
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of supported Ru–Triphos catalysts for continuous flow application in selective nitrile reduction†
Robert
Konrath
 ab,
Frank J. L.
Heutz
a,
Norbert
Steinfeldt
b,
Nils
Rockstroh
b and
Paul C. J.
Kamer
*b
ab,
Frank J. L.
Heutz
a,
Norbert
Steinfeldt
b,
Nils
Rockstroh
b and
Paul C. J.
Kamer
*b
aSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: rk49@st-andrews.ac.uk
bLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein Straße 29a, 18059 Rostock, Germany. E-mail: Paul.Kamer@catalysis.de
First published on 23rd July 2019
Abstract
The selective catalytic hydrogenation of nitriles represents an important but challenging transformation for many homogeneous and heterogeneous catalysts. Herein, we report the efficient and modular solid-phase synthesis of immobilized Triphos-type ligands in very high yields, involving only minimal work-up procedures. The corresponding supported ruthenium–Triphos catalysts are tested in the hydrogenation of various nitriles. Under mild conditions and without the requirement of additives, the tunable supported catalyst library provides selective access to both primary amines and secondary imines. Moreover, the first application of a Triphos-type catalyst in a continuous flow process is presented demonstrating high catalyst life-time over at least 195 hours without significant activity loss.
Introduction
Amines represent essential platform chemicals pivotal for industrial synthesis of both bulk and fine chemicals as well as for pharmaceuticals.1–3 Common methods towards the catalytic production of primary amines include amination of alcohols,4 reductive amination,5 and reduction of nitro compounds6–9 and amides.10,11 Atom-economical hydrogenation of nitriles using inexpensive molecular hydrogen can offer an alternative route to primary amines. While conventional but less benign procedures often use stoichiometric amounts of metal hydrides, heterogeneous catalysts are widely applied in manufacture of bulk chemicals, e.g. reduction of adiponitrile to hexamethylenediamine in the production of nylon-66.12 Heterogeneous catalysts are commonly based on noble (Pd) or base metals (RANEY®-Ni, Co and iron), which often suffer from reduced selectivity combined with low functional group tolerance.13–16 Base metals usually require harsh conditions and the use of additives, such as ammonia and mineral acids, to suppress side reactions.13,17 Milder reaction conditions can be applied when utilizing supported Pd catalysts, however, these are still reliant on acidic conditions to produce the terminal amines selectively.18–20 On the other hand, there has been a growing interest to develop highly selective homogeneous catalysts for applications in nitrile reduction under very mild conditions. Most transition-metal complexes are based on precious metals featuring well-defined ligand structures,17,21,22 but more recently also base metals23,24 have been employed. Tripodal phosphorus-based ligands, such as Triphos (Fig. 1, I), remain less studied in nitrile reduction despite their successful application in hydrogenations of challenging carboxylic acids derivatives.25–29 Initial attempts by Suarez and Fontal using a Ru–Triphos system in pure benzonitrile led to an unselective mixture of amines.30 Beller and co-workers reported on the tripodal phosphorus ligand II employed in Ru-catalyzed nitrile hydrogenation achieving high selectivities for a range of aliphatic and aromatic monoamines under mild and additive-free conditions,31 whereas no activity in this reaction was observed for a cobalt–Triphos catalyst.32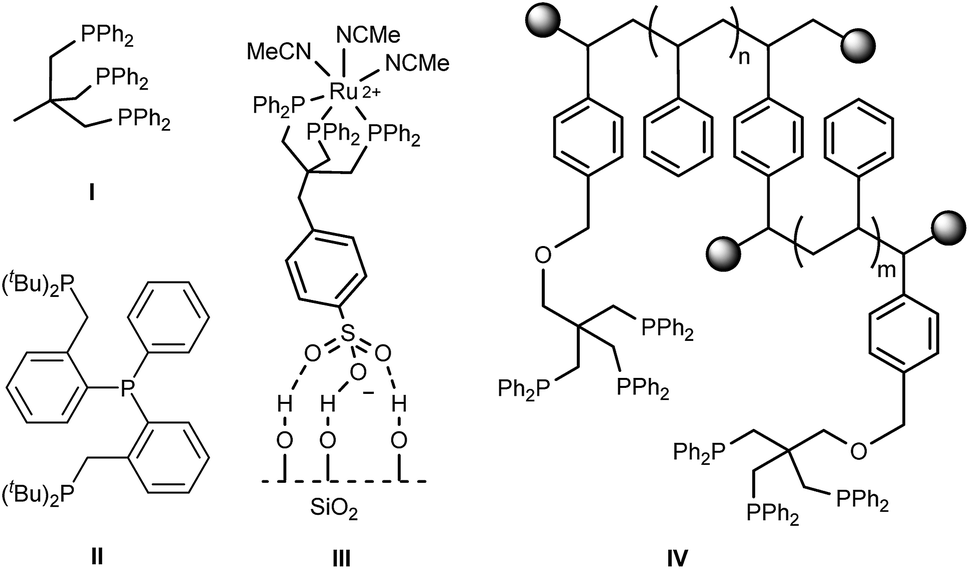 | ||
| Fig. 1 Tripodal ligands I and II31 and examples of immobilized Triphos on silica (III)33 and polystyrene (IV).34 | ||
