
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotent and selective in vitro and in vivo antiproliferative effects of metal–organic trefoil knots†
Farah
Benyettou‡
a,
Thirumurugan
Prakasam‡
 a,
Anjana
Ramdas Nair
b,
Ini-Isabee
Witzel
c,
Marwa
Alhashimi
a,
Tina
Skorjanc
a,
John-Carl
Olsen
d,
Kirsten C.
Sadler
b and
Ali
Trabolsi
*a
a,
Anjana
Ramdas Nair
b,
Ini-Isabee
Witzel
c,
Marwa
Alhashimi
a,
Tina
Skorjanc
a,
John-Carl
Olsen
d,
Kirsten C.
Sadler
b and
Ali
Trabolsi
*a
aProgram in Chemistry, New York University Abu Dhabi, UAE. E-mail: Ali.trabolsi@nyu.edu
bProgram in Biology, New York University Abu Dhabi, UAE
cCore Technology Platform, New York University Abu Dhabi, UAE
dDepartment of Chemistry, University of Rochester, Rochester, New York, USA
First published on 22nd May 2019
Abstract
A set of metal–organic trefoil knots (M-TKs) generated by metal-templated self-assembly of a simple pair of chelating ligands were well tolerated in vitro by non-cancer cells but were significantly more potent than cisplatin in both human cancer cells––including those resistant to cisplatin––and in zebrafish embryos. In cultured cells, M-TKs generated reactive oxygen species that triggered apoptosis via the mitochondrial pathway without directly disrupting the cell-membrane or damaging nuclear DNA. The cytotoxicity and wide scope for structural variation of M-TKs indicate the potential of synthetic metal–organic knots as a new field of chemical space for pharmaceutical design and development.
Introduction
Intensifying effort in medicinal inorganic chemistry1–6 promises to yield new cancer therapies that join the suite of organoplatinum complexes (cisplatin, carboplatin, and oxaliplatin) currently used to treat nearly half of all cancer patients undergoing chemotherapy.7 In this area, screening rather than rational design remains the primary method for identifying lead compounds. For example, a landmark study8 published by the National Cancer Institute tested 1100 different metal-based compounds against a diverse panel of cancer cell lines and categorized them according to their putative modes of action––binding to sulfhydryl groups, chelation, generation of reactive oxygen species (ROS), and production of lipophilic ions. However, few of these insights have translated to improved clinical care. More recent work highlights the anti-proliferative properties and modes of action of compounds that contain copper,9,10 iron,11 ruthenium,12–14 and other metals.15–18 Therefore, metals are now a promising frontline chemotherapy against many cancer types.Metals are stereochemically versatile and allow for the synthesis of complexes that have good shape complementarity to biological targets.19 They can also provide redox functionality that contributes to cytotoxicity.20 Furthermore, organometallic compounds and coordination complexes are easier to synthesize and modify than organic molecules, which facilitates the production of derivatives that have fewer side effects and a wider range of activities. For example, optimization of this type led to the development of the second generation platinum-based drugs carboplatin and oxaliplatin,21 and similar improvement is being made to ruthenium-based agents, with new drug candidates currently undergoing clinical trials.12–14
An effective feature of metal–organic complexes as anticancer agents is the acid-lability of their metal–ligand bonds. The extracellular environment of malignant tissue is often more acidic than normal tissue,22 providing an opportunity for localized metal release from metal–organic compounds. This could increase cancer-selective toxicity and minimize off target toxic effects.
Like some mononuclear complexes, discrete polymetallic coordination compounds including metallocycles, cages, and helices have exhibited in vitro cytotoxicities that rival or exceed that of cisplatin.23 Their relatively large size and rigidity may facilitate binding to the large flat surfaces of proteins, so-called “undruggable” targets.24,25 Moreover, efficient binding to cell-surface proteins may trigger active transport mechanisms that result in intracellular delivery of the agents and their inhibition of specific protein–protein interactions. Alternatively, binding and intracellular transport may be followed by metal release within lysosomes and subsequent metal-mediated damage of multiple intracellular targets.
Metal–organic knots and links are relatively large, often polymetallic species, similar to the more traditional metal–organic structures. However, being topologically non-trivial, they possess unique structural complexities and potentially unique binding affinities. Nevertheless, their biological activities have yet to be explored. Here, we describe potent in vitro and in vivo anti-proliferative properties of metal–organic trefoil knots (M-TK). The mechanism of cancer-toxicity appears to involve binding to the cell surface, intracellular delivery by active transport mechanisms, and the generation of reactive oxygen species (ROS) that damage mitochondria but spare nuclear DNA and the cell membrane. Scheme S1† illustrates the general strategy followed in this study.
Results & discussion
Design, synthesis and characterization
Previously, we reported a convenient protocol for the synthesis of zinc- and cadmium-containing trefoil knots (Zn-TK and Cd-TK, respectively).26–28 The procedure involves mixing diamino pyridine (DAB), 2,6-diformyl pyridine (DFP), and the corresponding metal(II) acetate in isopropanol solvent at 65 °C for 5 hours. We expanded on this work by isolating copper- and manganese-based trefoil knots (Cu-TK and Mn-TK, respectively), as well as an iron-containing complex that could not be accessed directly but could be obtained by transmetalation of Cd-TK.26 For this study, metal–organic trefoil knots (M-TKs) incorporating Zn(II), Cd(II), Cu(II) and Mn(II) were synthesized in 56, 59, 78 and 58% yields, respectively (Fig. S1A†). To prepare Fe-TK, Cd-TK was combined with 10 equivalents of iron(II) acetate in a mixed solvent of CH3OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN (1:1) at room temperature (Fig. S1B†). Metal-ion exchange occurred within 30 minutes and yielded Fe-TK in 82%. All newly prepared knotted structures were fully characterized by HR-MS, 1H and 13C NMR spectroscopies (ESI). Zn-TK, Cu-TK, and Cd-TK were also characterized by single crystal X-ray diffraction studies (Fig. 1).
:CH3CN (1:1) at room temperature (Fig. S1B†). Metal-ion exchange occurred within 30 minutes and yielded Fe-TK in 82%. All newly prepared knotted structures were fully characterized by HR-MS, 1H and 13C NMR spectroscopies (ESI). Zn-TK, Cu-TK, and Cd-TK were also characterized by single crystal X-ray diffraction studies (Fig. 1).
