
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVery short highly enantioselective Grignard synthesis of 2,2-disubstituted tetrahydrofurans and tetrahydropyrans†
Claudio
Monasterolo
 ,
Helge
Müller-Bunz‡
and
Declan G.
Gilheany
*
,
Helge
Müller-Bunz‡
and
Declan G.
Gilheany
*
Centre for Synthesis and Chemical Biology, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: claudio.monasterolo@ucd.ie; declan.gilheany@ucd.ie
First published on 27th May 2019
Abstract
Phenones with elongated chains are shown to be excellent substrates for ligand-promoted asymmetric Grignard synthesis of tertiary alcohols. In turn this enables the simple, short and highly enantioselective (up to 96% ee) preparation of chiral 2,2-disubstituted THFs and THPs. Thus, asymmetric addition of Grignard reagents to γ-chlorobutyrophenones and δ-chlorovalerophenones takes place in the presence of a chiral diaminocyclohexyl-derived tridentate ligand and subsequent base-promoted intramolecular cyclisation occurs with complete retention of asymmetry. As examples of the methodology, we report the shortest syntheses of gossonorol, γ-ethyl-γ-phenylbutyrolactone and δ-methyl-δ-tolylvalerolactone, the joint-shortest and flexible synthesis of boivinianin A and the shortest formal syntheses of boivinianin B and yingzhaosu C.
Introduction
One of the driving forces for improvement in organic synthesis is the relentless pressure for shorter routes to the desired compounds. However, ultimately, there must be a limit to that development when the shortest possible route is approached. The question then arises as to when to cease development of new routes to the target as the number of steps becomes very low. In that context the Grignard synthesis of alcohols is a powerful tool because of the inherent flexibility of its disconnections but, until recently, it lacked a crucial aspect, namely a complementary flexible stereoselectivity. Recently we,1 and others,2,3 have made progress towards a general asymmetric Grignard synthesis of tertiary alcohols. Here we present a notable expansion of the substrate scope of our methodology, applied in a series of very short syntheses.Our search for a test bed for the above ideas within our own work led us to consider chiral 2,2-disubstituted tetrahydrofurans (THFs) and tetrahydropyrans (THPs). The former have come to prominence recently through the work of List and co-workers4 but more generally THFs, THPs and their derivatives are found as widespread structural motifs in many classes of bioactive compounds showing a range of biological activities (e.g.Fig. 1).5 The challenge to develop efficient methods for their asymmetric construction has led to the devising of many diverse, highly stereoselective synthetic strategies.6–10 However the 2,2-disubstituted cases remain particularly recalcitrant. There are only a handful of asymmetric syntheses reported, all of which suffer from limitations of very narrow scope, use of complex substrates and/or low selectivity.11
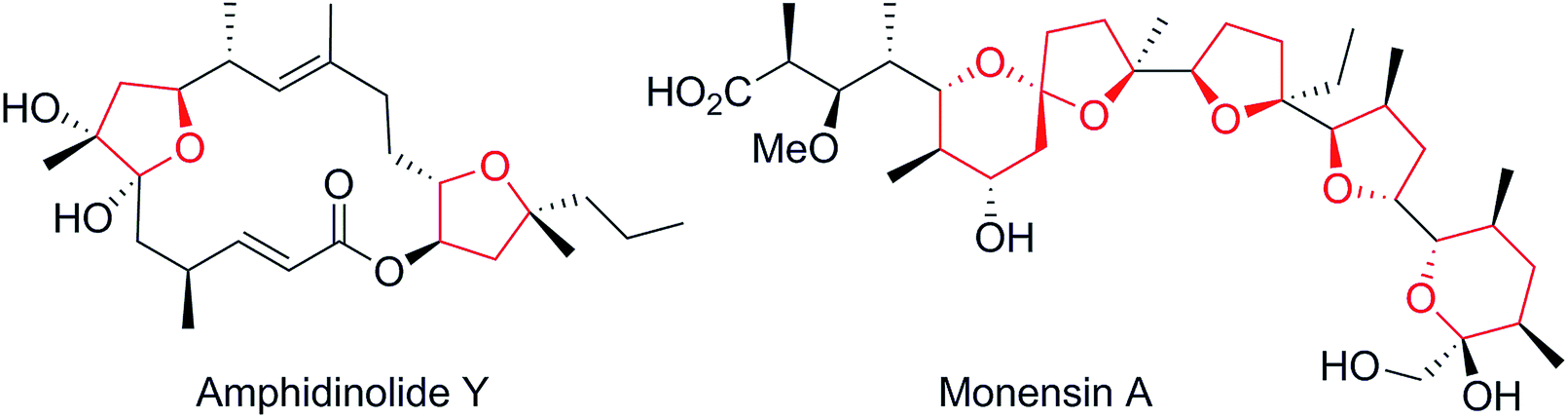 | ||
| Fig. 1 Examples of chiral bioactive 2,2-disubstituted THF. | ||
We now report a novel general asymmetric synthesis of both the THFs and the THPs that is complementary to those of List and co-workers and which enables a number of extremely short syntheses of products of biological interest.