Moreover, the intrinsic separation issue of homogeneous catalysts remains a limiting factor for many applications.35 To overcome this problem a tremendous research effort has been devoted to heterogenization of molecular catalysts.35–41 Thus, the ideal catalyst would combine the best of both worlds, i.e. high activity, selectivity and tunability of homogeneous catalysis and the ease of separation and recycling of heterogeneous catalysts. The enhanced stability of supported tridentate ligand-based catalysts compared to mono- and bidentate analogues can prevent metal leaching from the support. Approaches towards immobilization of modified Triphos ligands on dendrimers,42 in ionic liquids43 and in aqueous biphasic systems44–46 have been explored. Furthermore, SiO2 grafted Ru–Triphos (Fig. 1, III) has been applied in the reduction of benzonitrile to the corresponding secondary imine.33 Immobilization of Triphos onto polymeric supports was accomplished by a following post-modification47 as well as a bottom-up approach (Fig. 1, IV).34 However, all of the above mentioned strategies are cases where a single modified ligand is tethered to a support. Often these synthetic alterations require multiple steps and can lead to low yields due to troublesome purifications or functional group incompatibility which in turn hampers the creation of ligand diversity on the support.48 Moreover, tuning ligand properties by modifying the phosphorus donor moieties remains challenging. Opposed to the uncomplicated synthesis of C3V symmetrical arylphosphine tripods,49 unsymmetrical, mixed phosphorus donor ligands rely on less straightforward synthetic protocols.50–52 Different leaving groups bound to the tripodal backbone to facilitate selective phosphine substitution as well as additional borane protection and removal steps are often necessary. In case of a C1 symmetric Triphos analogue modified with one di(p-tolyl)phosphine and two PPh2 groups an overall yield of 21% starting from 3-methyl-3-oxetanemethanol was obtained.52 This in turn calls for a more efficient modular approach. Solid-phase synthesis (SPS) provides a promising alternative offering the combinatorial synthesis of libraries of tripodal ligands on a support in high yields requiring only minimal workup procedures.53–55 Moreover, SPS has proven to be an invaluable tool for automated parallel synthesis of ligand libraries in terms of application in high throughput experimentation.56–58 Since covalently bound to the support, recycling of the corresponding heterogenized catalysts can be vastly facilitated making them suitable candidates for application in continuous flow catalysis. Continuous processes allow exploring the long-term catalyst performance but also offer environmentally benign and safe processing, facile optimization for scale-up and high process reliability for multiphasic hydrogenation reactions, which in general are a prerequisite for economical process development.59–61 Herein, we report a modular SPS protocol towards recyclable Ru–Triphos complexes and their application in highly selective nitrile reduction. Furthermore, the first application of Triphos-based ligands under continuous flow processing is presented.
Results and discussion
Solid-phase synthesis of supported Triphos ligands
The SPS of supported tripodal phosphorus ligands was adapted from literature procedures in solution-phase.51,62 The secondary phosphines 1a–d immobilized on Merrifield resin cross-linked with 1% and 4% divinylbenzene (DVB, MF), JandaJel™ resin (JJ) and polystyrene (PS) were prepared as we reported previously.63,64 Deprotonation of 1a–d by using an excess of lithium diisopropylamide (LDA) yielded the corresponding supported lithium phosphides (Scheme 1, step 1). | ||
| Scheme 1 SPS approach towards supported Triphos ligands L1–L5. | ||
In the gel-phase 31P NMR spectrum, phenyl substituted lithiated phosphines (Li·1a–c) exhibit signals at δ = −40 ppm (see Fig. 2 for a representative example), while for tBu bearing lithium phosphide Li·1d supported on PS a shift of around Δδ = 7 ppm is observed. Next, Li·1a–d were reacted at room temperature with a slight excess of 1,1,1-tris(chloromethyl)ethane readily proceeding to the supported phosphine dimethylenechloride intermediates 2a–d (Scheme 1, step 2). Quantitative conversion was confirmed by 31P NMR by the appearance of a single peak at around δ = −30 ppm in cases of 2a–c and δ = −11.4 ppm for 2d respectively. High purity of representative compound 2a was confirmed by determination of the P and Cl loading by elemental analysis. Subsequently, intermediates 2a–d were treated with a secondary lithium phosphide to obtain the desired supported Triphos ligands L1–L5. By using an excess of LiPPh2 (for L1–L4) or LiP(o-Tol)2 (for L5) at 60 °C, full conversions to the corresponding ligands were achieved leading to a library of 5 supported Triphos-type ligands (Scheme 1, step 3).