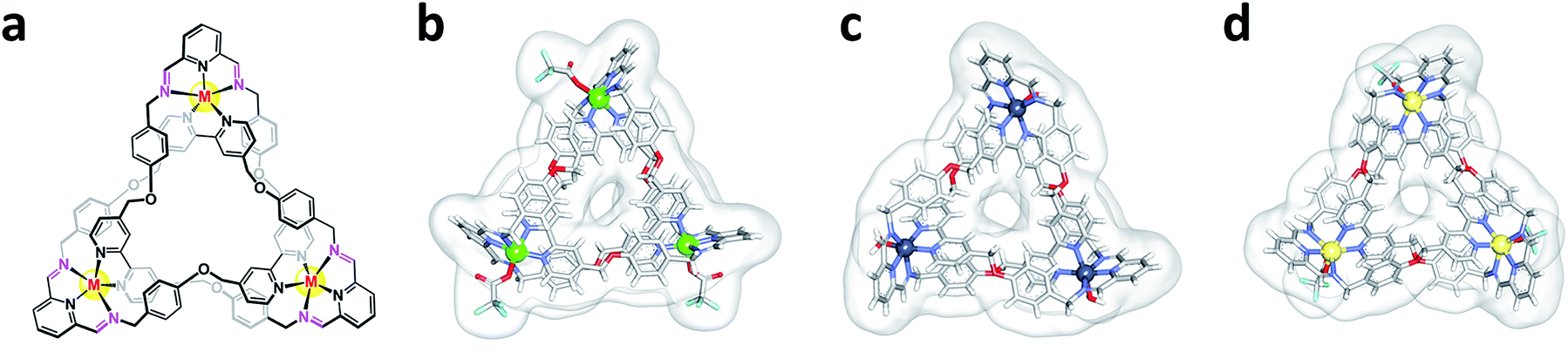 | ||
| Fig. 1 Structures of metal–organic trefoil knots. (a) General chemical structure of the metal–organic knots and X-ray crystal structures of (b) Cu-TK, (c) Zn-TK, and (d) Cd-TK. | ||
Biological activity and selectivity
M-TKs have a unique topology, stability under physiological conditions (Fig. S2†), and metal–organic composition which suggests they will be effective in delivering toxic metals to cells. We thus evaluated the toxicity of these complexes in vitro using cancer cells and in vivo using zebrafish embryos. Cadmium is a known carcinogen,29 so Cd-TK served only as an intermediate in the synthesis of Fe-TK and its in vitro and in vivo activities are presented for the sake of comparison.The anticancer activity of M-TKs (Cu-TK, Zn-TK, Fe-TK, Mn-TK and Cd-TK) was investigated against six cancer cell lines: HeLa (human cervical epithelial carcinoma), A2780 (human ovarian carcinoma), A2780/cis (human ovarian carcinoma (multi-drug resistant)), MDAMB-231 (human breast adenocarcinoma), PC3 (human prostate adenocarcinoma), and MCF-7 (human breast adenocarcinoma). All these cell lines represent cancers that are highly aggressive with a high risk of drug resistance.30 We compared the activity of the M-TKs to that of the commercially available drug cisplatin as a positive control (Table 1 and Fig. S5†). A2780 and A2780/cis cancer cells are human ovarian cancer cell lines that are genetically identical (isogenic), except that A2780/cis cells exhibit resistance to many conventional chemotherapeutic agents, especially cisplatin. Metal-free TK, obtained by the reduction of the imine bonds of Cd-TK (Fig. S1†), the individual organic ligands (DFP and DAB), and the acetate salts of all metals except Cd(II) exhibited minimal toxicities in all cell lines (Fig. S6†). In contrast, the M-TKs effectively induced cell death, several in the sub micromolar ranges (Table 1). This demonstrates that the potency of the complexes depends on their being wholly composed 3D structures.
| Cell lines | Zebrafish embryo | |||||||
|---|---|---|---|---|---|---|---|---|
| HeLa | A2780 | A2780/cis | MDAMB | PC3 | MCF-7 | HEK-293 | ||
| Cu-TK | 13.3 ± 0.1 | 3.2 ± 0.6 | 1.3 ± 0.1 | 2.4 ± 0.3 | 27.7 ± 1.9 | 4.8 ± 0.3 | 20.4 ± 0.5 | 4 |
| Zn-TK | 5.4 ± 0.5 | 8.3 ± 0.7 | 5.7 ± 1.0 | 6.0 ± 0.5 | 44.5 ± 3.2 | 17.7 ± 0.2 | 11.8 ± 0.2 | 8.8 |
| Fe-TK | 1.3 ± 0.6 | 5.2 ± 0.8 | 2.3 ± 1.2 | 3.1 ± 0.2 | 0.9 ± 0.1 | 9.1 ± 0.7 | ≫100 | 8 |
| Cd-TK | 1.5 ± 0.2 | 2.1 ± 0.3 | 0.6 ± 0.2 | 1.9 ± 0.1 | 11.7 ± 1.8 | 7.8 ± 2.5 | 16.8 ± 0.1 | 8 |
| Mn-TK | 3.3 ± 0.2 | 4.1 ± 0.1 | 4.2 ± 0.2 | 0.8 ± 0.05 | 7.5 ± 0.2 | 3.4 ± 0.2 | 24.3 ± 0.7 | 4.8 |
| TK | ≫100 | ≫100 | 54.6 ± 1.4 | ≫100 | ≫100 | 27.4 ± 0.3 | ≫100 | ≫100 |
| Cisplatin | 25.7 ± 4.3 | 11.2 ± 0.2 | 28.1 ± 1.0 | 16.5 ± 0.6 | 15.4 ± 1.3 | 5.8 ± 2.8 | 1.7 ± 0.5 | 250 |
M-TKs were more cytotoxic to the multidrug-resistant A2780/cis cells than cisplatin. The resistance factor (Rf), which compares the activity (IC50) of a compound in resistant cells versus wild-type (Rf = IC50(A2780/cis)/IC50(A2780)), was 2.5 for cisplatin, 1.0 for Mn-TK, and less than 1 for Cd-TK, Fe-TK, Zn-TK, and Cu-TK (Table S1†). The lack of cross-resistance exhibited by A2780/cis suggests that M-TKs and cisplatin induce cytotoxicity by different mechanisms.
Elevated cellular glutathione (GSH) levels are implicated in cisplatin-resistance, presumably through reduction or sequestration of cisplatin.31 Samples of M-TKs in the presence of 10 mM of GSH, at pH 7.4, and in D2O:DMSO (1:1), exhibited no significant 1H NMR spectral changes and generated no glutathione disulfide (GSSG) (Fig. S3†), indicating that the complexes are stable to GSH reduction and may therefore circumvent GSH-mediated resistance, unlike cisplatin.32
The in vitro selectivity index (SI, Table S1†) of each compound was assessed by activity in cancer cells versus activity in human embryonic kidney (HEK-293) cells (Table 1, Fig. S7†). HEK-293 cells are immortalized cells that are considered non- or low-tumorigenic.33 The SI was significantly less than 1 for cisplatin in all cancer cell lines, indicating non-specificity of this toxin. In contrast, all M-TKs were found to have SIs above 1 in all tested cancer cell lines, indicating selectivity. This could be attributed to different routes of internalization of cisplatin and M-TKs, as cisplatin penetrates both cancerous and non-cancer-derived cells via passive routes such as diffusion,32,34,35 whereas M-TKs could utilize an alternative transport mechanism.