Results and discussion
The route is based on our ligand-promoted asymmetric Grignard addition to ketones.1 Previously we reported that acetophenones gave only moderate selectivity but we have now discovered that chain extension in phenones renders them especially good substrates (illustrative examples in Scheme 1). In turn, this allowed us to explore their application in synthesis and we now report powerful ring-closure protocols using the resultant alcohols.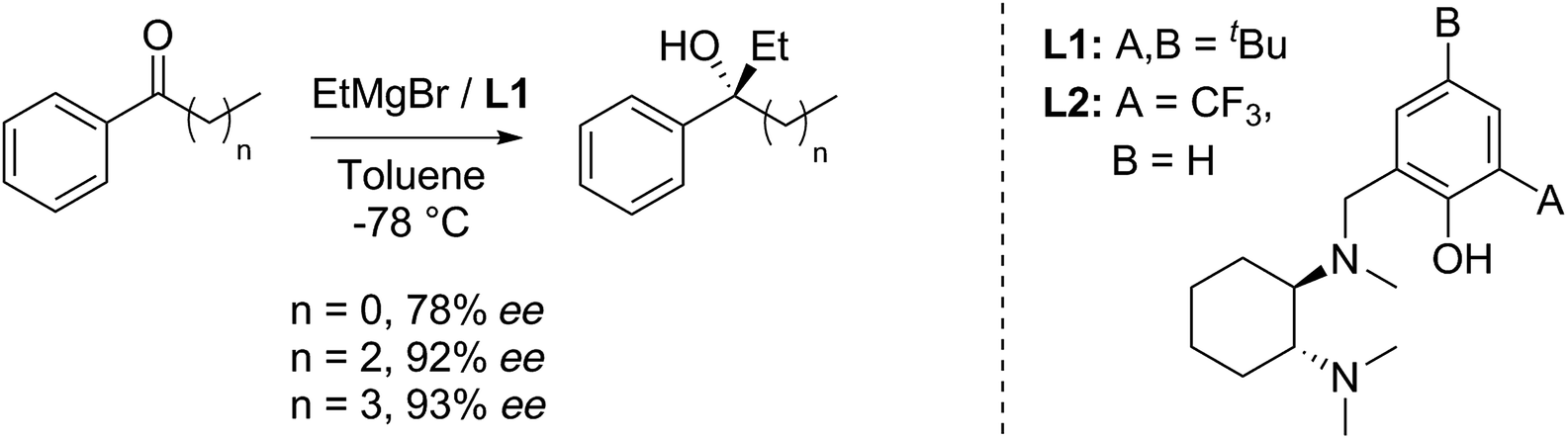 | ||
| Scheme 1 Side chain influence on asymmetric Grignard addition to phenones. | ||
Tertiary alcohols have not been commonly used for ring closure before but an exception is the use of the sesquiterpenoid (S)-gossonorol as an intermediate in the syntheses of a number of biologically interesting products such as boivinianin B.12 Our method does indeed provide the shortest route to date to (S)-gossonorol (2 steps, Scheme 2) from ketone 1, itself prepared by alkylation of p-methylacetophenone with 3,3-dimethylallyl bromide. Asymmetric addition of MeMgBr in the presence of ligand (S,S)-L1 delivered (S)-(−)-gossonorol in 66% yield and 93% ee.
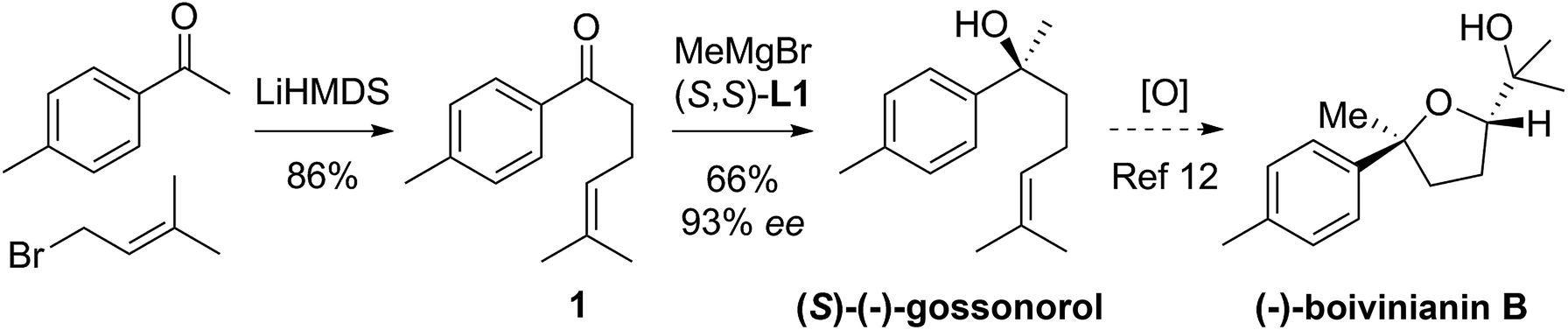 | ||
| Scheme 2 Two-step asymmetric synthesis of (S)-(−)-gossonorol and formal synthesis of (−)-boivinianin B. | ||
Previous syntheses of gossonorol12,13 show reduction in step count with time from 7-12b and 6-13a to 3-steps:13b that via functionalization of ketone 1 with dimethylzinc showing relatively lower selectivity,13a whereas Aggarwal and co-workers employed the lithiation/borylation strategy, achieving high stereoselectivity (98% ee) in 3-steps.13b That a further step reduction was possible demonstrates the power of the Grignard synthesis. Analogous arguments apply to the resultant formal synthesis of boivinianin B.
Turning now to the main focus of our work, which is the complementary and more general ring-closing of the ω-chloro species 2/3 to THFs and THPs as shown in Scheme 3. This had been reported in the racemic THF series by Capriati and co-workers14 and we thought it a good test of our methodology as a short route to these challenging classes of heterocycles. In particular, it was important to determine if our asymmetric Grignard reagents could tolerate the alkyl halide functionality.
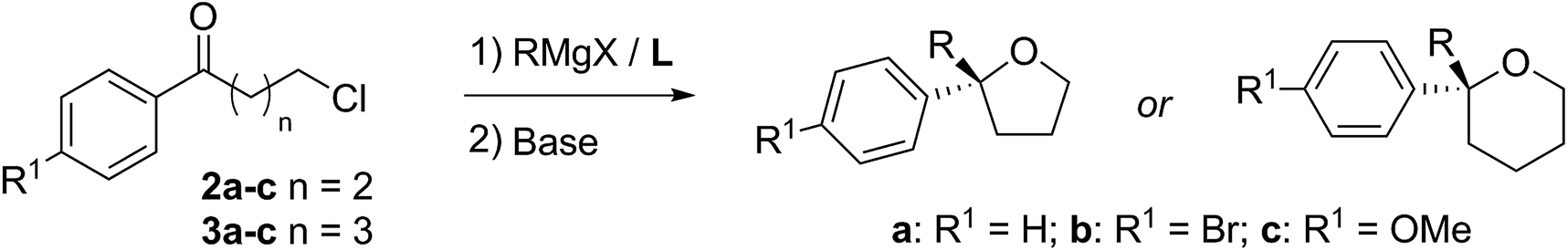 | ||
| Scheme 3 Asymmetric synthesis of 2,2-disubstituted THFs and THPs. | ||
As a test, we tried the reaction of 4-chlorobutyrophenone 2a with methylmagnesium bromide in the presence of ligand L1 (Table 1). To our delight, the reaction furnished alcohol 4aa in high enantioselectivity (Table 1, entry 1). Notably, only product 4aa was present in the crude reaction mixture, along with unreacted 2a, presumably derived from enolisation. In exploring the best conditions for the process, we found that, as we had previously observed,1a toluene is the solvent of choice to deliver high enantioselectivity (Table 1, entries 1–3). But in contrast to the previous work, in this case, it is the tert-butyl motif (L1) that gives the superior selection compared to the trifluoromethyl substituted ligand (L2, entry 4). Also in contrast, switching to the iodo Grignard reagent reduced selectivity (entry 5). Finally, variation of concentration and reactant equivalents allowed an increase in conversion without affecting the enantioselectivity (entries 6–9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Ligand (equiv.) | MeMgX (equiv.) | Conversionb (%) | ee (%) |
a On 0.1 mmol scale using (R,R)-ligand. MeMgX solution in Et2O, diluted in toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). Overall concentration 0.05 M. Quenched at −78 °C with IPA/H2O and NH4Cl sat (see ESI for details).