 | ||
| Fig. 2 31P NMR (THF) monitoring of the representative SPS of supported Triphos-ligand L1. | ||
The Triphos analogue L1 supported on MF exhibits two single peaks (δ = −25.4 and −28.1 ppm) in the 31P NMR spectrum, occurring in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. 2). For JJ-supported ligand L2 as well as L3 immobilized on the MF 4% DVB, nearly identical spectra were obtained. In case of PS supported L4, two peaks were observed for the PPh2 groups (δ = −25.0 and −25.8 ppm) which are present in an approximate ratio of 1:1:1 with the PtBu moiety (δ = −8.8 ppm). This could be attributed to the presence of the bulky tBu group leading to sterically hindered rotation which allows for the detection of the two inequivalent diastereotopic phosphorus nuclei on the NMR time scale. Similarly, L5 shows a signal for the PPh group (δ = −28.3 ppm) together with two signals for the diastereotopic P(o-Tol)2 moieties (δ = −51.9 and −52.0 ppm). The supported tripodal ligands L1–L5 were further characterized using FTIR and Raman spectroscopy and the exact phosphorus loading was determined by elemental analysis (see ESI†).
:1 ratio (Fig. 2). For JJ-supported ligand L2 as well as L3 immobilized on the MF 4% DVB, nearly identical spectra were obtained. In case of PS supported L4, two peaks were observed for the PPh2 groups (δ = −25.0 and −25.8 ppm) which are present in an approximate ratio of 1:1:1 with the PtBu moiety (δ = −8.8 ppm). This could be attributed to the presence of the bulky tBu group leading to sterically hindered rotation which allows for the detection of the two inequivalent diastereotopic phosphorus nuclei on the NMR time scale. Similarly, L5 shows a signal for the PPh group (δ = −28.3 ppm) together with two signals for the diastereotopic P(o-Tol)2 moieties (δ = −51.9 and −52.0 ppm). The supported tripodal ligands L1–L5 were further characterized using FTIR and Raman spectroscopy and the exact phosphorus loading was determined by elemental analysis (see ESI†).
Solid-phase synthesis of supported ruthenium complexes
The corresponding immobilized Ru–Triphos complexes C1–C5 were synthesized by treatment of L1–L5 with [RuHCl(PPh3)3CO] in toluene at 80 °C (Scheme 2). | ||
| Scheme 2 Synthesis of supported complexes C1–C5. | ||
The quantitative disappearance of the free ligand peaks was monitored by 31P NMR until full complexation was achieved. C1 shows three new broad signals appearing in an approximate 1:1:1 ratio (Fig. 3). While two sharper single resonances are observed at δ = 15.3 and 1.2 ppm for the remote PPh2 moieties, the broader signal of the phosphorus atom bound to the polymeric support exhibits a splitting (δ = 49.8 and 41.8 ppm) which could be attributed to the presence of isomeric Ru-complexes as observed in non-symmetrical homogeneous systems.52 The gel-phase 31P NMR of JandaJel™-supported complex C2 shows an analogous spectrum compared to C1 (Fig. S13, see ESI†). In this case however, much sharper signals are observed as a consequence of enhanced solution-like behavior of the JJ resin. Coupling constants cannot be assigned due to peak broadening which is commonly observed for molecular complexes heterogenized on solid supports.34,63,65,66 However, the chemical shifts of C1 are in line with those obtained for its solution-phase counterpart 3 (δ = 48.9, 12.2 and −0.4 ppm, Fig. 4a). 3 solely differs in a Ph group attached to the phosphorus atom instead of a benzyl group belonging to the MF support. Moreover, the supported complexes were characterized by solid-state 31P, 1H and 13C MAS NMR. In the case of C1, the 31P MAS NMR spectrum exhibits two signals appearing in a 1:2 ratio (Fig. 4c). The presence of a CO ligand and Ru–H was confirmed by FTIR and Raman spectroscopy (see ESI†). For CO and Ru–H, bands are observed at 1975–1968 cm−1 and 1925–1920 cm−1, respectively.