In vitro toxicity mechanism
We next investigated the mechanism of toxicity of M-TKs in HeLa cancer cells. We selected these cells for all future studies because they have been used as a model in cancer research, especially, tumor cell migration and invasion, drug development, and cell death pathways. We first evaluated the mechanism of uptake and then asked whether the cell killing effects of M-TKs were due to (i) necrosis, in which cells lose membrane integrity and die rapidly as a result of cell lysis; (ii) cytostatis, in which cells stop actively growing and dividing; and (iii) apoptosis, in which cells can activate a program of controlled cell death either through a mitochondrial induced pathway or extracellular signalling pathway.36–39Intracellular uptake and observation of morphological changes: we studied the internalization of M-TKs into HeLa cells using Fe-TK as a model, as this allowed use of Prussian blue staining for iron to identify the internalization of Fe (Fig. S9†). We found blue iron dots in cells treated with Fe-TK for 24 hours, while control cells remained unchanged.
In order to identify the mechanism by which M-TKs trigger cell death, we first incubated the cells with the complexes and examined the morphological changes produced. After 24 hours of incubation, control and TK-treated cells exhibited normal (flattened) morphology and normal proliferation (Fig. S10†). Cells treated with CdSO4, a known necrosis inducer,40 became swollen and necrotic. Treatment of HeLa cells with cisplatin and camptothecin (CPT), both apoptosis inducers, or any of the M-TKs, reduced proliferation and adhesion and caused the cells to shrink and become rounded (Fig. S10†), consistent with apoptosis and not necrosis.41
Mechanism of cell death
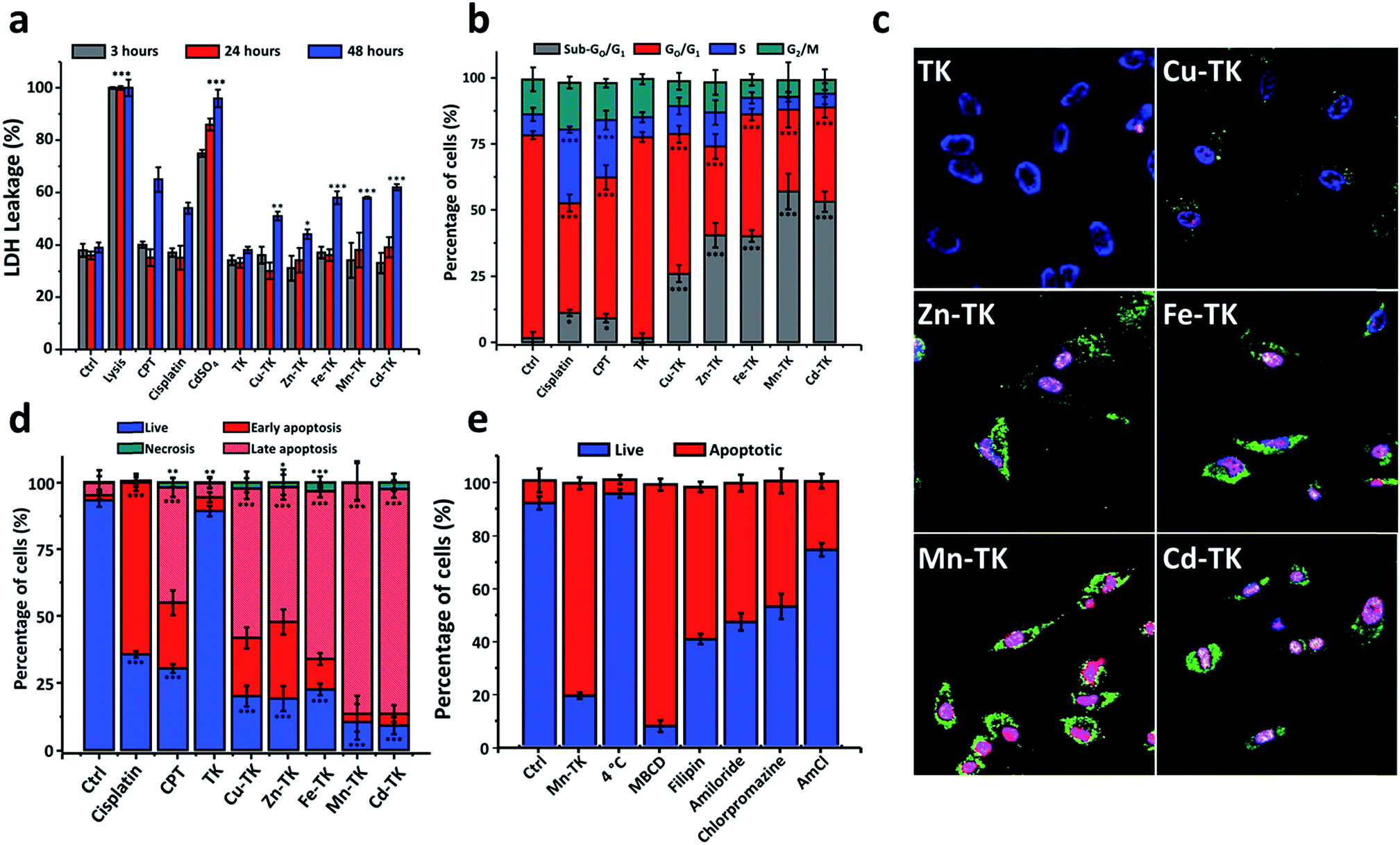 | ||
| Fig. 2 M-TKs induce apoptosis in HeLa cells. (a) Lactate dehydrogenase (LDH) levels in HeLa cells after 3 (grey), 24 (red) and 48 (blue) hours of incubation with no additives (control) or with lysis buffer (positive control), CPT, cisplatin, CdSO4, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK, or Cd-TK, each at 10 μM. (b) Cell cycle analysis of HeLa cells after 24 hours of incubation in the presence of no additives (control), or cisplatin, CPT, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK or Cd-TK, each at 10 μM. Percentages of cells in sub-G0/G1 (green), G0/G1 (blue), S (red), and G2/M (gray) phases were measured. (c) Confocal images of HeLa cells incubated for 3 hours with TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK or Cd-TK, each at 10 μM, followed by staining with Annexin V-FITC (green), DAPI (blue) and propidium iodide (red). (d) Fates of cells treated with no additives (control) or with cisplatin, CPT, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK or Cd-TK (10 μM) after 24 hours of incubation. Staining with Annexin V-FITC and PI was used to determine the percentages of cells that were alive (blue, Annexin V-/PI−), necrotic (green, Annexin V-/PI+), early apoptotic (red, Annexin V+/PI−), or late apoptotic (salmon, Annexin V+/PI+). (e) Quantitative comparison of Mn-TK internalization (10 μM, 3 hours) by different internalization routes in HeLa cells by studying the effect of endocytotic inhibitors (chlorpromazine, methyl-B-cyclodextrin (MBCD), filipin, amiloride and ammonium chloride (AmCl)) using Annexin V-FITC and PI co-staining. Error bars represent standard deviations of triplicate measurements. All values are expressed as mean ± STD. (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 compared to the control. | ||
Cell cycle analysis performed on cells treated for 24 hours revealed that while the majority of control cells and TK-treated cells were in the G0/G1-phase, cells treated with cisplatin or CPT accumulated in the S-phase, consistent with activation of the DNA damage checkpoint (Fig. 2b and S11†).46–49 M-TKs caused a significant increase in the proportion of cells in the sub-G0/G1 phase and reduced the number of cells in G1 and G2/M-phases (Fig. 2b). These results suggest that M-TKs perturb the cell cycle and are consistent with the possibility that M-TK treated cells undergo apoptosis.