b Determined by 1H-NMR analysis of the crude reaction mixture after work-up, only product and returned starting material were present.
c Determined by HPLC on chiral stationary phase.
d Overall concentration 0.08 M.
e Overall concentration 0.1 M. :5). Overall concentration 0.05 M. Quenched at −78 °C with IPA/H2O and NH4Cl sat (see ESI for details).
b Determined by 1H-NMR analysis of the crude reaction mixture after work-up, only product and returned starting material were present.
c Determined by HPLC on chiral stationary phase.
d Overall concentration 0.08 M.
e Overall concentration 0.1 M.
|
|||||
| 1 | Toluene | L1 (1.0) | Br (2.1) | 71 | 91 |
| 2 | Et2O | L1 (1.0) | Br (2.1) | 65 | 53 |
| 3 | THF | L1 (1.0) | Br (2.1) | 87 | 0 |
| 4 | Toluene | L2 (1.0) | Br (2.1) | 78 | 80 |
| 5 | Toluene | L1 (1.0) | I (2.1) | 73 | 78 |
| 6d | Toluene | L1 (1.0) | Br (2.1) | 74 | 88 |
| 7e | Toluene | L1 (1.0) | Br (2.1) | 74 | 85 |
| 8 | Toluene | L1 (1.3) | Br (2.4) | 78 | 91 |
| 9 | Toluene | L1 (1.3) | Br (2.6) | 86 | 83 |
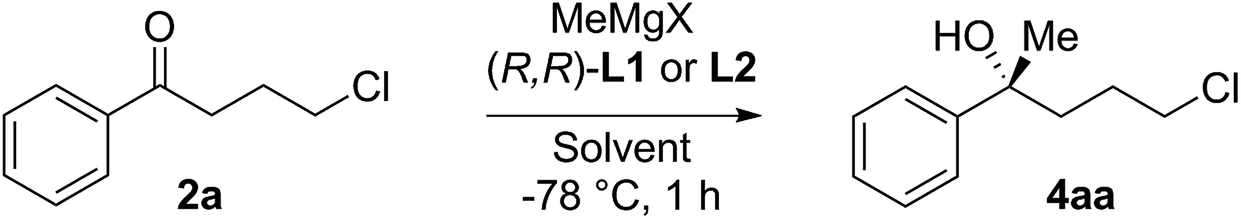
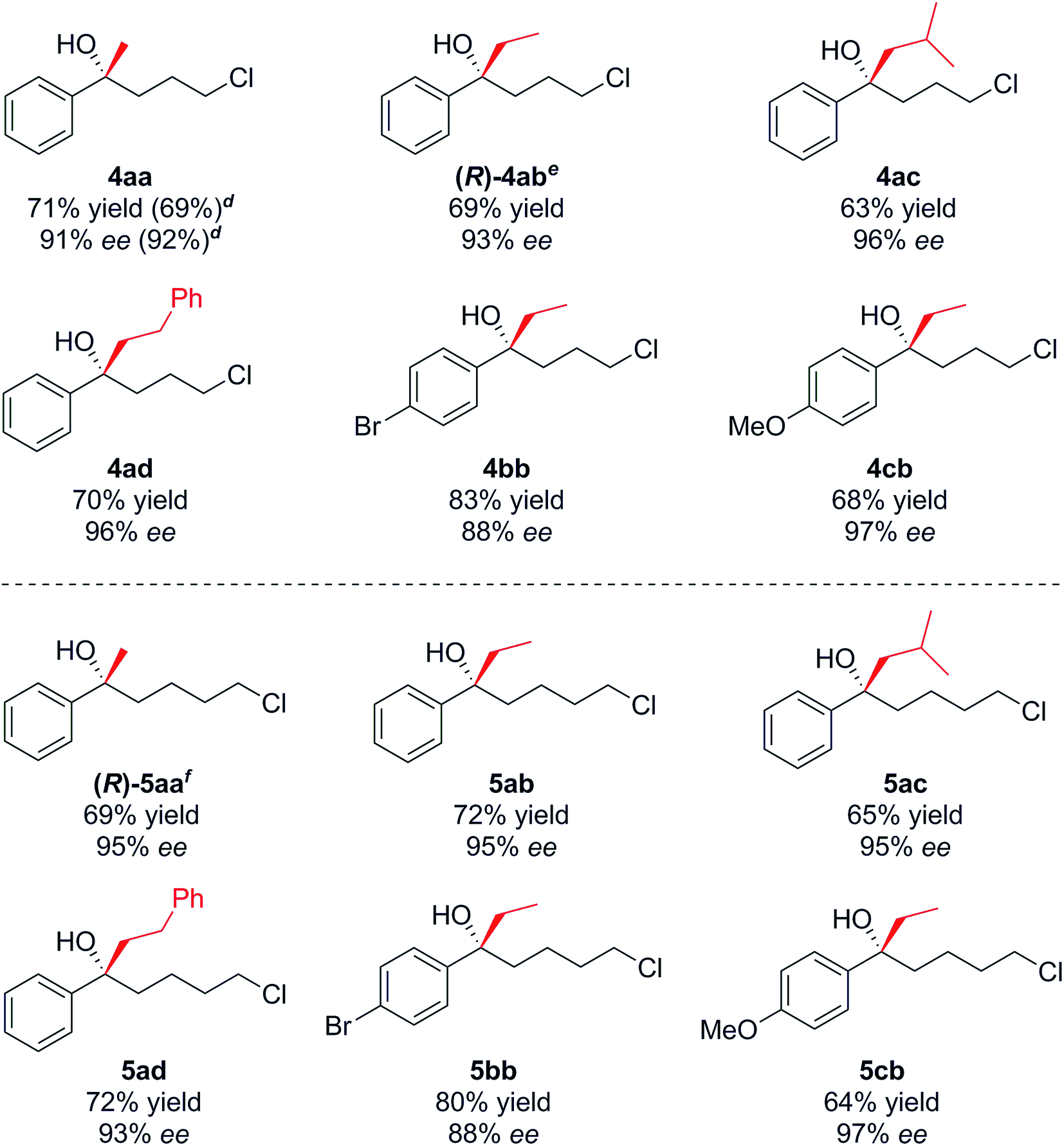
With optimised conditions in hand, we investigated the scope of the Grignard reagent and the substitution pattern on both γ-chlorobutyrophenones 2a–c and δ-chlorovalerophenones 3a–c (Table 2, R1 = H, Br, OMe). Various linear and branched Grignard reagents reacted well, producing the corresponding tertiary alcohols 4ab–4ad and 5aa–5ad in good yields and enantioselectivities. Similarly, electronic variation on the substrate was well tolerated for 4/5bb–cb, with just slight ee decreases for the more activated para-bromo cases 4bb and 5bbversus the deactivated para-methoxy cases 4cb and 5cb, which gave higher selectivity.15