 | ||
| Fig. 3 31P NMR (THF) monitoring of the representative SPS of supported Ru–Triphos complex C1. | ||
 | ||
| Fig. 4 (a) 31P NMR spectrum of solution-phase analogue 3, (b) gel-phase 31P NMR spectrum of supported complex C1 and (c) solid-state 31P MAS NMR spectrum of C1. Rotational sidebands are denoted by asterisks (*). | ||
Application in Ru-catalyzed nitrile reduction
Subsequently, the supported Ru–Triphos library was screened in the hydrogenation of nitriles to primary mono- and diamines. Prior to catalyst screening, benzonitrile (S1) and C1 were used to determine optimized reaction conditions. 1,4-Dioxane was chosen as solvent providing sufficient polymer swelling properties opposed to iPrOH, which was found to be the ideal solvent in homogeneous Ru–Triphos catalyzed nitrile reduction.31 After 18 h at 10 bar H2 pressure and 100 °C, S1 was readily converted with excellent selectivity towards the desired benzylamine (A) using 1.0 mol% of C1 (Table 1, entry 1). A similar performance was obtained when reducing the temperature to 80 °C (Table 1, entry 2) whereas 0.5 mol% gave 47% conversion with 97% selectivity to A (Table 1, entry 3). Further optimization conditions are listed in Table S1.† For selectivity enhancement towards the primary amine, both homogeneous and heterogeneous catalysts often require the use of base additives.13,18–20,67,68 Remarkably, mild and additive-free conditions can be applied for this highly selective heterogeneous Ru–Triphos system similar to the homogeneous system of Beller and co-workers.31 When screening the whole supported catalyst library at 100 °C and 1.0 mol% catalyst loading, JJ-supported C2 and PS-bound C4 performed analogously compared to C1 (Table 1, entries 4 and 6). Notably, C3 immobilized on MF 4% DVB, which is lacking solvent dependent swelling properties opposed to C1, gave 62% conversion and 97% selectivity (Table 1, entry 5) which could be driven to full conversion after 50 h.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst [mol%] | T [°C] | Conv.b [%] | Selectivityb [%] | ||
| A | B | C | ||||
| a Conditions: substrate (0.5 mmol), dioxane (1.0 mL), H2 (10 bar), 18 h. b Conversion and selectivity determined by GC using dodecane as internal standard. c Data taken from ref. 33; conditions: substrate (2.15 mmol), n-octane (30 mL), H2 (30 bar), 12 h. | ||||||
| 1 | C1 (1.0) | 100 | >99 | 99 | 1 | <1 |
| 2 | C1 (1.0) | 80 | >99 | 98 | 1 | <1 |
| 3 | C1 (0.5) | 80 | 48 | 97 | 3 | <1 |
| 4 | C2 (1.0) | 100 | >99 | 99 | 1 | <1 |
| 5 | C3 (1.0) | 100 | 62 | 97 | 3 | <1 |
| 6 | C4 (1.0) | 100 | >99 | 99 | 1 | <1 |
| 7 | C5 (1.0) | 100 | 86 | 5 | 92 | 2 |
| 8 | 3 (1.0) | 100 | >99 | 30 | 68 | 2 |
| 9c | III (0.2) | 100 | 95 | 3 | 88 | 5 |

Unexpectedly, C5 bearing more bulky P(o-Tol)2 groups resulted in 86% conversion with 92% selectivity towards the secondary imine B (Table 1, entry 7) which could be increased to >99% selectivity at 56% conversion after 8 hours (Table S1,† entry 10). In case of the solution-phase combination of [Ru(cod)(methallyl)2] (cod = cyclooctadiene) and II, no activity was observed when using 1,4-dioxane as a solvent.31 When applying homogeneous catalyst 3 under the same conditions as for its supported counterparts, an unselective mixture of A (30%), B (68%) and C (2%) was obtained (Table 1, entry 8). The superior selectivity of the developed heterogeneous catalyst can be attributed to the high local catalyst to substrate concentration within the confined space of a resin bead suppressing non-catalytic side-reactions. Moreover, the SiO2 supported system III (Fig. 1) gave 88% selectivity towards B instead of A (Table 1, entry 9).33 This showcases both the competitively high selectivity towards primary amines and the facile tunability of the resin-bound Triphos using SPS. Next, C1 was employed to determine the substrate scope. Electron-withdrawing para-bromo (S2) and para-fluoro-benzonitrile (S3) were converted selectively whereas NO2-substituted S4 led to no activity accompanied by the formation of ruthenium black as observed in homogeneous deaminative nitrile hydrogenation to primary alcohols (Fig. 5).69 This may be attributed to phosphine oxidation followed by deposition of the metal. More electron-donating para-methoxy groups (S5) resulted in 79% conversion and 97% selectivity. Methyl substituents in ortho-position (S6) proved to be more challenging with only 45% conversion but with excellent selectivity. At 120°C, S7 bearing an ortho-methoxy group was fully converted with 79% selectivity. Finally, the 1,3-disubstituted benzonitrile S8 was quantitatively converted into the corresponding diamine after 24 h at 100 °C and 30 bar of H2. Aliphatic cyclohexanecarbonitrile (S9) gave 46% conversion with high selectivity at 120 °C. Heptanenitrile (S10) was fully converted with 87% selectivity to heptylamine. Mainly partial hydrogenation of adiponitrile (S11) was observed with 92% selectivity to ω-aminocapronitrile which can serve as a precursor for the synthesis of nylon-6.70 Benzylic nitriles were selectively hydrogenated with excellent to moderate conversions (99% for S12 and 72% for S13).
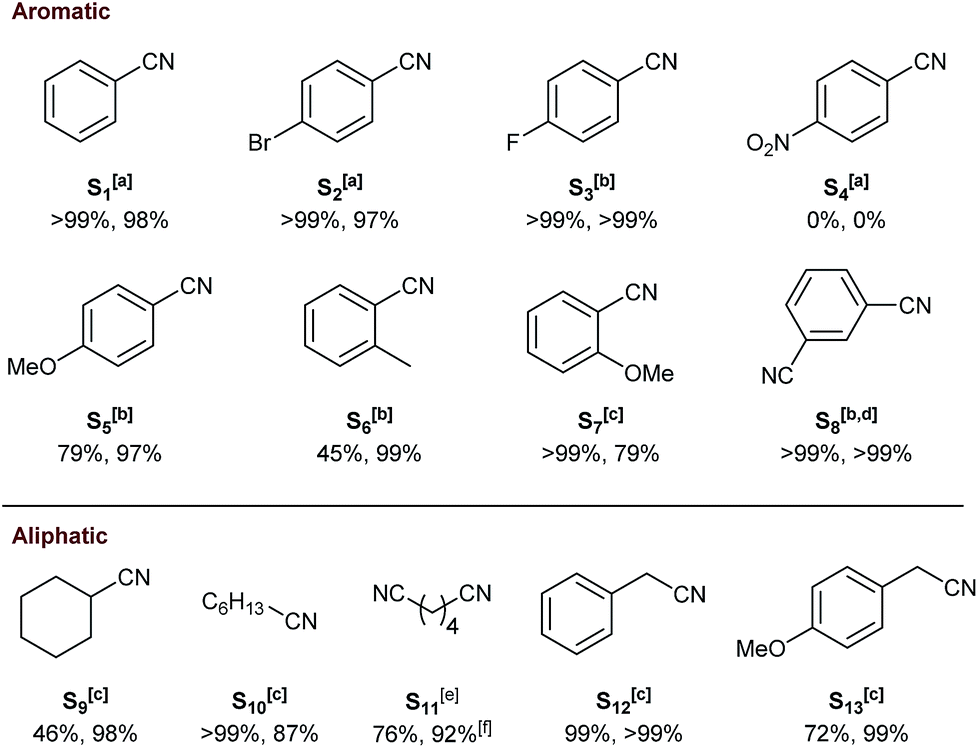 | ||
| Fig. 5 Substrate scope using supported catalyst C1 (conversion and selectivity to mono- or diamine indicated below structures). aReaction conditions: substrate (0.5 mmol), [Ru] (1.0 mol%), dioxane (1.0 mL), 80 °C, H2 (10 bar), 18 h. b100 °C. c120 °C. dH2 (30 bar). 24 h, e100 °C, H2 (30 bar), 24 h. fSelectivity towards ω-aminocapronitrile. | ||
Finally, the recyclability of the resin-bound catalyst C1 was examined. However, under batch conditions, a loss in activity was observed after the fourth run due to mechanical abrasion of the support (Table S2†). Hence, the novel supported catalyst was employed in continuous flow hydrogenation to determine the long-term catalyst performance. The system was initially studied under various flow rates, temperatures and pressures reaching up to 84% selectivity towards the primary amine A with full conversion at 150 °C (Table S3, Fig. S2†). While the secondary imine B was observed as the sole by-product, increasing amounts of secondary amine C formed when exceeding a process temperature of 120 °C.