We investigated the mechanism of cellular uptake of M-TKs by selectively blocking potential internalization pathways prior to exposure to Mn-TK. After Mn-TK exposure for 3 hours, we stained HeLa cells with Annexin V-FITC/PI to determine the mechanism of cell death (Fig. 2e and S15†). Mn-TK was chosen because it induced signs of apoptosis after only a few hours. To determine the relative amounts of active versus passive mechanisms involved in internalization, we measured the apoptosis rate in cells incubated with Mn-TK for 3 hours at either 4 °C or 37 °C. A few apoptotic cells were identified in cells kept at 4 °C and this increased to 80.1% at 37 °C (Fig. 2e and S15†). This difference indicates that active mechanisms of internalization, such as endocytosis, are likely at play.
To test this directly, cells were treated with several endocytotic inhibitors (chlorpromazine, methyl-B-cyclodextrin, filipin and amiloride)53 prior to addition of Mn-TK, followed by Annexin V-FITC/PI analysis. Inhibitors of macropinocytosis and both caveolin- and clathrin-mediated endocytosis blocked Mn-TK-induced apoptosis, suggesting that Mn-TK is likely internalized by these pathways (Fig. 2e and S15†). We speculate that the organic framework of Mn-TK not only stabilizes the metals in the +2 oxidation state, but also provides a lipophilic surface that may assist passage across cell membranes and play a role in biological recognition processes. A similar role for the ligand framework has been observed in the cases of ruthenium- and osmium-arene complexes.54
Inhibition of lysosome acidification in HeLa cells with ammonium chloride prior to Mn-TK treatment decreased the percentage of apoptotic cells to 10.6%, suggesting that lysosomal function is essential for M-TK toxicity (Fig. 2e and S15†). Tumor cell environments with pH as low as 5.5 have been reported, whereas pH 7.4 is typical for healthy cells.55 Proton 1H NMR studies in aqueous solution (Fig. S4†) showed that acidification (from 7.4 to 5.4) destabilizes the metal complexes by hydrolyzing their imine bonds and releasing their sub-components (metal ions and linkers) into the solution. We hypothesize that M-TK degradation occurs in the acidic environment of endosomes and lysosomes, with the released metals then bonding to endogenous ligands and ultimately triggering apoptosis by redox processes.
Mechanism of M-TK toxicity in zebrafish embryos
The promising in vitro activity of M-TKs prompted us to investigate the in vivo toxicity of the complexes using zebrafish embryos. Embryos were treated during early stages of development (from 10–24 hours post-fertilization; hpf, Fig. 3a), a period of rapid cell division. We assessed embryo viability at 24 hpf and identified the half-maximum lethal doses (LD50) of M-TKs as an order of magnitude lower (4–8 μM) than cisplatin (250 μM; Table 1), with Cu-TK having the highest potency (LD50 = 4.0 μM) (Fig. 3c). The purely organic TK had no toxicity or any observable effects on development at any concentrations tested (Fig. 3c), indicating that the toxicity was due to the metal component of the M-TKs.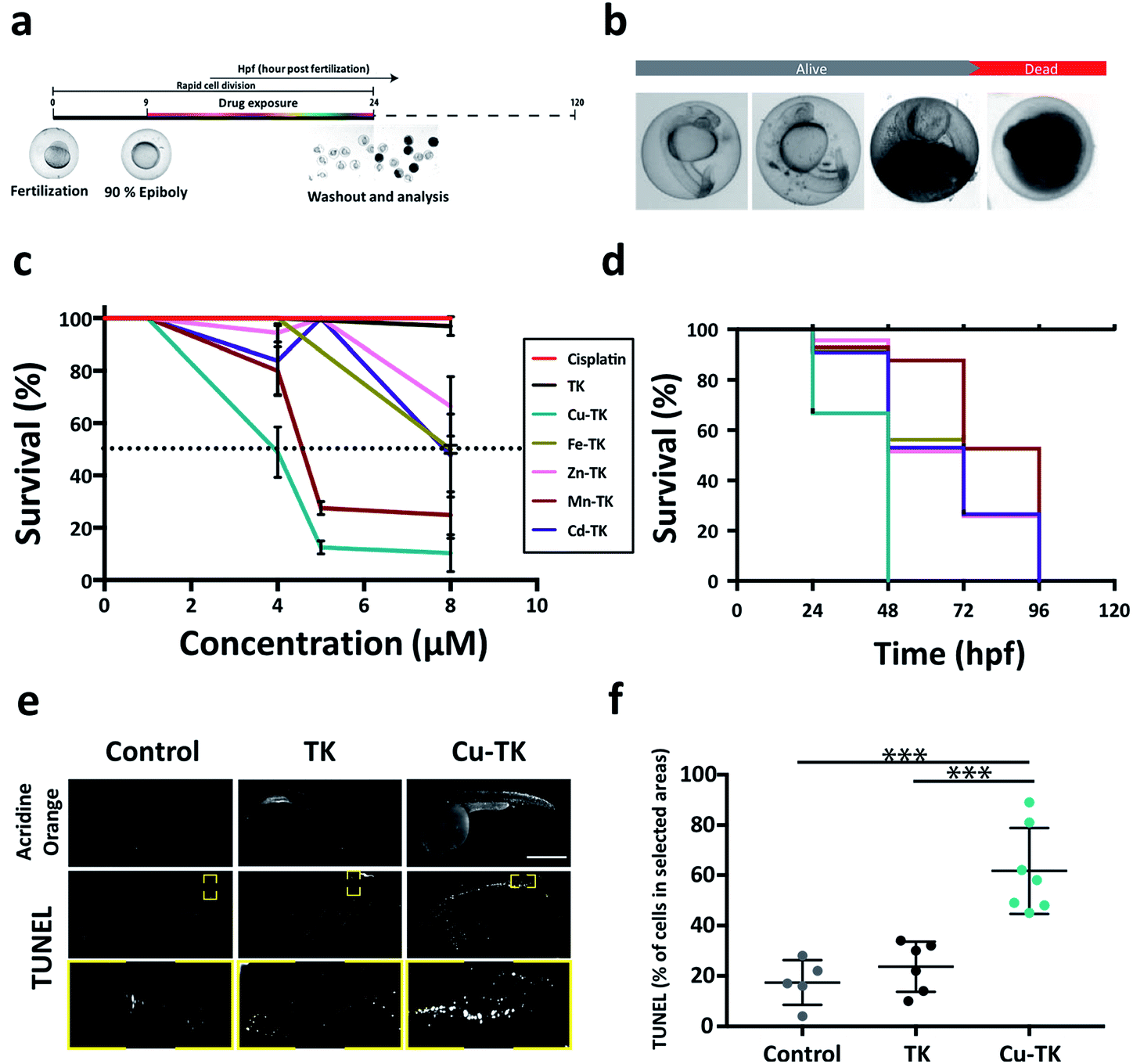 | ||