|
|
|---|
| a Procedure as for Table 1, entry 8. b Isolated yields. c Enantiomeric excess determined by HPLC analysis on chiral stationary phase. d Results in brackets refers to the opposite enantiomer obtained by using (S,S)-L1. e Absolute configuration from derived compound 9. f Absolute configuration determined by conversion to urethane 8 – see text and Fig. 2, configurations of other compounds shown assumed by analogy, see text. |
|
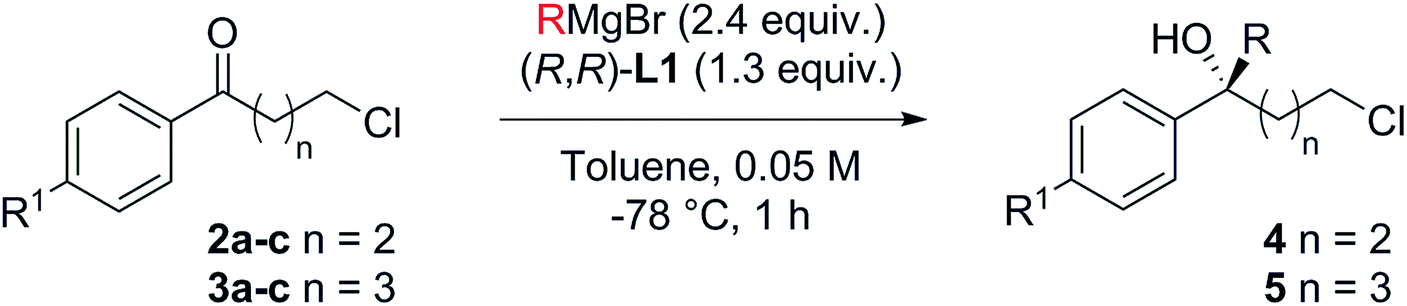
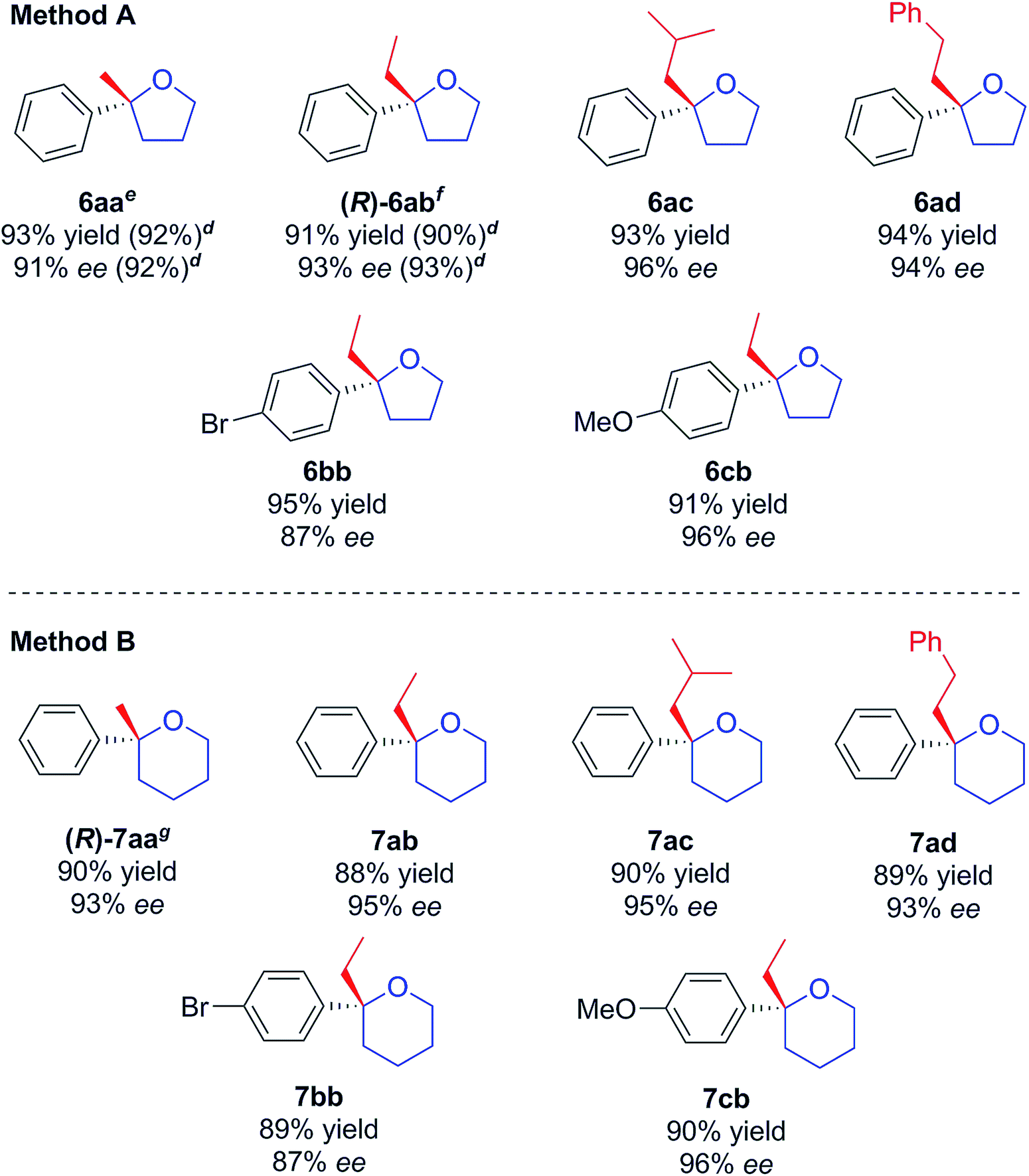
With a successful asymmetric synthesis of chiral, non-racemic, γ- and δ-chloro tertiary alcohols in hand, it was of great interest to see if they could be cyclised to the corresponding THF/THP derivatives (6 and 7) without loss of asymmetry. Again, we were delighted to find that this was so. Two slightly different methods (A & B) had to be used as detailed in Table 3. For the THFs, to avoid substitution side-products, sodium hydride was chosen as strong, non-nucleophilic base,16 with which alcohols 4 delivered the desired 2,2-disubstituted THFs 6 in excellent yields retaining the initial enantiomeric excesses (Table 3, Method A). For the THPs, NaH was much less effective and, after screening several bases (see Table 1, ESI†), we settled on KHMDS, with which chiral tertiary δ-chloro-alcohols 5 were smoothly cyclised delivering the corresponding THP derivatives 7 in high yields, again without loss of asymmetry and with only minor amounts of elimination to undesired terminal alkenes (Table 3, Method B). A one-pot addition/cyclisation strategy proved to be unsuccessful, resulting in complete recovery of tertiary alcohols.
