B, 1929, 62, 920 CrossRef; (b) R. M. Peltzer, O. Eisenstein, A. Nova and M. Cascella, J. Phys. Chem. B, 2017, 121, 4226 CrossRef CAS PubMed.
- E. Shirakawa, Y. Hayashi, K. Itoh, R. Watabe, N. Uchiyama, W. Konagaya, S. Masui and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 218 CrossRef CAS PubMed.
- B. E. Haines and O. Wiest, J. Org. Chem., 2014, 79, 2771 CrossRef CAS PubMed.
- (a) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; (b) J. F. Bunnett, Acc. Chem. Res., 1978, 11, 413 CrossRef CAS; (c) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71 CrossRef CAS PubMed.
- (a) M. F. Silva Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135 CrossRef CAS PubMed; (b) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360 CrossRef CAS PubMed; (c) J. Monot, A. Solovyev, H. Bonin-Dubarle, E. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 49, 9166 CrossRef CAS PubMed.
- X. Pan, A.-L. Vallet, S. Schweizers, K. Dahbi, B. Delpech, N. Blanchard, B. Graff, S. J. Geib, D. P. Curran, J. Lalevée and E. Lacôte, J. Am. Chem. Soc., 2013, 135, 10484 CrossRef CAS PubMed.
- M. Kamimori, H. Sakuragi, T. Suehiro, K. Tokumaru and M. Yoshida, Bull. Chem. Soc. Jpn., 1977, 50, 1195 CrossRef CAS.
- D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed., 1996, 35, 2703 Search PubMed.
- D. P. Curran, N. A. Porter and B. Giese, in Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, VCH, Weinheim, 2008 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data and theoretical investigations; X-ray data for compounds 4a, 4b and 4d. CCDC 1856403, 1857908 and 1857909. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01454c |
| ‡ The diastereomeric ratio was determined for the crude by 1H NMR (in CD2Cl2), and for the diastereotopic CH2 protons of the boracyclic moiety. |
| § Silanes or boranes were also used as hydride donors in FLP systems, but as depicted in the literature the hydride is, at best, distributed between the borenium and the hydride source in the absence of an unsaturated acceptor. In these cases, the borane is not regenerated in the presence of a silane source and in the absence of a Lewis base (see ESI p. S82–S85 for spectra with PhSiH3 and Et3SiH as hydride sources). |
| This journal is © The Royal Society of Chemistry 2019 |