| Fig. 3 M-TKs induce apoptosis in zebrafish embryos. (a) Treatment timeline for zebrafish embryos. (b) Sequence of death in 24 hpf live zebrafish embryos post treatment with M-TKs. (c) Dose–response curve for M-TKs, cisplatin, and metal-free TK. Dotted line indicates LD50. (d) Kaplan–Meier plot displaying the survival trend after treatment with M-TKs, cisplatin, and metal-free TK at a fixed concentration of 4 μM. (e) Representative images of control, TK- and Cu-TK-treated embryos. Top row – acridine orange assay. Middle row – TUNEL assay. Yellow boxes indicate area analyzed for TUNEL-positive cells. Bottom row – higher magnification inserts corresponding to the yellow boxes in the middle row. (f) Quantification of TUNEL-positive cells in control, TK-, and Cu-TK-treated zebrafish embryos. All values are expressed as mean ± STD. (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 compared to the control. | ||
Early life exposure to metals has been shown to have long term effects in many organisms, including zebrafish.56 We tested if M-TK exposure during early development affected survival at later times by washing out each compound and assessing embryos for mortality up to 120 hpf (Fig. 3c). All embryos pre-exposed to Cu-TK were dead by 48 hpf, whereas the mortality rate of embryos subjected to all other M-TKs was delayed until later time periods (Fig. 3c). Embryos treated with the organic TK at concentrations up to 100 μM remained alive even at 120 hpf. This suggests that subtoxic exposures to M-TKs during early development can have lethal consequences later in life. Our preliminary studies suggest that the metals alone are much less toxic than the corresponding M-TK, indicating that the M-TK confers an advantage over simple administration of metals as a cell killing approach.
Many embryos treated with concentrations just below the LD50 appeared morphologically abnormal, with smaller heads, eyes and body length (Fig. 3b). We asked whether this was attributed to cell death, as was observed for M-TK treated cancer cells. We assessed this in embryos treated with the maximal tolerable dose (MTD, the maximal concentration allowing >80% survival) using acridine orange and TUNEL assays (Fig. 3e and f). Embryos treated with the most potent complex (Cu-TK, 4 μM) (i) showed intense staining with acridine in the brain and the trunk of the embryos. These regions also contained TUNEL-positive cells, with almost 50% more apoptotic cells than untreated or metal-free TK-treated embryos. This indicates that M-TK induces apoptosis in vivo, consistent with in vitro results.
Mechanism of apoptosis induction
Chemotherapeutics cause cellular stresses that lead to apoptosis. Identification of the initial targets and effects of chemotherapeutic agents provides insight into the mechanism of apoptosis induction. We therefore assessed plasma membrane damage, nuclear DNA damage, reactive oxygen species (ROS) generation, and mitochondrial function in cells treated with TK and M-TKs.Lysis buffer causes extensive membrane damage and thus was used as a positive control, and all lysis buffer treated HeLa cells were stained blue (Fig. 4a and S16†). In the CdSO4-treated sample, 83.2% were stained due to membrane damage caused by necrosis. After three hours of treatment with CPT and cisplatin, 14.1% and 23.0% of the cells, respectively, exhibited membrane disruption, confirming that both drugs induce membrane damage as part of their toxic mechanism (Fig. 4a and S16†).59,60 In contrast, the staining that occurred in cells treated with any of the TKs was comparable to that found in untreated cells. This demonstrates that M-TKs cause minimal disruption of cell membranes.