|
|
|---|
| a Reactions were run on 0.1 mmol scale. Method A: NaH (5.0 equiv.), THF, 0 °C to r.t., 18 h. Method B: KHMDS (1.0 equiv.), THF, 0 °C to r.t., 1 h. b Isolated yields. c Enantiomeric excess determined by HPLC analysis on chiral stationary phase. d Results in brackets refer to the opposite enantiomer obtained by using (S,S)-L1. e Absolute configuration by comparison to literature (ref. 4a). f Absolute configuration from derived compound 9. g Absolute configuration based on that of compound 8 – see text and Fig. 2, configurations of other compounds shown assumed by analogy, see text. |
|
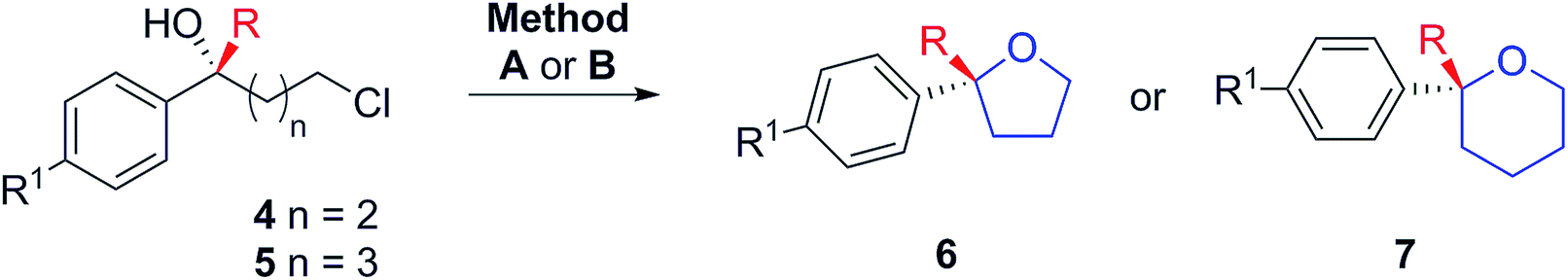
Regarding the absolute configurations of our produced materials, after much experimentation, we were able to unequivocally determine this to be (R) for (6-chloro-2-phenylhexan-2-ol) 5aa (obtained with R,R-L1) by single crystal X-ray crystallographic analysis of its urethane derivative 8 (Fig. 2) prepared by reaction of 5aa with p-bromophenyl isocyanate17 (see ESI†). The functionalization of chiral tertiary alcohols as urethanes to establish absolute configuration is particularly effective because the neutral conditions used avoid the risk of acid-catalysed racemisation.
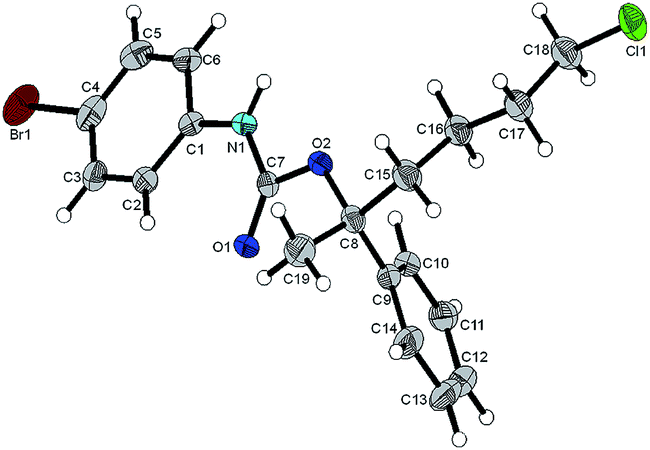 | ||
| Fig. 2 ORTEP diagram of 8 (thermal ellipsoids at 50% probability level). | ||
The (R)-enantiomer obtained for compound 5aa is consistent with the configurations observed in our preparations of (S)-gossonorol (Scheme 2), (S)-(−)-boivinianin A and lactones 9 & 12 (vide infra) and is consistent with our previous observations for asymmetric Grignard addition to phenones.1 The results suggest that the selectivity for asymmetric Grignard addition is such that (R,R)-L1 promotes addition of RMgBr to the phenone Si face and we have assumed that in showing the rest of the configurations in Tables 2 and 3. However this is still a tentative conclusion and more data are needed to build a definitive selectivity model.