Next, constant conditions of 100 °C and 20 bar pressure at a flow rate of 0.1 mL min−1 were chosen to provide moderate conversion and selectivity to enable monitoring of actual catalyst stability during the flow experiment. An initial incubation time followed by an increase in activity and selectivity were observed attaining a steady conversion (75%) and selectivity towards A (60%) after 100 h (Fig. 6). This could be attributed to the slow formation of the potentially active Ru-hydrido species analogous to the [Ru(cod)(methallyl)2]/II system reported by the group of Beller.31 Hence, pre-activation of the supported catalyst C1 with H2 or the addition of base to the feed could possibly diminish this activation period. In addition to A, the product mixture contained 40% of secondary imine B and 0.5% of secondary amine C after 100 h time-on-stream (TOS). The amount of C remained constant throughout the experiment. The difference in selectivity compared to the batch process is a consequence of the presence of the higher concentration of the imine intermediate resulting from the low conversion level (75%) needed to monitor catalyst stability. Conversion of the imine intermediate to the primary amine is prevented once the imine has left the catalyst bed.
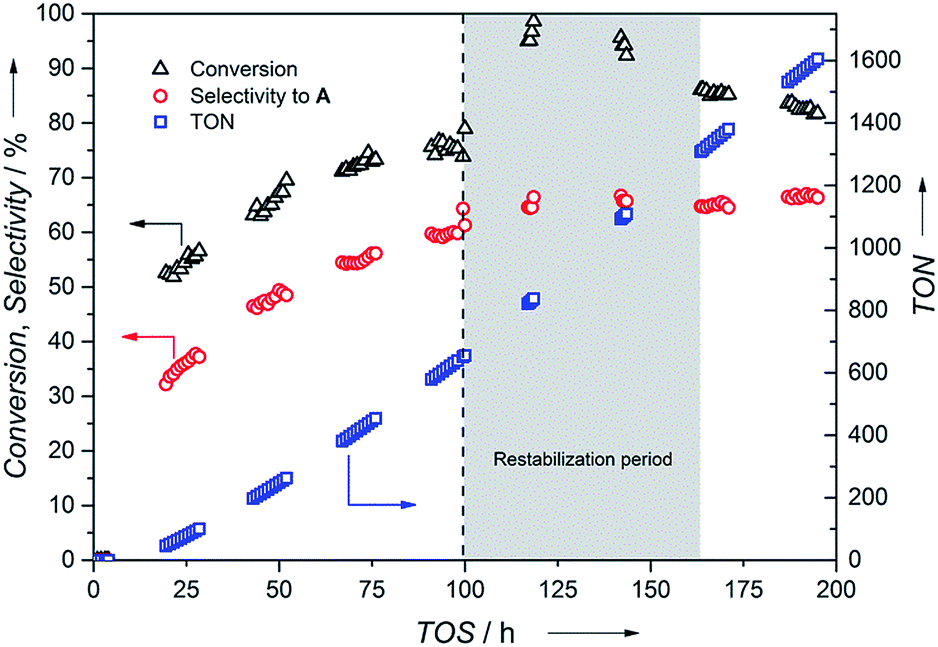 | ||
Fig. 6 Continuous flow hydrogenation of S1 using C1. Conditions: ncat = 0.134 mmol, T = 100 °C, 20 bar, 0.25 M S1 in dioxane at 0.1 mL min−1, ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) (H2) = 2.5 mL min−1. Change of substrate feed at 100 h caused malfunctioning of HPLC pump for 1 h (dashed line). Grey area indicates catalyst restabilization period. Conversion determined by GC using dodecane as internal standard. Selectivity is reported towards formation of primary amine A. (H2) = 2.5 mL min−1. Change of substrate feed at 100 h caused malfunctioning of HPLC pump for 1 h (dashed line). Grey area indicates catalyst restabilization period. Conversion determined by GC using dodecane as internal standard. Selectivity is reported towards formation of primary amine A. | ||
Replacing the feed solution after 100 h caused a flow outage for 1 h followed by an activity increase of the catalyst to nearly full conversion requiring a subsequent restabilization period. Steady conversion (86%) and selectivity towards benzylamine A (65%) were recovered after 171 h followed by a small drop in activity (3%) within the next 24 h. Over 195 h TOS, a remarkable catalyst life-time and a total TON of 1605 were achieved together with no Ru metal contamination observed in the product stream indicated by ICP-OES analysis.