Significant DNA fragmentation was detected in control cells that were treated for two minutes with H2O2, whereas no fragmentation was found in cells treated with cisplatin, CPT, TK, or any of the M-TKs (p > 0.005, Fig. 4b and S17†). Thus, at the tested concentrations and incubation times, M-TKs do not appear to cause apoptosis by fragmenting DNA. Cells were also treated for a longer time (12 hours) with 5 μM Zn-TK or 5 μM cisplatin to allow for DNA crosslinking. The alkaline comet assay detected no cross-linking in either of these samples. (Cisplatin is known to induce crosslinking at higher concentrations.61)
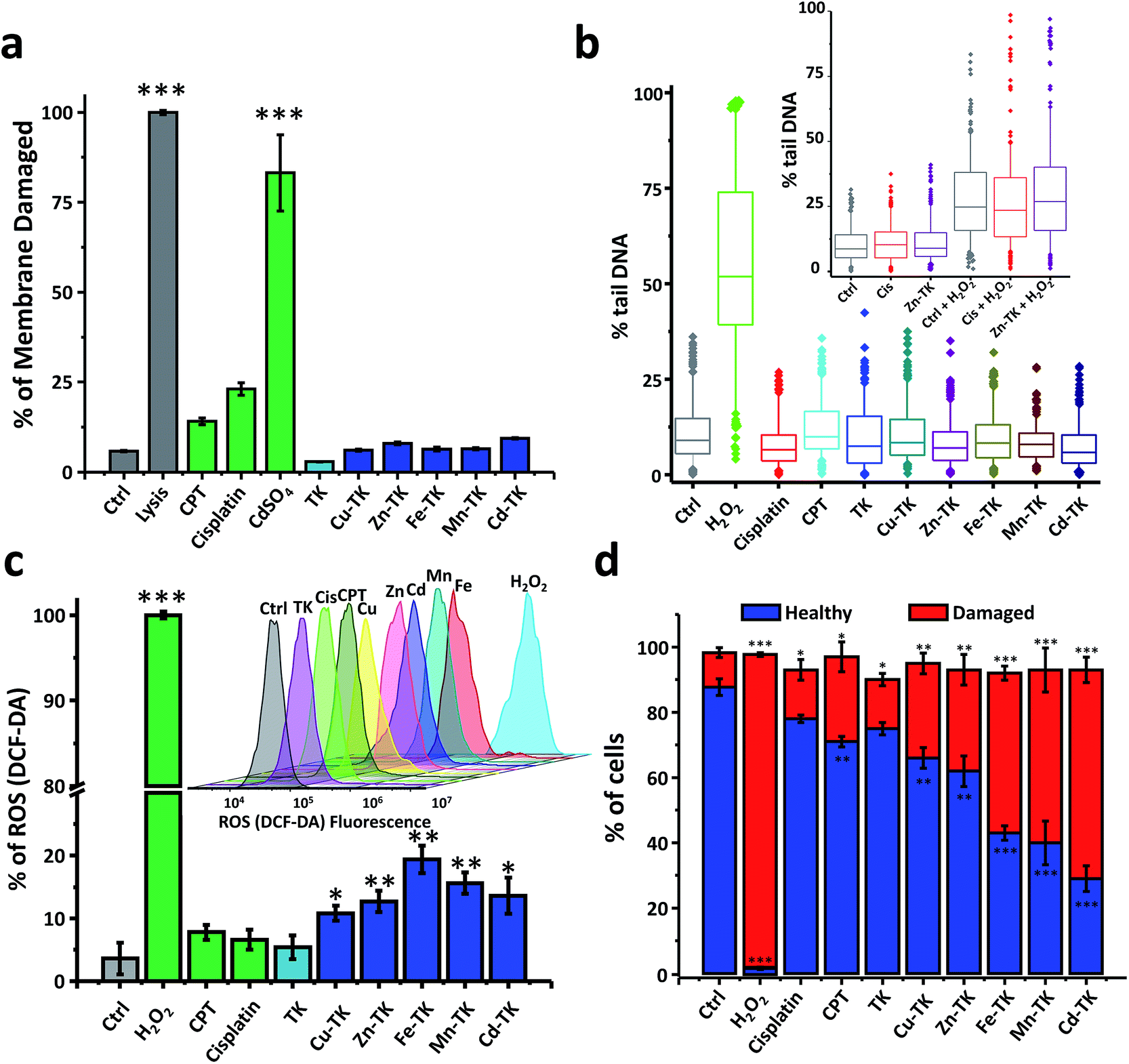 | ||
| Fig. 4 M-TKs induce ROS that damage mitochondria but avoid damaging nuclear DNA and leave the plasma membrane intact. (a) Measurement of cell membrane integrity. Trypan blue exclusion staining of HeLa cells treated with lysis buffer, CPT, cisplatin, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK, or Cd-TK (10 μM) for 3 hours, and of untreated cells (control) showed that M-TKs are well tolerated. Quantification involved counting clearly separated individual cells. (b) Assessment of DNA damage by the alkaline comet assay. HeLa cells were left untreated (control) or were treated with H2O2 at 10 μM for 2 min, or with cisplatin, CPT, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK or Cd-TK, each at 10 μM for 10 min. Inset: testing for crosslinking effects of Zn-TK by a crosslinking alkaline comet assay. HeLa cells treated with 5 μM cisplatin or Zn-TK for 12 hours were subsequently treated with H2O2 (2 min, 10 μM); control cells were only treated with H2O2. Quantification of comet assays was based on triplicate measurements of the percent of comet tail DNA; boxes display the 25–75th percentiles, with a median line, and whiskers extending from the 5th-95th percentiles. (c) Quantitative analysis of ROS generation in HeLa cells after 3 hours of treatment with no additives (control), or H2O2, CPT, cisplatin, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK or Cd-TK (10 μM). H2O2 was used as a positive control. Inset: 3D flow cytometry histograms of cells treated after 3 hours of incubation. Each peak represents 50000 cells. (d) Mitochondrial membrane potential changes (ΔΨm) in HeLa cells after 1 hour exposure to no additives (control), H2O2, cisplatin, CPT, TK, Cu-TK, Zn-TK, Fe-TK, Mn-TK or Cd-TK (10 μM), measured using the JC-1 probe. H2O2 was used as a positive control. Error bars represent standard deviations of triplicate measurements. All values are expressed as mean ± STD. (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 compared to the control. | ||
We examined the mitochondrial membrane potential (Δψm) of cells using the membrane-permeant JC-1 dye (5,5′,6,6′-tetraethyl-benzimidazolylcarbocyanine iodide) and flow cytometry. We measured Δψm using the membrane-permeant JC-1 dye which, in healthy cells with intact mitochondria, forms aggregates that fluoresce red; whereas, in early apoptotic cells, it remains primarily monomeric in form and emits green fluorescence (Fig. 4d and S19†). Control cells exhibited strong red fluorescence (87.7%) and weak green fluorescence (10.6%). Weak red fluorescence intensity (1.9%) and strong green fluorescence intensity (95.8%) were observed after H2O2 treatment. Cells treated with cisplatin or CPT also showed lower red fluorescence (71.4% and 78.0%, respectively) and increased green fluorescence (15.2% and 28.1%, respectively) relative to healthy control cells.66,67 Finally, cells treated with M-TKs exhibited decreased red fluorescence (29.1% to 66.6%) and increased green monomer fluorescence (29.2% and 64.7%) relative to healthy control cells, indicating depolarization of the mitochondrial membrane and increased mitochondrial permeability, a potent trigger for the mitochondria-mediated apoptotic pathway.