We were now in a position to apply the novel asymmetric methodology for very short syntheses of suitable targets. γ-Ethyl-γ-phenylbutyrolactone 9, is an anticonvulsant and hypnotic drug, and is a candidate as a potential treatment for refractive epilepsy and Parkinson's disease.18 Our 3-step synthesis utilised compound (S)-6ab (Table 3, entry 2, 93% ee, prepared using (S,S)-L1), which was oxidised with ruthenium oxide and sodium periodate (Scheme 4) to (S)-9 in 66% yield without loss of enantiopurity. Its configuration was determined from the literature optical rotation.20
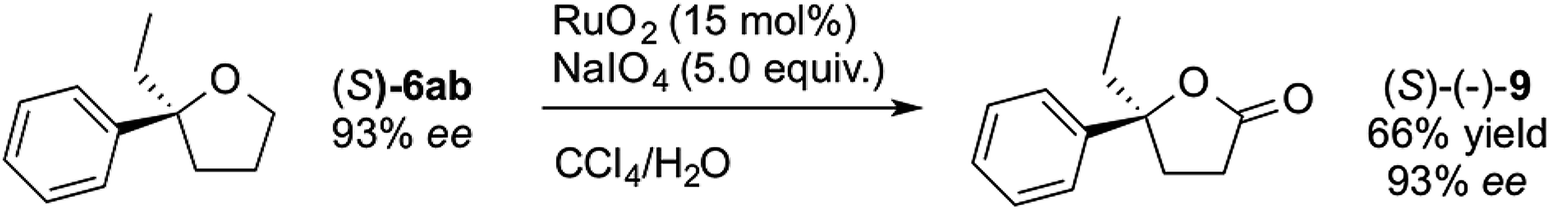 | ||
| Scheme 4 Preparation of γ-ethyl-γ-phenylbutyrolactone 9. | ||
A 2-step preparation of 9 has been reported19 but this was with moderate enantioselectivity (70% ee) while a previous 4-step route (with a BINOL-derived chiral auxiliary) gave 9 with good selectivity (92% ee).20
The method was then applied to the preparation of 2-methyl-2-tolyltetrahydrofuran 10, by addition of methylmagnesium bromide to commercially available 4′-methyl-4-chlorobutyrophenone 11, followed by cyclisation. Compound 10 could then undergo oxidation, thereby producing (S)-(−)-boivinianin A in three steps. (Scheme 5). Its configuration was determined from both the literature optical rotation and CSP-HPLC comparison.4
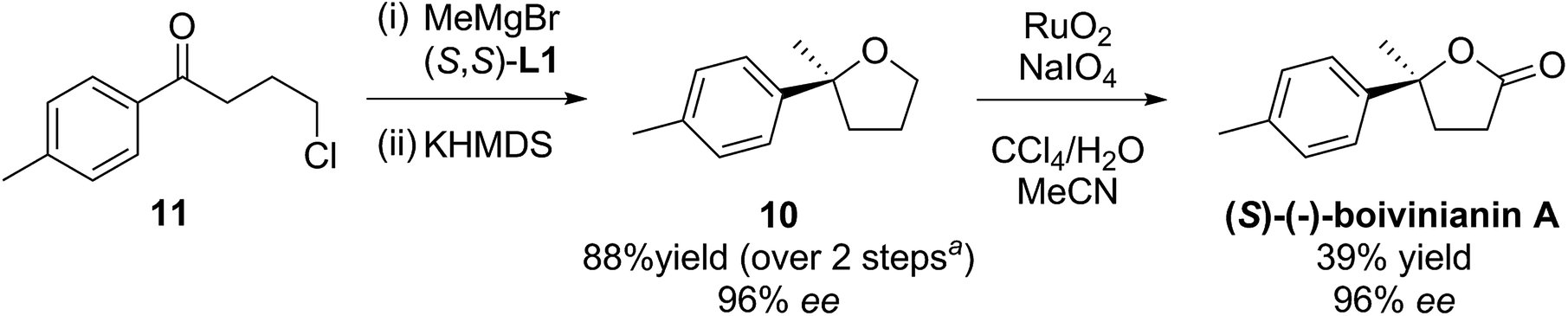 | ||
| Scheme 5 Preparation of (S)-(−)-boivinianin A. aAfter correcting for recovered starting material 11 in step (i). | ||
It is interesting that there are four previous asymmetric syntheses of boivinianin A, all of which are also 3-steps. List and co-workers reported two highly enantioselective (96% ee) preparations, the earlier involving resolution/oxidation,21 while this year's4 used the intramolecular hydroalkoxylation strategy. Alternative 3-step methods were reported by Shi and co-workers22 and Yeung and co-workers23 but these had relatively lower selectivities (79% & 82% ee respectively).
We also wished to check if a similar Grignard addition/cyclisation/oxidation strategy would work for THP analogues. We chose lactone (S)-12, previously used as an intermediate in the synthesis of (S)-gossonorol on route to boivinianin B12b and the anti-malarial yingzhaosu C.24 Addition of MeMgBr to commercially available ketone 13 in the presence of (S,S)-L1, followed by cyclisation gave THP 14, which could be oxidised with potassium permanganate and iron(III) chloride25 to lactone (S)-12 with excellent enantiopurity (Scheme 6). Its configuration was determined from the literature optical rotation.12b
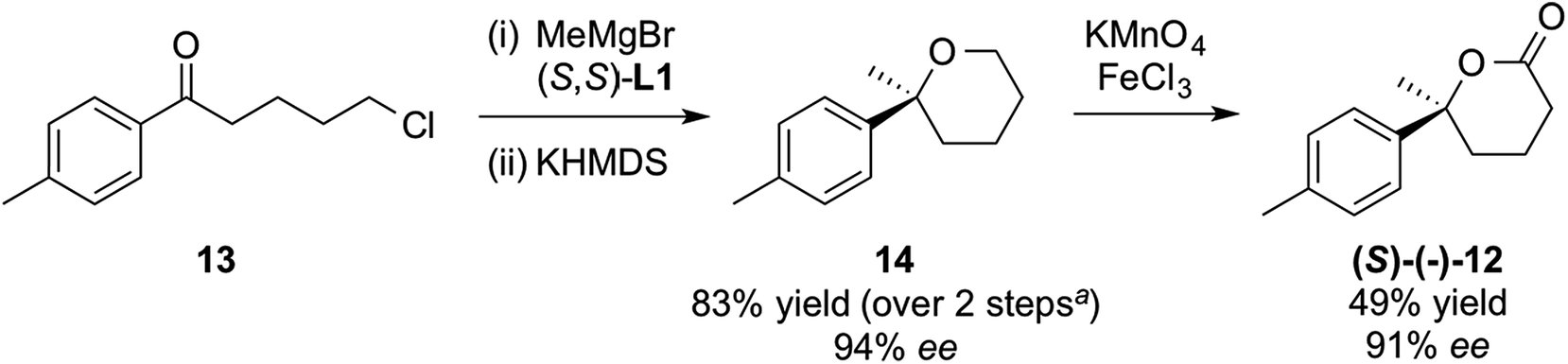 | ||
| Scheme 6 Application of methodology to THPs. aAfter correcting for recovered starting material 13 in step (i). | ||
It seems that para-substitution on the phenyl ring (at least with methyl) does not change the face-selectivity on the phenone. This 3-step synthesis of lactone 12 marks a notable improvement compared to other strategies reported so far, the shortest of which were both 5-steps.12b,26
Finally we briefly examined the utility of the method in the preparation of chiral 2,2-disubstituted oxetanes by addition of EtMgBr to 3-chloropropiophenone. The corresponding tertiary alcohol was obtained with reasonable stereocontrol (85% ee) but was contaminated with a significant amount of elimination side-product acrylophenone.