Conclusions
In summary, we developed an efficient and modular solid-phase synthesis protocol to access diverse supported Triphos-type ligands (L1–L5) and their corresponding Ru-complexes (C1–C5) requiring only minimal work-up steps. The solid phase approach opens the way to automated synthesis of large ligand libraries enabling a fine-tuned design of catalysts that allow transformations with high selectivity. The library was successfully applied in highly selective nitrile reduction under mild conditions in the absence of any performance enhancing additive as reported for many monophasic and heterogeneously catalyzed liquid/solid reactions. Opposed to homogeneous counterparts, the robust heterogeneous system provided highly selective access to both primary amines and secondary imines due to the modular and hence easily tunable ligand structure. Moreover, a novel Triphos-type catalyst was applied for the first time in a continuous flow process proving high catalyst stability without significant loss in performance for at least 195 hours. Screening of conditions leading to a shorter catalyst incubation time is currently ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the University of St Andrews, the Engineering and Physical Sciences Research Council (Award Reference 1658187) and the European Union (Marie Curie ITN SusPhos, Grant Agreement No. 317404). We thank A. Simmula for ICP- OES analysis and Dr D. M. Dawson for solid-state NMR measurements.Notes and references
- B. R. Brown, The organic chemistry of aliphatic nitrogen compounds, Oxford University, New York, 1994 Search PubMed.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed.
- P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Amines, Aliphatic: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015 Search PubMed.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS.
- A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035–2052 CrossRef CAS PubMed.
- P. Loos, H. Alex, J. Hassfeld, K. Lovis, J. Platzek, N. Steinfeldt and S. Hübner, Org. Process Res. Dev., 2015, 20, 452–464 CrossRef.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed.
- A. A. N. Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC.
- E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS PubMed.
- B. D. Herzog and R. A. Smiley, in Hexamethylenediamine: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- S. Nishimura, in Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons, New York, 2001, ch. 7, pp. 254–285 Search PubMed.
- T. G. Dewdney, D. A. Dowden, B. D. Hawkins and M. Wyndham, Ger. Offen., DE 2429293 A1 19750320, 1975.
- A. F. Elsasser, WO98/19991, 1998.
- P. Kukula, M. Studer and H.-U. Blaser, Adv. Synth. Catal., 2004, 346, 1487–1493 CrossRef CAS.
- C. de Bellefon and P. Fouilloux, Catal. Rev., 1994, 36, 459–506 CrossRef CAS.
- L. Hegedűs and T. Máthé, Appl. Catal., A, 2005, 296, 209–215 CrossRef.
- Y. Saito, H. Ishitani, M. Ueno and S. Kobayashi, ChemistryOpen, 2017, 6, 211–215 CrossRef CAS PubMed.
- M. Yoshimura, A. Komatsu, M. Niimura, Y. Takagi, T. Takahashi, S. Ueda, T. Ichikawa, Y. Kobayashi, H. Okami, T. Hattori, Y. Sawama, Y. Monguchi and H. Sajiki, Adv. Synth. Catal., 2018, 360, 1726–1732 CrossRef CAS.
- D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Holscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed.
- X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed.
- Y. Li, C. Topf, X. Cui, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 5196–5200 CrossRef CAS PubMed.
- I. Mellone, F. Bertini, L. Gonsalvi, A. Guerriero and M. Peruzzini, Chimia, 2015, 69, 331–338 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, E. Alberico, K. Junge, H. Junge and M. Beller, Chem. Sci., 2016, 7, 3432–3442 RSC.
- T. Suarez and B. Fontal, J. Mol. Catal., 1988, 45, 335–344 CrossRef CAS.
- R. Adam, C. B. Bheeter, R. Jackstell and M. Beller, ChemCatChem, 2016, 8, 1329–1334 CrossRef CAS.
- R. Adam, C. B. Bheeter, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, ChemSusChem, 2017, 10, 842–846 CrossRef CAS PubMed.
- C. Bianchini, V. Dal Santo, A. Meli, W. Oberhauser, R. Psaro and F. Vizza, Organometallics, 2000, 19, 2433–2444 CrossRef CAS.
- C. Bianchini, M. Frediani and F. Vizza, Chem. Commun., 2001, 479–480 RSC.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345–3384 CrossRef CAS PubMed.
- R. van Heerbeek, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Rev., 2002, 102, 3717–3756 CrossRef CAS PubMed.
- D. J. Cole-Hamilton and R. P. Tooze, in Catalyst Separation, Recovery and Recycling: Chemistry and Process Design, ed. D. J. Cole-Hamilton and R. P. Tooze, Springer Netherlands, Dordrecht, 2006, pp. 1–8 Search PubMed.
- P. J. C. Hausoul, C. Broicher, R. Vegliante, C. Göb and R. Palkovits, Angew. Chem., Int. Ed., 2016, 55, 5597–5601 CrossRef CAS PubMed.
- P. C. J. Kamer, D. Vogt and J. W. Thybaut, Contemporary Catalysis: Science, Technology, and Applications, The Royal Society of Chemistry, 2017 Search PubMed.
- R. A. Findeis and L. H. Gade, Eur. J. Inorg. Chem., 2003, 2003, 99–110 CrossRef.
- L. Harmand, S. Samer, J. Andrieu, H. Cattey, M. Picquet and J.-C. Hierso, Open Org. Chem. J., 2012, 6, 1–11 CrossRef CAS.
- C. Bianchini, A. Meli, V. Patinec, V. Sernau and F. Vizza, J. Am. Chem. Soc., 1997, 119, 4945–4954 CrossRef CAS.
- I. Rojas, F. L. Linares, N. Valencia and C. Bianchini, J. Mol. Catal. A: Chem., 1999, 144, 1–6 CrossRef CAS.
- C. Bianchini, A. Meli and W. Oberhauser, New J. Chem., 2001, 25, 11–12 RSC.
- P. Schober, G. Huttner, L. Zsolnai and A. Jacobi, J. Organomet. Chem., 1998, 571, 279–288 CrossRef CAS.
- R. A. Findeis and L. H. Gade, Dalton Trans., 2003, 249–254 RSC.
- W. Hewertson and H. R. Watson, J. Chem. Soc., 1962, 1490–1494 RSC.
- H. Heidel, G. Huttner and G. Helmchen, Z. Naturforschung B, 1993, 48, 1681–1692 CAS.
- L. Söncksen, C. Gradert, J. Krahmer, C. Näther and F. Tuczek, Inorg. Chem., 2013, 52, 6576–6589 CrossRef PubMed.
- N. Nakagawa, E. J. Derrah, M. Schelwies, F. Rominger, O. Trapp and T. Schaub, Dalton Trans., 2016, 45, 6856–6865 RSC.
- D. Obrecht and J. M. Villalgordo, in Solid-Supported Combinatorial and Parallel Synthesis of Small-Molecular-Weight Compound Libraries, Elsevier Science ltd., Oxford, 1998, ch. 1, pp. 1–184 Search PubMed.
- K. Burgess, Solid-Phase Organic Synthesis, John Wiley & Sons, Inc., New York, 2002 Search PubMed.
- M. C. Samuels, B. H. G. Swennenhuis and P. C. J. Kamer, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. v. Leeuwen, John Wiley & Sons, Ltd, Chichester, 2012, ch. 16, pp. 463–479 Search PubMed.
- S. E. Booth, C. M. Dreef-Tromp, P. H. H. Hermkens, J. A. P. A. de Man and H. C. J. Ottenheijm, in Combinatorial Chemistry, ed. G. Jung, Wiley-VCH Verlag GmbH, Weinheim, 1999, ch. 2, pp. 35–76 Search PubMed.
- C. Gennari and U. Piarulli, Chem. Rev., 2003, 103, 3071–3100 CrossRef CAS PubMed.
- M. Renom-Carrasco and L. Lefort, Chem. Soc. Rev., 2018, 47, 5038–5060 RSC.
- A. Kirschning, W. Solodenko and K. Mennecke, Chem.–Eur. J., 2006, 12, 5972–5990 CrossRef CAS PubMed.
- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- A. Muth, O. Walter, G. Huttner, A. Asam, L. Zsolnai and C. Emmerich, J. Organomet. Chem., 1994, 468, 149–163 CrossRef CAS.
- F. J. L. Heutz, M. C. Samuels and P. C. J. Kamer, Catal. Sci. Technol., 2015, 5, 3296–3301 RSC.
- M. C. Samuels, F. J. L. Heutz, A. Grabulosa and P. C. J. Kamer, Top. Catal., 2016, 59, 1793–1799 CrossRef CAS.
- T. T. Adint and C. R. Landis, J. Am. Chem. Soc., 2014, 136, 7943–7953 CrossRef CAS PubMed.
- X. Wang, E. A. P. Ling, C. Guan, Q. Zhang, W. Wu, P. Liu, N. Zheng, D. Zhang, S. Lopatin, Z. Lai and K.-W. Huang, ChemSusChem, 2018, 11, 3591–3598 CrossRef CAS PubMed.
- S. Enthaler, D. Addis, K. Junge, G. Erre and M. Beller, Chem.–Eur. J., 2008, 14, 9491–9494 CrossRef CAS PubMed.
- S. Enthaler, K. Junge, D. Addis, G. Erre and M. Beller, ChemSusChem, 2008, 1, 1006–1010 CrossRef CAS PubMed.
- I. G. Molnár, P. Calleja, M. Ernst, A. S. K. Hashmi and T. Schaub, ChemCatChem, 2017, 9, 4175–4178 CrossRef.
- A. J. M. van Dijk, R. Duchateau, E. J. M. Hensen, J. Meuldijk and C. E. Koning, Chem.–Eur. J., 2007, 13, 7673–7681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization, continuous flow optimization. See DOI: 10.1039/c9sc01415b |
| This journal is © The Royal Society of Chemistry 2019 |