Anti-metastatic properties
During cancer progression, deregulation of cell growth leads to escape of cancer cells from the primary tumor and establishment of metastases.68,69 The ability of M-TKs to inhibit cancer cell migration was evaluated in a scratch wound healing assay (Fig. S20 and S21†). The wounds produced in control HeLa cells and in TK-treated HeLa cells were completely healed after 24 hours. In contrast, cell migration and wound healing were significantly suppressed in HeLa cells treated with M-TKs, especially with Cu-TK or Cd-TK, which each suppressed migration by approximately 60% relative to control. We next examined cell adhesion and found that while cisplatin and CPT inhibited adhesion minimally,49,70 and TK had no effect relative to untreated cells, M-TKs drastically reduced the number of adherent cells (Fig. S22 and S23†). Therefore, we conclude that M-TK treatment exhibits both the cell killing and antimetastatic properties which are sought in effective chemotherapeutics.Conclusions
We report the development of five metal-templated molecular TKs (Cu-TK, Zn-TK, Fe-TK, Mn-TK and Cd-TK) as vehicles for the delivery of metals to cancer cells. These nanoscale, water-soluble complexes showed high potency in vitro against six cancer cell lines (HeLa, A2780, A2780/cis, MDAMB-231, PC3, and MCF-7) and in vivo in zebrafish embryos. The potency of the compounds was in many cases higher than that of cisplatin and other previously reported metal complexes. Their main routes of delivery were found to be macropinocytosis and both caveolin- and clathrin-mediated endocytosis, which are all more active in cancer cells than in normal cells.35 In contrast, cisplatin and other small molecules penetrate cells by diffusion, which is less cancer-selective in vitro. Thus, we hypothesize that M-TKs are less toxic to HEK-293 cells because they are internalized less. Notably, the M-TKs were toxic in cisplatin-resistant A2780 cells, suggesting that M-TKs and cisplatin induce cytotoxicity via different mechanisms.Another favorable feature of the M-TKs is their constituent imine bonds, which provide a means for rapid hydrolysis of the complexes and release of the metals and organic linkers at the low pH inside cancer cells.71 This acid-sensitivity is also consistent with the selective toxicity of the complexes toward cancer cells. At higher pH, the M-TKs remain rigidly stable and resistant to thermal, chemical and enzymatic attack, with their metal centers being protected extracellularly until delivered intracellularly.
Our work shows that M-TKs induce ROS that damage mitochondria, while nuclear DNA and the plasma membrane remain intact. Compounds that damage DNA, a ubiquitous target, tend to be more systemically toxic and cause more side-effects than those that are more cancer-specific.
Future investigations will focus on the mechanism of action of the M-TKs and determine whether their ROS-mediated toxicity involves specific intracellular targets. Our work in vivo confirms the viability of studying the effects of these compounds in whole vertebrates, as the M-TKs were well tolerated by zebrafish and appeared to selectively attack dividing cells. Finally, we anticipate that the simple, modular synthesis of the knotted trefoil scaffold will facilitate systematic structural modification and derivative screening for optimization of the potency and selectivity of the complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank New York University Abu Dhabi (NYUAD), UAE, for its generous support of this research, which was carried out using the Core Technology Platform resources at NYUAD. We also acknowledge the Al Jalila Foundation (AJF 201646), the New York University Abu Dhabi Research Enhancement Fund (REF AY_2017-2018), and the National Institute of Health (NIH 5R01AA018886 to KCS) for funding. The authors also thank Miss Jumaanah Alhashemi for TOC realization.Notes and references
- T. W. Hambley, Dalton Trans., 2007, 43, 4929–4937 RSC.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598, 10.1039/b905798f.
- M. Frezza, S. Hindo, D. Chen, A. Davenport, S. Schmitt, D. Tomco and Q. P. Dou, Curr. Pharm. Des., 2010, 16, 1813–1825 CrossRef CAS PubMed.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 RSC.
- C. Hu, X. Li, W. Wang, R. Zhang and L. Deng, Curr. Med. Chem., 2014, 21, 1220–1230 CrossRef CAS PubMed.
- T. C. Johnstone, G. Y. Park and S. J. Lippard, Anticancer Res., 2014, 34, 471–476 CAS.
- R. Huang, A. Wallqvist and D. G. Covell, Biochem. Pharmacol., 2005, 69, 1009–1039 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Metallomics, 2015, 7, 1459–1476 RSC.
- W. A. Wani, U. Baig, S. Shreaz, R. A. Shiekh, P. F. Iqbal, E. Jameel, A. Ahmad, S. H. Mohd-Setapar, M. Mushtaque and L. Ting Hun, New J. Chem., 2016, 40, 1063–1090 RSC.
- R. F. S. Lee, S. Escrig, C. Maclachlan, G. W. Knott, A. Meibom, G. Sava and P. J. Dyson, Int. J. Mol. Sci., 2017, 18, 1869 CrossRef PubMed.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS PubMed.
- H. Khan, A. Badshah, M. Said, G. Murtaza, J. Ahmad, B. J. Jean-Claude, M. Todorova and I. S. Butler, Appl. Organomet. Chem., 2013, 27, 387–395 CrossRef CAS.
- A. Jurowska, K. Jurowski, J. Szklarzewicz, B. Buszewski, T. Kalenik and W. Piekoszewski, Curr. Med. Chem., 2016, 23, 3322–3342 CrossRef CAS PubMed.
- B. Ali and M. A. Iqbal, ChemistrySelect, 2017, 2, 1586–1604 CrossRef CAS.
- I. E. Leon, J. F. Cadavid-Vargas, A. L. Di Virgilio and S. B. Etcheverry, Curr. Med. Chem., 2017, 24, 112–148 CrossRef CAS.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS PubMed.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling, 2011, 15, 1085–1127 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 89 CrossRef CAS PubMed.
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K. W. Chi, Cell Death Differ., 2013, 46, 2464–2474 CAS.
- B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, J. Med. Chem., 2016, 59, 2312–2327 CrossRef CAS PubMed.
- A. E. Modell, S. L. Blosser and P. S. Arora, Trends Pharmacol. Sci., 2016, 37, 702–713 CrossRef CAS.
- T. Prakasam, M. Lusi, M. Elhabiri, C. Platas-Iglesias, J. C. Olsen, Z. Asfari, S. Cianferani-Sanglier, F. Debaene, L. J. Charbonniere and A. Trabolsi, Angew. Chem., Int. Ed., 2013, 52, 9956–9960 CrossRef CAS.
- T. Prakasam, R. A. Bilbeisi, M. Lusi, J.-C. Olsen, C. Platas-Iglesias and A. Trabolsi, Chem. Commun., 2016, 52, 7398–7401 RSC.
- R. A. Bilbeisi, T. Prakasam, M. Lusi, R. El Khoury, C. Platas-Iglesias, L. J. Charbonnière, J.-C. Olsen, M. Elhabiri and A. Trabolsi, Chem. Sci., 2016, 7, 2524–2531 RSC.
- J. Liu, W. Qu and M. B. Kadiiska, Toxicol. Appl. Pharmacol., 2009, 238, 209–214 CrossRef CAS PubMed.
- M. Mimeault, R. Hauke, P. P. Mehta and S. K. Batra, J. Cell. Mol. Med., 2007, 11, 981–1011 CrossRef CAS PubMed.