Conclusions
We have shown (i) that phenones with extended chains are excellent substrates for our ligand-promoted asymmetric Grignard synthesis of tertiary alcohols and (ii) that the methodology tolerates primary alkyl halides. This has enabled us to develop a new, very short, highly enantioselective method for the preparation of both 2,2-disubstituted THFs and THPs. We believe this is the first asymmetric preparation of these challenging oxygen heterocycle classes by Grignard synthesis from simple commercially available ketones. The avoidance of transition metals, together with the straightforward preparation and reuse of the ligand, ensures wide applicability of the method. This is demonstrated by the extremely short and selective syntheses of lactones 9 and 11 and natural compounds boivinianin A and gossonorol, the latter also providing formal syntheses of boivinianin B and yingzhaosu C. In each of these cases, our methodology is shorter and/or complementary to existing methods and easily allows analogue generation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Enterprise Ireland is acknowledged for postdoctoral funding for CM (grant CF/2013/3321). Arran Chemicals, Athlone, Ireland are gratefully acknowledged for gifts of resolved trans-1,2-diaminocyclohexane. We thank Dr Kirill Nikitin for valuable discussions.Notes and references
- (a) B. Bieszczad and D. G. Gilheany, Angew. Chem., Int. Ed., 2017, 56, 4272 CrossRef CAS PubMed; (b) B. Bieszczad and D. G. Gilheany, Org. Biomol. Chem., 2017, 15, 6483 RSC.
- A. V. R. Madduri, S. R. Harutyunyan and A. J. Minnaard, Angew. Chem., Int. Ed., 2012, 51, 3164 CrossRef CAS PubMed; A. V. R. Madduri, A. J. Minnaard and S. R. Harutyunyan, Chem. Commun., 2012, 48, 1478 RSC; J. F. Collados, R. Solà, S. R. Harutyunyan and B. Maciá, ACS Catal., 2016, 6, 1952 CrossRef.
- M. R. Luderer, W. F. Bailey, M. R. Luderer, J. D. Fair, R. J. Dancer and M. B. Sommer, Tetrahedron: Asymmetry, 2009, 20, 981 CrossRef CAS; Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876 CrossRef.
- (a) N. Tsuji, J. L. Kennemur, T. Buyck, S. Lee, S. Prévost, P. S. J. Kaib, D. Bykov, C. Farès and B. List, Science, 2018, 359, 1501 CrossRef CAS PubMed; (b) S. Lee, H. Y. Bae and B. List, Angew. Chem., Int. Ed., 2018, 57, 12162 ( Angew. Chem. , 2018 , 130 , 12339 ) CrossRef CAS PubMed.
- Leading references: (a) Nucleosides: B. Roy, A. Depaix, C. Périgaud and S. Peyrottes, Chem. Rev., 2016, 116, 7854 CrossRef CAS PubMed; (b) Macrolides: A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Álvarez, Chem. Rev., 2013, 113, 4567 CrossRef CAS PubMed; (c) Lignans: R. B. Teponno, S. Kusari and M. Spiteller, Nat. Prod. Rep., 2016, 33, 1044 RSC; (d) Acetogenins: C.-C. Liaw, J.-R. Liou, T.-Y. Wu, F.-R. Chang and Y.-C. Wu, in Progress in the Chemistry of Organic Natural Products 101, ed. A. D. Kinghorn, H. Falk, S. Gibbons and J. i. Kobayashi, Springer International Publishing, Cham, 2016, pp. 113–230 Search PubMed (e) Ionophores: A. Huczyński, Bioorg. Med. Chem. Lett., 2012, 22, 7002 CrossRef PubMed.
- For general reviews on oxygen heterocycles see: (a) M. C. Elliott, J. Chem. Soc., Perkin Trans. 1, 2002, 2301 RSC; (b) R. Jacques, R. Pal, N. A. Parker, C. E. Sear, P. W. Smith, A. Ribaucourta and D. M. Hodgson, Org. Biomol. Chem., 2016, 14, 5875 RSC, specifically for THFs, see: (c) J. P. Wolfe and M. B. Hay, Tetrahedron, 2007, 63, 261 CrossRef CAS PubMed; (d) G. Jalce, X. Franck and B. Figadère, Tetrahedron: Asymmetry, 2009, 20, 2537 CrossRef CAS; (e) A. de la Torre, C. Cuyamendous, V. Bultel-Poncé, T. Durand, J.-M. Galano and C. Oger, Tetrahedron, 2016, 72, 5003 CrossRef CAS, for THPs, see: (f) P. A. Clarke and S. Santos, Eur. J. Org. Chem., 2006, 2006, 2045 CrossRef; (g) I. Larrosa, P. Romea and F. Urpí, Tetrahedron, 2008, 64, 2683 CrossRef CAS; (h) N. M. Nasir, K. Ermanisa and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC; (i) F. Vetica, P. Chauhan, S. Dochaina and D. Enders, Chem. Soc. Rev., 2017, 46, 1661 RSC.
- Metal-catalysed cyclisations: Pd: (a) A. F. Ward and J. P. Wolfe, Org. Lett., 2010, 12, 1268 CrossRef CAS PubMed; (b) J. Kim, W. Jeong and Y. H. Rhee, Org. Lett., 2017, 19, 242 CrossRef CAS PubMed, Rh: (c) S. M. Nicolle, W. Lewis, C. J. Hayes and C. J. Moody, Angew. Chem., Int. Ed., 2015, 54, 8485 CrossRef CAS PubMed, Au: (d) J. L. Mascareñas and F. López, in Au-Catalyzed Synthesis and Functionalization of Heterocycles, ed. M. Bandicni, Springer International Publishing, Cham, 2016, pp. 1–52 Search PubMed, Os: (e) H. Sugimoto, T. Kanetake, K. Maeda and S. Itoh, Org. Lett., 2016, 18, 1246 CrossRef CAS PubMed, Other metals: (f) L. Ferrand, Y. Tang, C. Aubert, L. Fensterbank, V. Mouriès-Mansuy, M. Petit and M. Amatore, Org. Lett., 2017, 19, 2062 CrossRef CAS PubMed.
- Organocatalysed cyclisations: (a) D. Enders, C. Wang and A. Greb, Adv. Synth. Catal., 2010, 352, 987 CrossRef CAS; (b) K. Asano and S. Matsubara, J. Am. Chem. Soc., 2011, 133, 16711 CrossRef CAS PubMed; (c) G. He, F. Wu, W. Huang, R. Zhou, L. Ouyang and B. Han, Adv. Synth. Catal., 2014, 356, 2311 CrossRef CAS; (d) Y. Xie, G.-J. Cheng, S. Lee, P. S. J. Kaib, W. Thiel and B. List, J. Am. Chem. Soc., 2016, 138, 14538 CrossRef CAS PubMed.
- Cycloadditions: (a) A. T. Parsons, M. J. Campbell and J. S. Johnson, Org. Lett., 2008, 10, 2541 CrossRef CAS PubMed; (b) C. Zhang, M. Xu, J. Ren and Z. Wang, Eur. J. Org. Chem., 2016, 2016, 2467 CrossRef CAS; (c) Y. Zhou, F.-L. Zhu, Z.-T. Liu, X.-M. Zhou and X.-P. Hu, Org. Lett., 2016, 18, 2734 CrossRef CAS PubMed; (d) A. Gupta, R. Kholiya and D. S. Rawat, Asian J. Org. Chem., 2017, 6, 993 CrossRef CAS.
- Sugar derivatisation: (a) L. V. R. Reddy, A. D. Roy, R. Roy and A. K. Shaw, Chem. Commun., 2006, 3444 RSC; (b) R. W. Foster, C. J. Tame, D.-K. Bučar, H. C. Hailes and T. D. Sheppard, Chem.–Eur. J., 2015, 21, 15947 CrossRef CAS PubMed.
- (a) A. Blanc and F. D. Toste, Angew. Chem., Int. Ed., 2006, 45, 2096 CrossRef CAS PubMed; (b) A. G. Smith, M. C. Slade and J. S. Johnson, Org. Lett., 2011, 13, 1996 CrossRef CAS PubMed; (c) N. Cox, M. R. Uehling, K. T. Haelsig and G. Lalic, Angew. Chem., Int. Ed., 2013, 52, 4878 CrossRef CAS PubMed; (d) B. M. Trost and D. A. Bringley, Angew. Chem., Int. Ed., 2013, 52, 4466 CrossRef CAS PubMed; (e) B. A. Hopkins, Z. J. Garlets and J. P. Wolfe, Angew. Chem., Int. Ed., 2015, 54, 13390 CrossRef CAS PubMed; (f) Y.-F. Cheng, X.-Y. Dong, Q.-S. Gu, Z.-L. Yu and X.-Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 8883 CrossRef CAS PubMed; (g) J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y.-Y. Yeung, ACS Catal., 2018, 8, 850 CrossRef CAS.
- (a) K. Abecassis and S. E. Gibson, Eur. J. Org. Chem., 2010, 2010, 2938 CrossRef; (b) M. Aursnes, J. E. Tungen and T. V. Hansen, J. Org. Chem., 2016, 81, 8287 CrossRef CAS PubMed.
- (a) S. González-López, M. Yus and D. J. Ramón, Tetrahedron: Asymmetry, 2012, 23, 611 CrossRef; (b) V. K. Aggarwal, L. T. Ball, S. Carobene, R. L. Connelly, M. J. Hesse, B. M. Partridge, P. Roth, S. P. Thomas and M. P. Webster, Chem. Commun., 2012, 48, 9230 RSC.
- L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192 RSC.
- However alcohol 4cb was configurationally unstable and slowly underwent racemisation at room temperature, unlike the other tertiary alcohols prepared. This can be attributed to stabilisation by the methoxy group of the corresponding carbocation formed during the racemisation process.
- (a) C. Gronnier, S. Kramer, Y. Odabachian and F. Gagosz, J. Am. Chem. Soc., 2012, 134, 828 CrossRef CAS PubMed; (b) B. Guo, G. Schwarzwalder and J. T. Njardarson, Angew. Chem., Int. Ed., 2012, 51, 5675 CrossRef CAS PubMed; (c) B. Guo and J. T. Njardarson, Chem. Commun., 2013, 49, 10802 RSC.
- Alcohol 5aa was reacted with p-bromophenylisocyanate in the presence of SnII Lewis acid to obtain urethane 8 with complete retention of configuration, see ESI.† T. Francis and M. P. Thorne, Can. J. Chem., 1976, 54, 24 CrossRef CAS . Crystallographic data for 8 have been deposited with the Cambridge Crystallographic Data Centre [CCDC 1883098].
- (a) V. Díaz, F. Rasgado and R. L. Y. Avila, Acta Med., 1994, 30, 9 Search PubMed; (b) A. V. L. Rasgado, I. Villanueva and V. D. Fernando, Acta Pharm., 2017, 67, 215 Search PubMed.
- (a) S. Fukuzawa, K. Seki, M. Tatsuzawa and K. Mutoh, J. Am. Chem. Soc., 1997, 119, 1482 CrossRef CAS; (b) K. Mikami and M. Yamaoka, Tetrahedron Lett., 1998, 39, 4501 CrossRef CAS; (c) N. J. Kerrigan, P. C. Hutchison, T. D. Heightman and D. J. Procter, Org. Biomol. Chem., 2004, 2, 2476 RSC.
- Y. Tamai, T. Hattori, M. Date, S. Koike, Y. Kamikubo, M. Akiyama, K. Seino, H. Takayama, T. Oyama and S. Miyano, J. Chem. Soc., Perkin Trans. 1, 1999, 1685 RSC.
- I. Čorić, S. Müller and B. List, J. Am. Chem. Soc., 2010, 132, 17370 CrossRef PubMed.
- B. Wang, Y.-M. Shen and Y. Shi, J. Org. Chem., 2006, 71, 9519 CrossRef CAS PubMed.
- X. Jiang, C. K. Tan, L. Zhou and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2012, 51, 7771 CrossRef CAS PubMed.
- J. Boukouvalas, R. Pouliot and Y. Fréchette, Tetrahedron Lett., 1995, 36, 4167 CrossRef CAS , this is racemic synthesis..
- The use of RuO2/NaIO4 resulted in degradation of THP derivative 14 to a mixture of unidentified products.
- K. Spielmann, R.-M. de Figueiredo and J.-M. Campagne, J. Org. Chem., 2017, 82, 4737 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1883098. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00978g |
| ‡ To whom correspondence about the single crystal X-ray diffraction analysis should be addressed E-mail: helge.muellerbunz@ucd.ie |
| This journal is © The Royal Society of Chemistry 2019 |