- H. H. W. Chen, I.-S. Song, A. Hossain, M.-K. Choi, Y. Yamane, Z. D. Liang, J. Lu, L. Y. H. Wu, Z. H. Siddik, L. W. J. Klomp, N. Savaraj and K. M. Tien, Mol. Pharmacol., 2008, 74, 697–704 CrossRef CAS PubMed.
- N. D. Eljack, H.-Y. M. Ma, J. Drucker, C. Shen, T. W. Hambley, E. J. New, T. Friedrich and R. J. Clarke, Metallomics, 2014, 6, 2126–2133 RSC.
- C. Shen, M. Gu, C. Song, L. Miao, L. Hu, D. Liang and C. Zheng, Biologicals, 2008, 36, 263–268 CrossRef CAS PubMed.
- D. P. Gately and S. B. Howell, Br. J. Cancer, 1993, 67, 1171–1176 CrossRef CAS PubMed.
- D. Li, Y.-T. Zhang, M. Yu, J. Guo, D. Chaudhary and C.-C. Wang, Biomaterials, 2013, 34, 7913–7922 CrossRef CAS PubMed.
- L. Bracci, G. Schiavoni, A. Sistigu and F. Belardelli, Cell Death Differ., 2014, 21, 15–25 CrossRef CAS PubMed.
- S. Orrenius, P. Nicotera and B. Zhivotovsky, Toxicol. Sci., 2011, 119, 3–19 CrossRef CAS PubMed.
- G. Majno and I. Joris, Am. J. Pathol., 1995, 146, 3–15 CAS.
- L. Bracci, G. Schiavoni, A. Sistigu and F. Belardelli, Cell Death Differ., 2014, 21, 15–25 CrossRef CAS PubMed.
- W. Fiers, R. Beyaert, W. Declercq and P. Vandenabeele, Oncogene, 1999, 18, 7719–7730 CrossRef CAS PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- F. K.-M. Chan, K. Moriwaki and M. J. De Rosa, Methods Mol. Biol., 2013, 979, 65–70 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, C. Gueydan and J. Han, Cell Res, 2018, 28, 9–21 CrossRef CAS PubMed.
- C. Rogers, T. Fernandes-Alnemri, L. Mayes, D. Alnemri, G. Cingolani and E. S. Alnemri, Nat. Commun., 2017, 8, 14128 CrossRef CAS PubMed.
- M. Kajstura, H. D. Halicka, J. Pryjma and Z. Darzynkiewicz, Cytometry, Part A, 2007, 71, 125–131 CrossRef PubMed.
- J. M. Wagner and L. M. Karnitz, Mol. Pharmacol., 2009, 76, 208–214 CrossRef CAS PubMed.
- V. Velma, S. R. Dasari and P. B. Tchounwou, Biomarker Insights, 2016, 11, 113–121 CrossRef CAS PubMed.
- P. Takahashi, A. Polson and D. Reisman, Apoptosis, 2011, 16, 950–958 CrossRef CAS PubMed.
- L. Ferrara and E. B. Kmiec, Nucleic Acids Res., 2004, 32, 5239–5248 CrossRef CAS PubMed.
- R. D. Phair and T. Misteli, Nature, 2000, 404, 604–609 CrossRef CAS PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 RSC.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- D. Dutta and J. G. Donaldson, Cell Logist, 2012, 2, 203–208 CrossRef PubMed.
- A. F. Peacock and P. J. Sadler, Chem.–Asian J., 2008, 3, 1890–1899 CrossRef CAS PubMed.
- M. Zaki, F. Arjmand and S. Tabassum, Inorg. Chim. Acta, 2016, 444, 1–22 CrossRef CAS.
- K. Bambino and J. Chu, Curr. Top. Dev. Biol., 2017, 124, 331–367 Search PubMed.
- H. H. Grunicke, Eur. J. Cancer Clin. Oncol., 1991, 27, 281–284 Search PubMed.
- R. J. Boohaker, M. W. Lee, P. Vishnubhotla, J. M. Perez and A. R. Khaled, Curr. Med. Chem., 2012, 19, 3794–3804 CrossRef CAS PubMed.
- A. Rebillard, D. Lagadic-Gossmann and M. T. Dimanche-Boitrel, Curr. Med. Chem., 2008, 15, 2656–2663 CrossRef CAS PubMed.
- A. Zare-Mirakabadi, A. Sarzaeem, S. Moradhaseli, A. Sayad and M. Negahdary, Iran. J. Cancer Prev., 2012, 5, 109–116 Search PubMed.
- G. G. Hovhannisyan, T. S. Haroutunyan and R. M. Arutyunyan, Exp. Oncol., 2004, 26, 240–242 CAS.
- S. Alarifi, D. Ali, S. Alkahtani, A. Verma, M. Ahamed, M. Ahmed and H. A. Alhadlaq, Int. J. Nanomed., 2013, 8, 983–993 CrossRef PubMed.
- P. Bragado, A. Armesilla, A. Silva and A. Porras, Apoptosis, 2007, 12, 1733–1742 CrossRef CAS PubMed.
- Q. Li, W. Qiu, Q. Zhu, Y. Zu, X. Deng, T. Zhao, C. Jiang and L. Zhang, Molecules, 2011, 16, 7803–7814 CrossRef CAS PubMed.
- J. F. Goossens, J. P. Henichart, L. Dassonneville, M. Facompre and C. Bailly, Eur. J. Pharm. Sci., 2000, 10, 125–131 CrossRef CAS PubMed.
- Y. R. Fu, Z. J. Yi, Y. R. Yan and Z. Y. Qiu, Mitochondrion, 2006, 6, 211–217 CrossRef CAS PubMed.
- R. Marullo, E. Werner, N. Degtyareva, B. Moore, G. Altavilla, S. S. Ramalingam and P. W. Doetsch, PLoS One, 2013, 8, e81162 CrossRef PubMed.
- W. Sun, Y. Han, Z. Li, K. Ge and J. Zhang, Langmuir, 2016, 32, 9237–9244 CrossRef CAS PubMed.
- J. A. Joyce and J. W. Pollard, Nat. Rev. Cancer, 2009, 9, 239–252 CrossRef CAS PubMed.
- S. Maubant, S. Cruet-Hennequart, L. Poulain, F. Carreiras, F. Sichel, J. Luis, C. Staedel and P. Gauduchon, Int. J. Cancer, 2002, 97, 186–194 CrossRef CAS PubMed.
- P. Swietach, R. D. Vaughan-Jones, A. L. Harris and A. Hulikova, Philos. Trans. R. Soc., B, 2014, 369, 20130099 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1549049. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01218d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |