
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigins of the odd optical observables in plutonium and americium tungstates†
Justin N.
Cross
a,
Tsung-Han
Lee
b,
Chang-Jong
Kang
 b,
Yong-Xin
Yao
c,
Samantha K.
Cary
a,
Jared T.
Stritzinger
a,
Matthew J.
Polinski
d,
Carla D.
McKinley
e,
Thomas E.
Albrecht Schmitt
*e and
Nicola
Lanata
*f
b,
Yong-Xin
Yao
c,
Samantha K.
Cary
a,
Jared T.
Stritzinger
a,
Matthew J.
Polinski
d,
Carla D.
McKinley
e,
Thomas E.
Albrecht Schmitt
*e and
Nicola
Lanata
*f
aChemistry Division and the Plutonium Science and Manufacturing Directorate, Los Alamos National Laboratory, PO Box 1663, Los Alamos, New Mexico, 87545 USA
bDepartment of Physics and Astronomy, Rutgers University, Piscataway, New Jersey 08856, USA
cDepartment of Physics and Astronomy and Ames Laboratory, U.S. Department of Energy, Iowa State University, Ames, Iowa 50011, USA
dDepartment of Chemistry and Biochemistry, Bloomsburg University of Pennsylvania, Bloomsburg, Pennsylvania 17815, USA
eDepartment of Chemistry and Biochemistry, Florida State University, 95 Chieftan Way, 310 DLC, Tallahassee, Florida 32306, USA. E-mail: albrecht-schmitt@chem.fsu.edu
fDepartment of Physics and Astronomy, Aarhus University, 8000, Aarhus C, Denmark. E-mail: lanata@phys.au.dk
First published on 21st May 2019
Abstract
A series of trivalent f-block tungstates, MW2O7(OH)(H2O) (M = La, Ce, Pr, Nd, and Pu) and AmWO4(OH), have been prepared in crystalline form using hydrothermal methods. Both structure types take the form of 3D networks where MW2O7(OH)(H2O) is assembled from infinite chains of distorted tungstate octahedra linked by isolated MO8 bicapped trigonal prisms; whereas AmWO4(OH) is constructed from edge-sharing AmO8 square antiprisms connected by distorted tungstate trigonal bipyramids. PuW2O7(OH)(H2O) crystallizes as red plates; an atypical color for a Pu(III) compound. Optical absorption spectra acquired from single crystals show strong, broadband absorption in the visible region. A similar feature is observed for CeW2O7(OH)(H2O), but not for AmWO4(OH). Here we demonstrate that these significantly different optical properties do not stem directly from the 5f electrons, as in both systems the valence band has mostly O-2p character and the conduction band has mostly W-5d character. Furthermore, the quasi-particle gap is essentially unaffected by the 5f degrees of freedom. Despite this, our analysis demonstrates that the f-electron covalency effects are quite important and substantially different energetically in PuW2O7(OH)(H2O) and AmWO4(OH), indicating that the optical gap alone cannot be used to infer conclusions concerning the f electron contribution to the chemical bond in these systems.
Introduction
The past two decades have given rise to a renaissance in our understanding of bonding in f-element compounds that has altered the simplistic view of the chemistry of these elements as being solely dictated by ionic radii and coulombic forces.1,2 For example, carbon K-edge X-ray absorption spectroscopy measurements on the actinocenes, Th(C8H8)2 and U(C8H8)2, demonstrated that thorocene, which is typically thought to exhibit almost exclusively ionic interactions between the ThIV center and the ligands, actually has larger orbital mixing than occurs in uranocene.3 Likewise, the 4f orbitals of lanthanides are primarily considered as being “core-like” and non-interacting with surrounding ligands, but a recent Ce(IV) nitrosyl complex, among the most stable Ce(IV) complexes reported to date, has covalent interactions involving the 4f orbitals.4 Furthermore, the array of research efforts directed toward understanding the general preference of soft-donor ligands for 5f over 4f elements has led to a description of a covalent bond with minimal orbital overlap, but little build-up of electron density between atoms in most cases.5–13 However, even this is currently undergoing revision in light of the high kinetic energy of electrons in 5f elements.14 We have added to this discourse on the unusual nature of f-block bonding with the characterization of trivalent lanthanide and actinide borates where the actinides structurally deviating from their lanthanide congeners and from each other.15 This deviation, especially with CfIII, appears to be derived from bonding differences that are characteristic of the individual actinide elements and does not seem to correspond to a monotonic extrapolation from pre-berkelium actinide elements.16In order to uncover the underlying ligand properties that determine whether or not significant differences between 4f and 5f elements are expressed, we have also prepared a number of actinide oxoanion compounds that span both the lanthanide and actinide series.17–20 Currently, some of the most promising oxoanions are those with large hyperpolarizabilities; a quantified value of the susceptibility of a M–O bond to the electric field from impinging light that is calculated from phase-matched second-harmonic generation data.21 These oxoanions possess strong second-order Jahn-Teller distortions leading to ligands that possess a wide variety of coordination environments.22–29 Specifically, d0 metal oxoanions are attractive ligands to explore bonding differences between the lanthanides and actinides because they have diverse coordination, can polymerize, have high hyperpolarizabilities, and their bonding is fairly well defined.29,30 There have been few previously described low-valent actinides complexed with d0 metal-containing oxoanions despite their potential importance to the nuclear fuel cycle.31–37 We have recently reported several plutonium molybdates and a family of chromates that have unusual electronic characteristics and display structural deviation from the lanthanides.14,38,39
Here we explore d0 metal oxoanions with trivalent actinides by synthesizing a series of f-block tungstates of the formula MW2O7(OH)(H2O) (M = Pu, La, Ce, Pr, Nd) and AmWO4(OH), where americium structurally diverges under similar reaction conditions. The electronic absorption spectrum of plutonium tungstate features a broad absorption from approximately 300 to 600 nm, giving the crystals an unusual red color. A similar feature is also observed in the spectrum of cerium tungstate. Interestingly, our analysis demonstrates that that the f-electron covalency effects are indeed quite important and substantially different energetically in PuW2O7(OH)(H2O) and AmWO4(OH). On the other hand, we also demonstrate that the unusual color of plutonium tungstate does not stem directly from the 5f electrons, as in both systems the valence band has mostly O-2p character and the conduction band has mostly W-5d character. We also find that a similar pattern is realized in the 4f and 5f chromates α-CsM(CrO4)2 (M = Sm, Eu, Am) recently synthesized in ref. 14. The fact that color and f-electron covalency are not directly connected in these systems poses general challenges in relation with the fundamental goal of characterizing and understanding bonding in f-element compounds, which can only be addressed by combining state-of-the art experimental and quantum-simulation techniques.
Experimental
Caution!
239Pu (t1/2 = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 065 year), 240Pu (t1/2 = 6537 year), and 243Am (t1/2 = 7380 year) represent potential health risks owing to their α and γ emission. All studies with plutonium and americium were conducted in a Category II Nuclear Hazard Facility. All experiments were carried out with approved safety operating procedures.
065 year), 240Pu (t1/2 = 6537 year), and 243Am (t1/2 = 7380 year) represent potential health risks owing to their α and γ emission. All studies with plutonium and americium were conducted in a Category II Nuclear Hazard Facility. All experiments were carried out with approved safety operating procedures.
Syntheses
WO3 (Alfa Aesar 99.8%), LaCl3–7H2O (99.9%, Alfa Aesar), CeCl3 (99.9%, Alfa Aesar), PrCl3·xH2O (99.9%, Alfa Aesar), NdCl3·xH2O (99.9%, Alfa Aesar), and hydrobromic acid (ACS reagent 48% Sigma Aldrich) were used as obtained. Plutonium (94% 239Pu, 6% 240Pu) in the form of PuCl3 was received from LANL. As it had oxidized to the red PuOCl2, reduction back to Pu3+ was accomplished by adding 50 μL of conc. HBr, and reducing to a residue in a furnace at 130 °C. The purple residue was then used as the starting Pu source. Reactions were run in Parr 4749 autoclaves with a 10 mL internal volume PTFE liner for Pu and Am, and 23 mL internal volume liners for the lanthanides. Deionized water was used in all reactions.PuW2O7(OH)(H2O)
PuOCl2 (0.0099 g, 0.0303 mmol) and conc. HBr (50 μL) were heated in a 10 mL Teflon liner at 130 °C until reduced to a solid residue. After cooling to room temperature, the liner was moved to an argon-filled glovebox. WO3 (0.0071 g, 0.0306 mmol) and 200 μL of argon-sparged DI H2O were added to the liner. The liner was sealed in a steel jacket and heated to 200 °C in a box furnace in the glovebox for three days then slowly cooled over two days. The resulting product was rinsed with DI water, and dark-red plates were isolated along with unreacted WO3. Repeating this reaction at 230 °C produced the same product, but with an increased yield and less unreacted WO3.AmWO4(OH)
A residue of AmCl3·nH2O was prepared from AmO2 and 5 M HCl to produce an approximately 0.010 g (0.0286 mmol) residue. The residue was dissolved in 200 μL of Millipore H2O and transferred to a 10 mL Teflon liner. WO3 (0.0070 g, 302 mmol) was added to the liner which was subsequently sealed in steel jacket and heated to 200 °C in a box furnace then slowly cooled over two days. It was found that after this treatment, the water had leaked out of the liner leaving an amorphous orange powder and unreacted WO3. DI H2O (200 μL) was added to the received material, the liner sealed in a steel jacket, and reheated in a box furnace to 230 °C for one day then slowly cooled over two days. After this treatment light-orange crystals of poor quality and no particular shape were isolated. Despite the ill-defined shape of the crystals, they still diffract remarkably well.LaW2O7(OH)(H2O)
LaCl3·7H2O (0.0743 g, 0.200 mmol), WO3 (0.0464 g, 0.200 mmol), and 2 mL of DI H2O were added to a 23 mL Teflon liner. The liner was sealed in a steel autoclave and heated to 200 °C for three days then slowly cooled over two days. Colorless plates and unreacted WO3 were isolated.CeW2O7(OH)(H2O)
CeCl3 (0.0493 g, 0.200 mmol), WO3 (0.0464 g, 0.200 mmol), and 2 mL of DI H2O were added to a 23 mL Teflon liner. The liner was sealed in a steel autoclave and heated to 200 °C for three days then slowly cooled over two days. Yellow plates and unreacted WO3 were isolated. Repeating the reaction at 230 °C for one day then slowly cooling over two days produced larger crystals of the complex.PrW2O7(OH)(H2O)
PrCl3·7H2O (0.0710 g, 0.200 mmol), WO3 (0.0464 g, 0.200 mmol), and 2 mL of DI H2O were added to a 23 mL Teflon liner. The liner was sealed in a steel autoclave and heated to 200 °C for three days then slowly cooled over two days. Light-green plates and unreacted WO3 were isolated. Repeating the reaction at 230 °C for one day then slowly cooling over two days produced larger crystals of the complex.NdW2O7(OH)(H2O)
NdCl3·xH2O (0.0717 g, 0.200 mmol), WO3 (0.0464 g, 0.200 mmol), and 2 mL of DI H2O were added to a 23 mL Teflon liner. The liner was sealed in a steel autoclave and heated to 200 °C for three days then slowly cooled over two days. Light-pink plates and unreacted WO3 were isolated. Repeating the reaction at 230 °C for one day then slowly cooling over two days produced larger crystals of the complex.Crystallographic studies
Single crystals of all products were placed on MiTeGen® mounts using viscous Immersion oil and optically aligned on a Bruker D8 Quest X-ray diffractometer using a digital camera. Initial intensity measurements were performed using a IμS X-ray source (Mo Kα, λ = 0.71073 Å) with high-brilliance and high-performance focusing multilayer optics. Standard Quest software was used for determination of the unit cells and data collection control. The intensities of reflections of a sphere were collected by a combination of multiple sets of exposures (frames). Each set had a different φ angle for the crystal and each exposure covered a range of 0.5° in ω. A total of 1464 images with an exposure time of 10 to 20 seconds were collected depending on the crystal. The Quest software was used for data integration including Lorentz and polarization corrections. Crystallographic data for all six compounds can be found in Table 1. Crystals of PuW2O7(OH)(H2O) were non-merohedrally twinned with a rotation angle of 180°. The program CELL_NOW was used to index the twinned crystal. Multi-scan absorption corrections were applied using the program SADABS or TWINABS.| a R(F) = ∑||Fo| − |Fc||/∑|Fo|. b R w(Fo2) = [∑[w(Fo2 − Fc2)2]/∑wFo4]1/2. | ||||||
|---|---|---|---|---|---|---|
| Compound | La | Ce | Pr | Nd | Pu | Am |
| Formula Mass | 653.61 | 654.81 | 652.61 | 655.61 | 755.76 | 507.91 |
| Color and habit | Colorless, plate | Yellow, plate | Green, plate | Pink, plate | Red, plate | Orange, plate |
| Space group | P21/m | P21/m | P21/m | P21/m | P21/m | P21/n |
| a (Å) | 5.991(3) | 5.943(2) | 5.9040(1) | 5.9121(5) | 5.8680(5) | 5.338(1) |
| b (Å) | 8.736(4) | 8.712(2) | 8.7129(1) | 8.7194(7) | 8.6979(7) | 12.626(3) |
| c (Å) | 7.165(3) | 7.118(2) | 7.0935(1) | 7.1024(6) | 7.0601(6) | 6.760(2) |
| β (°) | 106.044(4) | 105.854(3) | 105.695(1) | 105.700(1) | 105.388(2) | 111.475(6) |
| V (Å3) | 360.4(3) | 354.5(1) | 351.291(9) | 352.47(5) | 347.42(5) | 424.0(2) |
| Z | 2 | 2 | 2 | 2 | 2 | 4 |
| T (K) | 296 | 296 | 296 | 296 | 296 | 100 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Maximum 2θ (deg.) | 28.355 | 28.33 | 28.91 | 28.573 | 30.551 | 31.591 |
| ρ calcd (g cm−3) | 5.996 | 6.124 | 6.170 | 6.180 | 7.205 | 7.940 |
| μ (Mo Kα) (cm−1) | 376.78 | 386.91 | 395.07 | 398.29 | 424.22 | 449.66 |
| R(F) for Fo2 > 2 s (Fo2)a | 0.0330 | 0.0192 | 0.0103 | 0.0210 | 0.0200 | 0.0232 |
| R w(Fo2)b | 0.0814 | 0.0374 | 0.0259 | 0.0464 | 0.0462 | 0.0424 |
UV-vis-NIR spectroscopy
UV-vis-NIR data were acquired from single or twinned crystals of PuW2O7(OH)(H2O), CeW2O7(OH)(H2O), NdW2O7(OH)(H2O), and AmWO4(OH) using a Craic Technologies microspectrophotometer. Crystals were placed on quartz slides under immersion oil, and the data was collected from 200 to 1200 nm for each compound. Data shown are regions of interest.Results and discussion
Structure and topological description
MW2O7(OH)(H2O) (M = Pu, La, Ce, Pr, and Nd) crystallizes in the space group P21/m and is a dense 3D network composed of infinite chains of edge-sharing WO6 distorted octahedra, linked by MO8 polyhedra. The corrugated tungstate chains propagate parallel to the b axis. The f-element metal centers are approximated by bicapped trigonal prisms where seven of the eight O atoms are donated from tungstate, and the remaining O atom is a coordinating water molecule.40 The MO8 units are isolated from each other, but link three tungstate chains, building the network as shown in Fig. 1.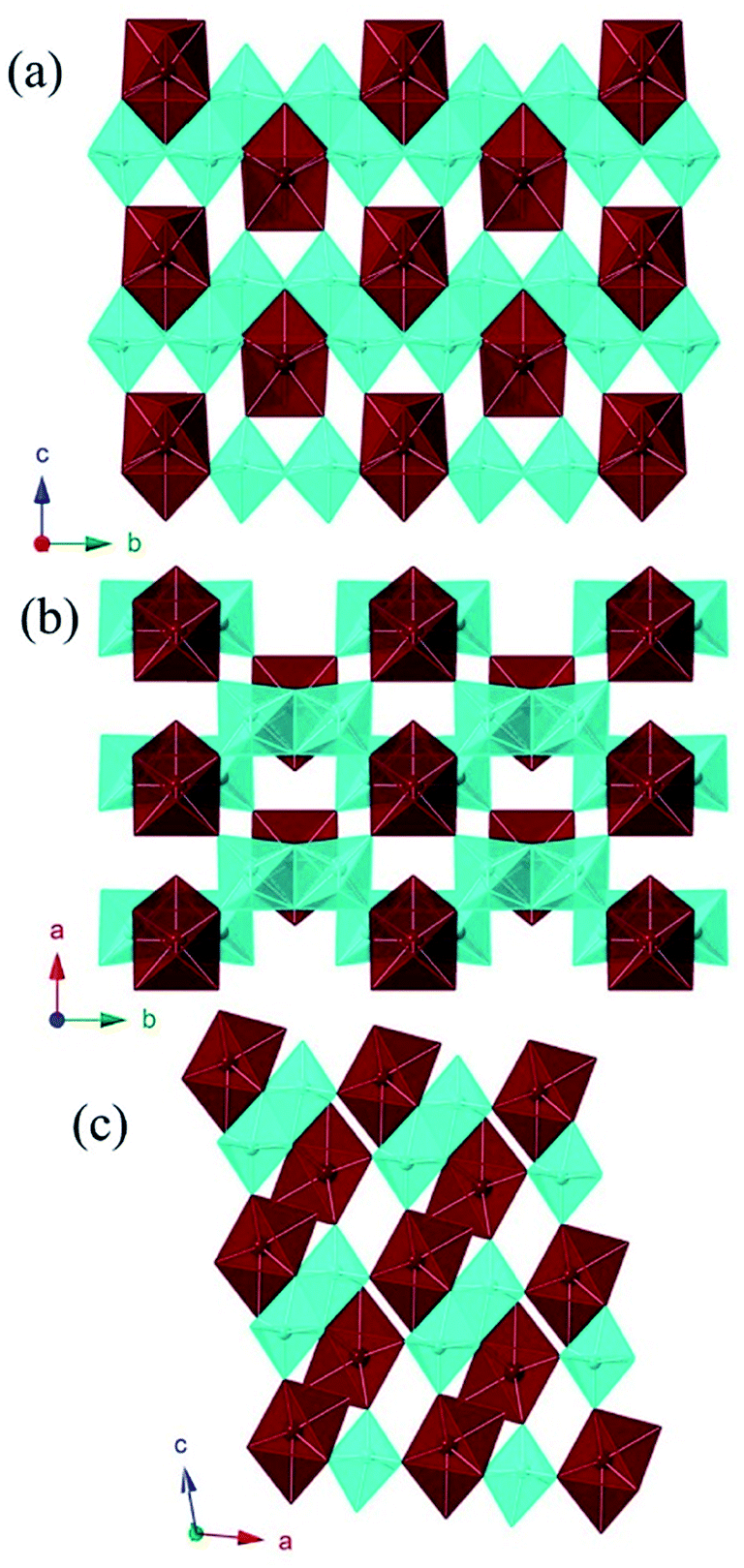 | ||
| Fig. 1 Polyhedral representations of PuW2O7(OH)(H2O) along each axis showing isolated Pu(III) bicapped trigonal prisms connecting chains of tungstate distorted octahedra. PuIIIO8 metal centers are within red polyhedra and tungstate metal centers are in blue distorted octahedra. | ||
Only one crystallographically unique M atom and one crystallographically unique W atom are found in the asymmetric unit. The distorted octahedra of tungstate have two short (∼1.75 Å), one intermediate (∼1.88 Å), two normal (∼2.0 Å), and one slightly long (∼2.20 Å) W–O bonds. This unit can be thought of in terms of a second-order Jahn-Teller distortion as having a C2 distortion or, in molecular terms, as the cis-tungstyl unit. This elongated W–O bond is protonated as indicated by bond valence sum (BVS) calculations providing the additional positive charge necessary to balance the formula.41 The trigonal prismatic O atoms are donated from the tungstate groups as shown in Fig. 2. These M–O bonds range from ∼2.37 Å to ∼2.46 Å as can be seen in Table 2 for Pu and Table 3 for the lanthanides. In isotypic structures of 4f and 5f compounds, comparisons of bond lengths can provide insight into covalency because the ionic radii match fairly well between certain lanthanides and actinides.17,41–43 For Pu(III) the corresponding lanthanide is Ce(III); although we have proposed Nd(III) as being a better match for higher coordination numbers. The average Pu–O bond differences are 0.029(5) Å and 0.018(6) Å shorter than the Ce and Nd bond distances, respectively. Taking into account standard error of the mean, Pu is still statistically shorter than both Ce and Nd. This structure type has not been previously reported as there are few hydrothermally-prepared f-block tungstates that are not polyoxometalate-type complexes.
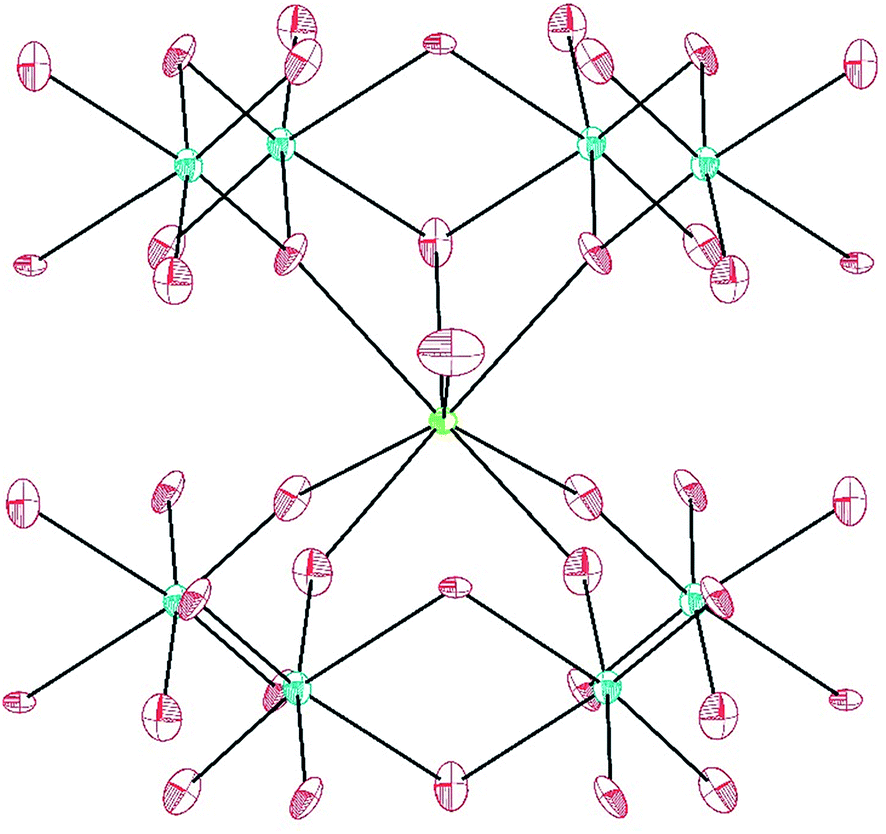 | ||
| Fig. 2 Ball-and-stick representation of the coordination environment of Pu(III) in PuW2O7(OH)(H2O). The lime-green sphere is Pu(III), blue spheres are W(VI), and red spheres are O atoms. The O atom at the center of the figure is the coordinating water molecule. | ||
| PuW2O7(OH)(H2O) | AmWO4(OH) | ||
|---|---|---|---|
| Pu(1)–O(2) | 2.374(4) | Am(1)–O(1) | 2.357(5) |
| Pu(1)–O(2) | 2.374(4) | Am(1)–O(5) | 2.396(5) |
| Pu(1)–O(3) | 2.396(5) | Am(1)–O(2) | 2.420(5) |
| Pu(1)–O(3) | 2.396(5) | Am(1)–O(3) | 2.449(5) |
| Pu(1)–O(4) | 2.428(5) | Am(1)–O(4) | 2.460(5) |
| Pu(1)–O(4) | 2.428(5) | Am(1)–O(4) | 2.473(5) |
| Pu(1)–O(5) | 2.535(8) | Am(1)–O(5) | 2.496(5) |
| Pu(1)–O(1) | 2.747(7) | Am(1)–O(2) | 2.503(5) |
| W(1)–O(4) | 1.756(5) | W(1)–O(1) | 1.763(5) |
| W(1)–O(3) | 1.768(5) | W(1)–O(3) | 1.764(5) |
| W(1)–O(2) | 1.884(5) | W(1)–O(4) | 1.813(5) |
| W(1)–O(1) | 1.962(3) | W(1)–O(2) | 1.818(5) |
| W(1)–O(2) | 2.157(5) | W(1)–O(5) | 2.398(5) |
| W(1)–O(6) | 2.199(4) | ||
| LaW2O7(OH)(H2O) | CeW2O7(OH)(H2O) | PrW2O7(OH)(H2O) | NdW2O7(OH)(H2O) | ||||
|---|---|---|---|---|---|---|---|
| La(1)–O(5) | 2.437(9) | Ce(1)–O(5) | 2.397(4) | Pr(1)–O(5) | 2.384(2) | Nd(2)–O(5) | 2.381(5) |
| La(1)–O(5) | 2.437(9) | Ce(1)–O(5) | 2.397(4) | Pr(1)–O(5) | 2.384(2) | Nd(2)–O(5) | 2.381(5) |
| La(1)–O(3) | 2.457(9) | Ce(1)–O(3) | 2.429(4) | Pr(1)–O(3) | 2.413(2) | Nd(2)–O(3) | 2.418(6) |
| La(1)–O(3) | 2.457(9) | Ce(1)–O(3) | 2.429(4) | Pr(1)–O(3) | 2.413(2) | Nd(2)–O(3) | 2.418(6) |
| La(1)–O(4) | 2.497(9) | Ce(1)–O(4) | 2.477(4) | Pr(1)–O(4) | 2.464(2) | Nd(2)–O(6) | 2.462(5) |
| La(1)–O(4) | 2.497(9) | Ce(1)–O(4) | 2.477(4) | Pr(1)–O(4) | 2.464(2) | Nd(2)–O(6) | 2.462(5) |
| La(1)–O(1) | 2.59(2) | Ce(1)–O(1) | 2.543(6) | Pr(1)–O(1) | 2.541(3) | Nd(2)–O(1) | 2.545(8) |
| La(1)–O(2) | 2.77(1) | Ce(1)–O(2) | 2.758(6) | Pr(1)–O(2) | 2.747(3) | Nd(2)–O(2) | 2.752(8) |
| W(1)–O(3) | 1.756(9) | W(1)–O(4) | 1.755(4) | W(1)–O(4) | 1.754(2) | W(1)–O(6) | 1.756(5) |
| W(1)–O(4) | 1.770(8) | W(1)–O(3) | 1.760(4) | W(1)–O(3) | 1.759(2) | W(1)–O(3) | 1.762(5) |
| W(1)–O(5) | 1.885(9) | W(1)–O(5) | 1.888(4) | W(1)–O(5) | 1.884(2) | W(1)–O(5) | 1.888(5) |
| W(1)–O(2) | 1.965(7) | W(1)–O(2) | 1.961(3) | W(1)–O(2) | 1.963(2) | W(1)–O(2) | 1.968(4) |
| W(1)–O(5) | 2.149(9) | W(1)–O(5) | 2.159(4) | W(1)–O(5) | 2.163(2) | W(1)–O(5) | 2.169(6) |
| W(1)–O(6) | 2.218(9) | W(1)–O(6) | 2.208(4) | W(1)–O(6) | 2.202(2) | W(1)–O(4) | 2.207(5) |
AmWO4(OH) crystallizes in the space group P21/n and is also a 3D network consisting of layers of edge-sharing Am polyhedra connected by tungstate square bipyramids. The eight coordinate Am metal centers are approximated by square antiprisms that edge share with three other Am polyhedra forming layers that propagate parallel to the [ac] plane. These layers are linked by the distorted WO4OH3− anions that donate all O atoms to Am as shown in Fig. 3. The tungstate geometry can be considered a 4 + 1 coordination where the environment goes from tetrahedral with four short bonds (ranging from ∼1.76 Å to 1.81 Å) to distorted trigonal bipyramid with the addition of the long bond (2.398(5) Å) to the OH− group. The Am–O bonds range from 2.357(5) Å to 2.503(5) Å. This structure type has been previously reported for Nd, however synthetic details are lacking.44
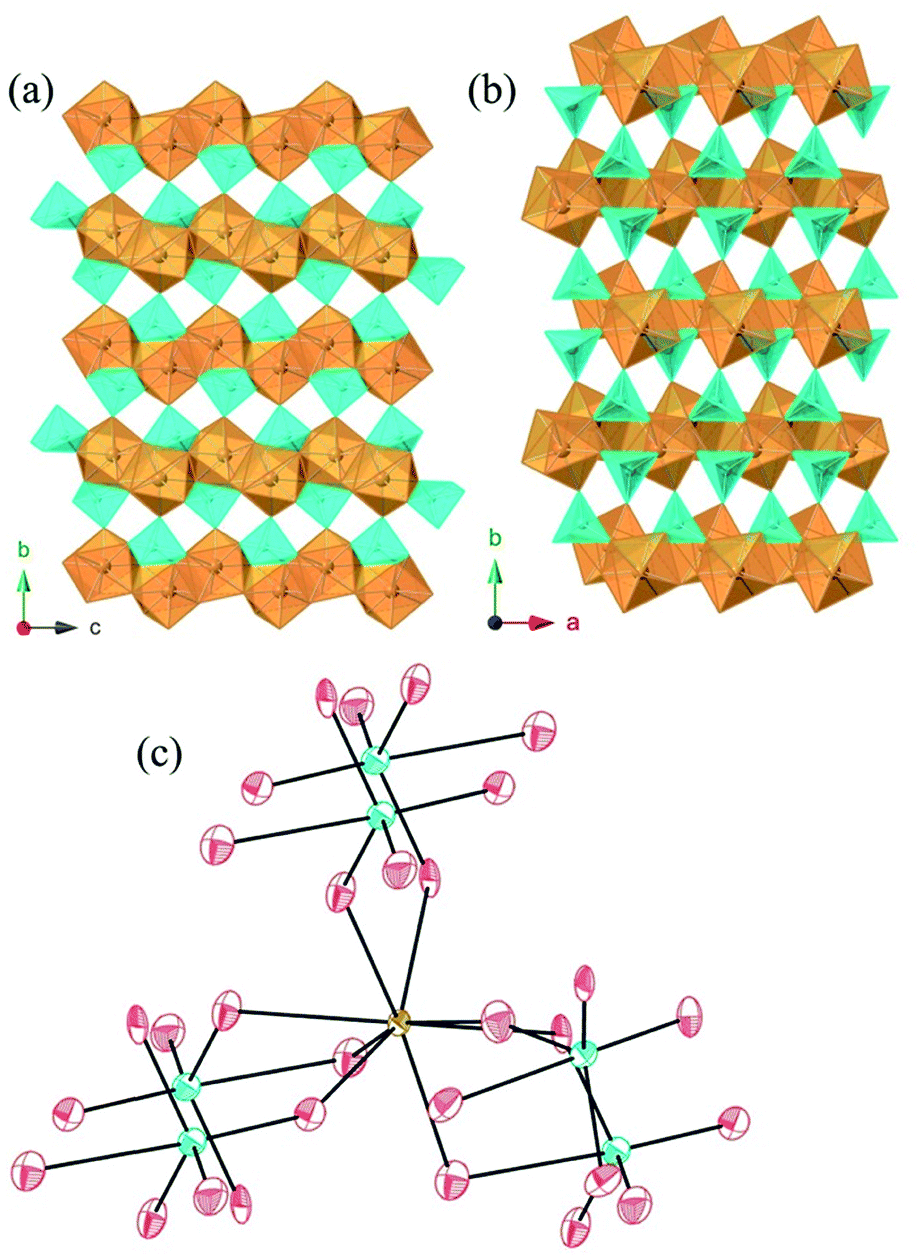 | ||
| Fig. 3 Polyhedral representation of AmWO4(OH) where Am(III) is represented in orange, W(VI) is represented in blue, and O atoms as red spheres. (a and b) Polyhedral representation along the a and c axes, respectively, displaying the edge sharing chains of Am(III)O8. (c) Ball-and-stick coordination of Am(III) showing all eight O atoms being donated from tungstate forming an approximate square antiprism. | ||
UV-vis-NIR spectroscopy
Absorption data were collected for MW2O7(OH)(H2O) (M = Pu, Nd, and Ce) and AmWO4(OH) on single crystals using a microspectrophotometer and are shown in Fig. 4. The Ce and Pu compounds both exhibit intense broadband features that extend into the UV. The band in the Ce complex leads to the observed yellow color. While yellow Ce(III) complexes are known, it is more typical for Ce(III) oxoanion complexes to be colorless.15,17,18,20,45,46 The broad absorption of the Pu complex proceeds into what appears to be the 4L13/2 and 4M15/2 peaks around 600 nm and the 6F3/2 peak ∼660 nm. The transitions at higher wavelengths are the typical ones observed for Pu(III).46–49 The broadbands and these transitions are responsible for the deep-red color of the crystals that is atypical for Pu(III) complexes. | ||
| Fig. 4 Electronic absorption spectra of CeW2O7(OH)(H2O) (top) and PuW2O7(OH)(H2O) (bottom). The typical f–f transitions of Pu(III) are observed starting around 600 nm and continuing into the lower energies. | ||
The intense broadband is distinctly absent in the AmWO4(OH) and NdW2O7(OH)(H2O) spectra and the absorption is generally weaker, but the f–f transitions of Am(III) and Nd(III) are observed as shown in Fig. 5. The major peak at 510 nm and the small transition at 520 nm in the Am tungstate can be attributed to the 5L6 and 5D2 states. The peaks from 790 to 825 nm are from the 7F5 and 7F6 transitions.
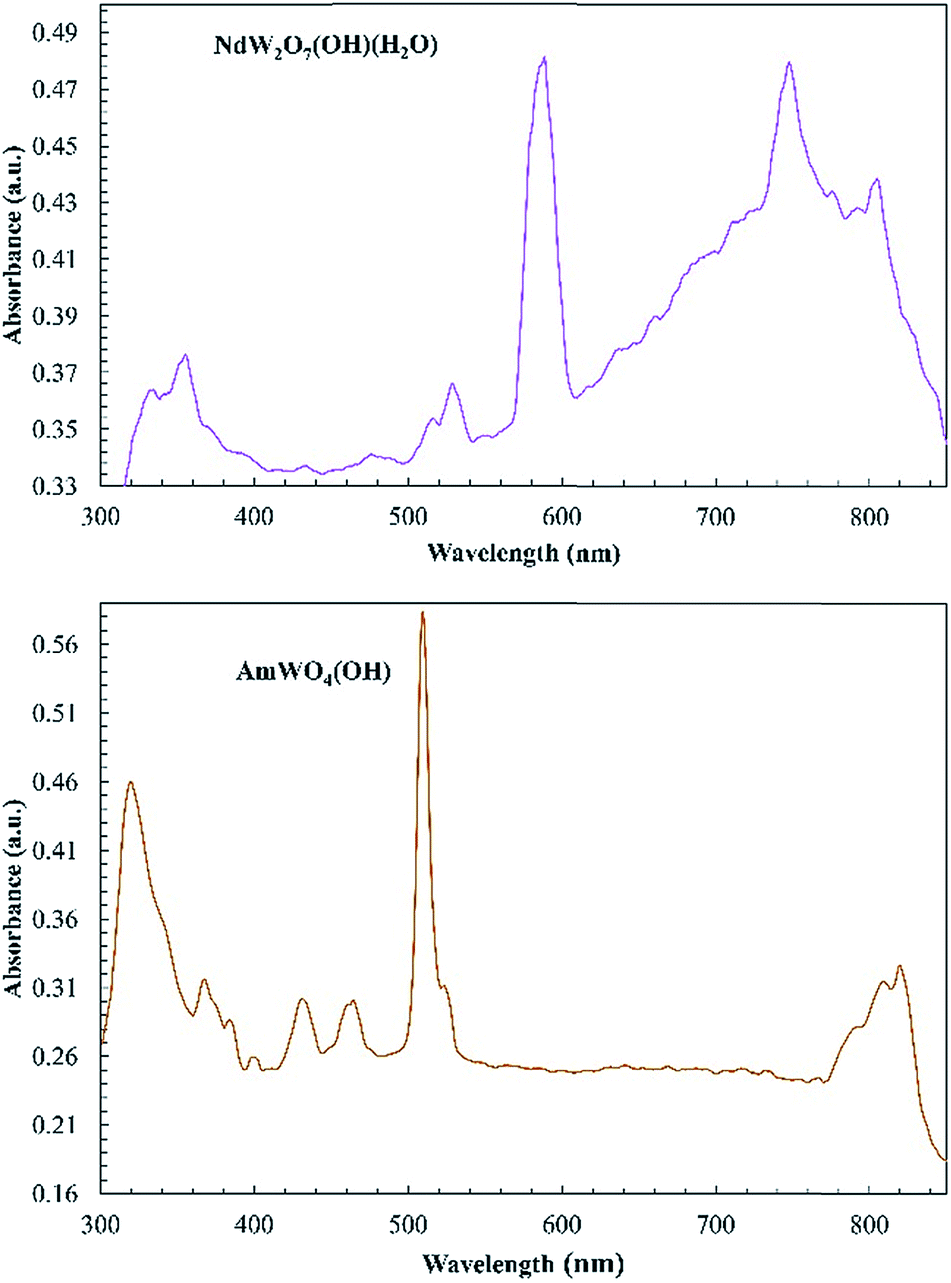 | ||
| Fig. 5 Electronic absorption spectra of NdW2O7(OH)(H2O) (top) and AmWO4(OH) (bottom). The absorption of these complexes is very weak, but the typical f–f transitions are observed. | ||
We have previously noted intense broadband features and unusual color in both Pu(III) and Ce(III) compounds coupled with d0 oxoanions.39,50 The fact that these are observed at low energies (∼500 nm) only in the Pu and Ce compounds and not in Am or Nd is noteworthy. The excited state of Ce(III) is known to be 5d and easily perturbed by ligand-field effects.51–54 Given this, there might be significant interactions between the frontier orbitals of the d0 metal anion and the 5d orbital on Ce(III) leading to the broad absorption observed. This was the case with the recently published Ce polyoxometallate, where the DFT calculations showed that charge transfer occurred between Ce(III) and W(VI) resulting in its photocatalytic properties.54 Am(III) and Nd(III) tungstate do not have the low energy CT band. The CT is shifted to higher energies that cannot be fully resolved with the microspectrophotometer. A possible explanation is that these features are related to the ease in accessing the M(IV) oxidation state. Both Ce(III) and Pu(III) are able to access tetravalent states fairly easily arise from redox instability of Ce(III) and Pu(III) where the absorptions seen are metal-to-ligand CT transitions. The features seen here are very similar to the features seen in the electronic spectra of transition metal tungstates.55 These spectra indicate a metal-to-metal charge transfer (MMCT) where the partially occupied An/Ln metal orbitals transfer to the empty tungsten 5d orbitals.55 Identifying exactly which orbitals are involved and how the orbital energies change going from 4f to 5f requires theoretical studies, which we discuss in the next section.56
Band structures of PuW2O7(OH)(H2O) and AmWO4(OH)
Rather than continuing to speculate on the origins of the unusual colors exhibited in these systems—and their possible connection with 5f-electron covalency,—here we calculate the electronic structure of PuW2O7 and AmWO4 employing the local density approximation (LDA)57 to density functional theory (DFT)58,59 in combination with dynamical mean field theory (LDA + DMFT)60–62 and in combination with the Gutzwiller approximation (LDA + GA),63–67 which are techniques specifically designed to capture the electron correlations beyond the single-particle picture underlining classic approximations to DFT.68–70Both GA and DMFT have been interfaced with the DFT code WIEN2k71 utilizing the implementation described in ref. 72 and employing the standard fully-localized limit form for the double-counting functional.73 Consistently with previous work,14 in our calculations we assume that the screened values of the Hund's coupling constant and the local Coulomb interaction strength are J = 0.7 eV and U = 6.0 eV, respectively. The LDA + DMFT calculations are performed at room temperature (T = 290 K) utilizing the continuous time quantum Monte Carlo (CTQMC) impurity solver,72,74 while the LDA + GA calculations are performed at T = 0 K.
The LDA + DMFT angle-resolved photoemission spectra (ARPES),75 the corresponding 5f-electron contribution to the quasi-particle density of states (DOS) and the j = 5/2 and j = 7/2 components of the self-energy of these two materials, where j is the single-particle total angular momentum, are shown in panels (a–c) of Fig. 6 and 7. The bare unpolarized LDA band structures are also reported in panel (d). Our simulations indicate that the quasiparticle gaps of PuW2O7 and AmWO4 are about 1.2 eV and 2.5 eV, respectively, in agreement with our UV-vis-NIR absorption experiments. The fact that the LDA + DMFT band structure displays pronounced incoherent features and is considerably different with respect to bare LDA indicates clearly that, as expected, the f-electron correlations influence dramatically the electronic structure of these materials. The different behavior of the j = 5/2 and j = 7/2 components of the self-energy and the resolved DOS indicate that also the SOC effect is very important in both systems.
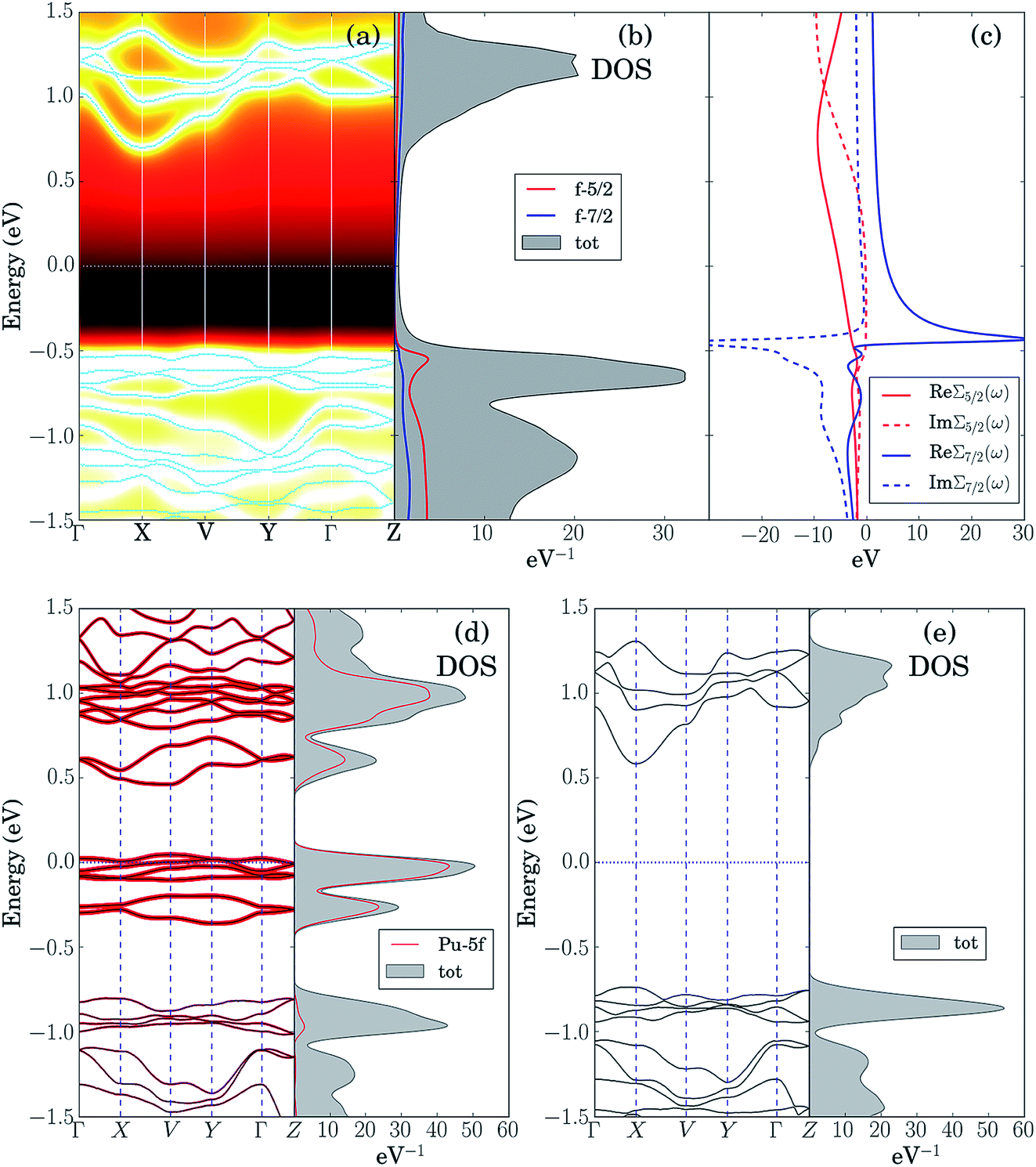 | ||
| Fig. 6 Theoretical analysis spectral properties of PuW2O7(OH)(H2O): (a) LDA + DMFT ARPES spectra computed at T = 290 K and corresponding quasi-particle bands. (b) Spectral contributions to the Pu-5f 5/2 and 7/2 contributions to the DOS. (c) 5/2 and 7/2 components of the self-energy. (d) Bare unpolarized LDA bands and corresponding Pu-5f 5/2 and 7/2 contributions to the DOS. (e) Modified LDA bands obtained by setting to 0 the hybridization between the 5f orbitals and their environment. | ||
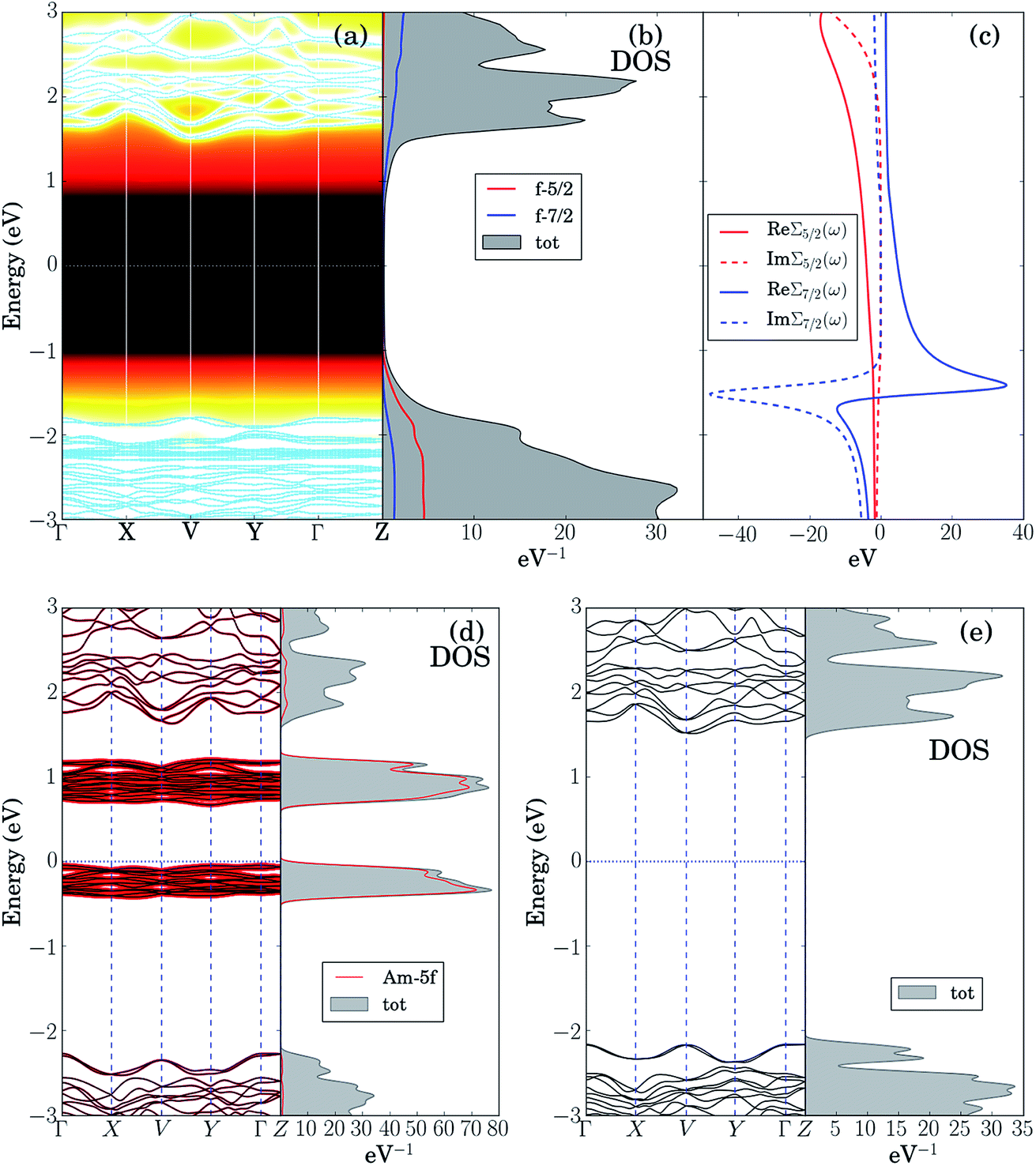 | ||
| Fig. 7 Theoretical analysis spectral properties of AmWO4(OH): (a) LDA + DMFT ARPES spectra computed at T = 290 K and corresponding quasi-particle bands. (b) Spectral contributions to the Pu-5f 5/2 and 7/2 contributions to the DOS. (c) 5/2 and 7/2 components of the self-energy. (d) Bare unpolarized LDA bands and corresponding Pu-5f 5/2 and 7/2 contributions to the DOS. (e) Modified LDA bands obtained by setting to 0 the hybridization between the 5f orbitals and their environment. | ||
In order to shed light on the physical origin of the pronounced differences between the optical properties of PuW2O7 and AmWO4, as well as on the influence of the f-electron correlations on the spectra of these 2 materials, it is very insightful to plot the LDA bands modified by removing artificially the contribution of the f-electron degrees of freedom in the LDA Kohn–Sham Hamiltonian,59i.e., by setting to 0 the hybridization between the 5f orbitals and their environment, see panels (e) of Fig. 6 and 7. Interestingly, we observe that the so-obtained low-energy band structures are very similar to the physical LDA + DMFT quasi-particle bands. Furthermore, in both systems the valence band has mostly O-2p character, while the conduction band has mostly W-5d character. This analysis demonstrates that the pronounced differences between the optical gaps of these 2 compounds do not stem directly from the 5f degrees of freedom. On the other hand, the pronounced incoherent features of the LDA + DMFT bands and the resolved DOS are an indirect indication of the fact that the O-2p and 5f degrees of freedom are hybridized, which suggests that the Am–O and Pu–O chemical bonds are not purely ionic. Here we quantify the importance of the 5f-electron covalency effects in the materials considered utilizing the procedure developed in ref. 14, i.e., we compare the physical LDA + GA ground-state energy of the systems with the energy that the system would have if the 5f degrees of freedom were entirely disentangled from the rest of the system, i.e., with the total energy minimum realizable in a quantum state with 5f valence degrees of freedom hosting exactly the nominal value of electrons (6 electrons for Am and 5 electrons for Pu), completely decoupled from the rest of the system. According to our calculations, the so-obtained measure of covalency is ΔEcov ≈ 0.72 eV f.u.−1 for PuW2O7 and ΔEcov ≈ 1.08 eV f.u.−1 for AmWO4.
A complementary indication of covalency is provided by the local 5f-electron reduced density matrix, which is formally obtained from the ground-state wavefunction of the systems by tracing out all degrees of freedom except those of the 5f shell. In Table 4 and Table 5 are shown the quantum labels of the eigenstates of the local Pu-5f and Am-5f electron reduced density matrix of PuW2O7 and AmWO4 with highest probability weights. Our calculations indicate that both systems are “mixed-valent compounds”, as they display non-negligible charge fluctuations. In particular, we note that the sum of the probability weights arising from other multiplets besides the highest ones (which correspond to the nominal atomic ground-state configurations) is about 24% for Pu and 13% for Am.
| i | 1 | 2 | 3 | 4 |
| w i | 0.76 | 0.061 | 0.059 | 0.017 |
| N i | 5 | 6 | 4 | 6 |
| J i | 2.5 | 0 | 4 | 6 |
| i | 1 | 2 | 3 | 4 |
| w i | 0.87 | 0.082 | 0.035 | 0.002 |
| N i | 6 | 7 | 5 | 6 |
| J i | 0 | 3.5 | 2.5 | 6 |
In summary, our analysis indicates that the f-electron covalency effects have a strong influence on the total energy and the chemical bonds in PuW2O7(OH)(H2O) and AmWO4(OH). Indirect signatures of f-electron covalency can be observed also in the ARPES spectra, which displays pronounced incoherent features and f-electron hybridization effects at low energies. On the other hand, in both systems the valence bands have mostly O-2p character and the conduction bands have mostly W-5d character. Furthermore, the quasi-particle gap is essentially unaffected by the 5f degrees of freedom. Consequently, while the 5f-covalency effects are energetically important, the pronounced differences observed between the optical properties of these compounds do not stem directly from the 5f degrees of freedom. Interestingly, as shown in the supplemental material in Fig. S5–S7,† a similar situation occurs also in 3 different 4f and 5f chromates: α-CsM(CrO4)2 (M = Sm, Eu, Am).
Conclusions
Atypical coloration of compounds has been a harbinger of the need for understanding electronic structure since the earliest days of chemistry. It is one of the driving forces behind the evolution of ligand-field theory from crystal-field theory because the latter does not allow for the correct prediction of the color of most transition metal complexes because it does not account for the role that the d orbitals play in covalent bonding.76 In the actinide compounds unusual coloration can indeed be indicative of unusual metal–ligand interactions.38,77,78 However, the tendency is to jump to the conclusion that the coloration of compounds and complexes that are different from that observed from the respective hydrated cations (e.g. [An(H2O)9]3+) can always be ascribed to significant utilization of the metal-based frontier orbitals in bonding. For example, [Pu(H2O)9]3+ both in concentrated solutions and the solid-state exhibits deep-purple coloration. Yet, both PuIII(Se2PPh2)3(py)2 and [PuIII(dpa)(H2O)4]Br (dpa = dipicolinate) are green. In both of these examples the green color is ascribed to ligand-to-metal charge transfer, and in the latter case it facilitates fascinating oxidation though mixed-valent compounds that can exhibit intervalence charge transfer.79 However, what these examples fail to capture is that color arises from many sources and is not always based on the electronic features that are created from single metal centers. In gemstones, coloration is commonly created by trace dopants and defects.The study of the 5f tungstates PuW2O7(OH)(H2O) and AmWO4(OH) addressed in this work, supplemented by the analysis of the 4f and 5f chromates α-CsM(CrO4)2 (M = Sm, Eu, Am)14 shown in the supplemental material, provides a new variation on this theme, where the f-orbitals are strongly entangled with O-2p electrons, but this hybridization plays no role in the odd colors of the compounds—whose variations are mostly dictated by the different character of the W-5d bands in the tungstates and in the Cr-3d bands in the chromates. These insights exemplify how combining state-of-the-art experimental and quantum-simulation techniques is essential for characterizing and understanding chemical bonds (and, in particular, their connection with the optical properties) in f-electron systems.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
All experimental studies were supported by the U.S. Department of Energy, Basic Energy Sciences, Heavy Elements Chemistry Program, U.S. Department of Energy, under Grant DE-FG02-13ER16414. Theoretical studies were supported by the U.S. Department of Energy as a part of the Center for Actinide Science and Technology (CAST) funded by the Energy Frontiers Research Program, under Award Number DE-SC0016568. N. L. was also supported by the VILLUM FONDEN via the Centre of Excellence for Dirac Materials (Grant No. 11744). T.-H. L and C.-J. K were supported by the Department of Energy under Grant No. DE-FG02-99ER45761. Y. Y. was supported by the U.S. Department of energy, Office of Science, Basic Energy Sciences, as a part of the Computational Materials Science Program. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) funded by NSF under Grants No. TG-DMR170121.Notes and references
- G. R. Choppin, J. Alloys Compd., 2002, 344, 55–59 CrossRef CAS.
- N. A. Edelstein, J. Fuger, J. J. Katz and L. R. Morss, in The Chemistry of the Actinides and Transactinide Elements, ed. L. R. Morss, N. A. Edelstein, J. Fuger and J. J. Katz, Springer, Berlin, 4th edn, 2011, vol. 3, pp. 1753–1835 Search PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC.
- J. A. Bogart, A. J. Lewis, S. A. Medling, N. A. Piro, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2013, 52, 11600–11607 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- I. Kirker and N. Kaltsoyannis, Dalton Trans., 2011, 40, 124–131 RSC.
- M. J. Tassell and N. Kaltsoyannis, Dalton Trans., 2010, 39, 6719–6725 RSC.
- I. D. Prodan, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 033101 CrossRef.
- A. J. Gaunt, S. D. Reilly, A. E. Enriquez, B. L. Scott, J. A. Ibers, P. Sekar, K. I. M. Ingram, N. Kaltsoyannis and M. P. Neu, Inorg. Chem., 2008, 47, 29–41 CrossRef CAS PubMed.
- M. P. Jensen and A. H. Bond, J. Am. Chem. Soc., 2002, 124, 9870–9877 CrossRef CAS PubMed.
- M. Mazzanti, R. L. Wietzke, J. Pecaut, J. M. Latour, P. Maldivi and M. Remy, Inorg. Chem., 2002, 41, 2389–2399 CrossRef CAS PubMed.
- T. Mehdoui, J. C. Berthet, P. Thuéry and M. Ephritikhine, Dalton Trans., 2004, 4, 579–590 RSC.
- A. J. Gaunt, B. L. Scott and M. P. Neu, Angew. Chem., Int. Ed., 2006, 45, 1638–1641 CrossRef CAS PubMed.
- S. S. Galley, A. A. Arico, T.-H. Lee, X. Deng, Y.-X. Yao, J. M. Sperling, V. Proust, J. S. Storbeck, V. Dobrosavljevic, J. N. Neu, T. Siegrist, R. E. Baumbach, T. E. Albrecht-Schmitt, N. Kaltsoyannis and N. Lanatà, J. Am. Chem. Soc., 2018, 140, 1674–1685 CrossRef CAS PubMed.
- M. J. Polinski, D. J. Grant, S. Wang, E. V. Alekseev, J. N. Cross, E. M. Villa, W. Depmeier, L. Gagliardi and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2012, 134, 10682–10692 CrossRef CAS PubMed.
- M. J. Polinski, E. B. Garner III, R. Maurice, N. Planas, J. T. Stritzinger, T. G. Parker, J. N. Cross, T. D. Green, E. V. Alekseev, S. M. Van Cleve, W. Depmeier, L. Gagliardi, M. Shatruk, K. L. Knappenberger, G. Liu, S. Skanthakumar, L. Soderholm, D. A. Dixon and T. E. Albrecht-Schmitt, Nat. Chem., 2014, 6, 387–392 CrossRef CAS PubMed.
- J. N. Cross, E. Villa, S. Wang, J. Diwu, M. J. Polinski and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 8419–8424 CrossRef CAS PubMed.
- J. Diwu, D. J. Grant, S. Wang, L. Gagliardi and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 6906–6915 CrossRef CAS PubMed.
- J. Lin, J. Diwu, J. N. Cross, E. M. Villa and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 10083–10085 CrossRef CAS PubMed.
- J. Lin, J. N. Cross, J. Diwu, N. A. Meredith and T. E. Albrecht-Schmitt, Inorg. Chem., 2013, 52, 4277–4281 CrossRef CAS PubMed.
- J. Goodey, J. Broussard and P. S. Halasyamani, Chem. Mater., 2002, 14, 3174–3180 CrossRef CAS.
- H. A. Jahn and E. Teller, Proc. R. Soc. London, Ser. A, 1937, 161, 220–235 CAS.
- R. A. Wheeler, M. H. Whangbo, T. Hughbanks, R. Hoffman, J. K. Burdett and T. A. Albright, J. Am. Chem. Soc., 1986, 108, 2222–2236 CrossRef CAS PubMed.
- R. G. Pearson, J. Mol. Struct.: THEOCHEM, 1983, 103, 25–34 CrossRef.
- S. K. Kang, H. Tang and T. A. Albright, J. Am. Chem. Soc., 1993, 115, 1971–1981 CrossRef CAS.
- R. E. Cohen, Nature, 1992, 358, 136–138 CrossRef CAS.
- J. K. Burdett, Molecular Shapes, Wiley-Interscience, New York, 1980 Search PubMed.
- M. Kunz and I. D. Brown, J. Solid State Chem., 1995, 115, 395–406 CrossRef CAS.
- P. S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef CAS.
- S. G. Minasian, J. M Keith, E. R. Batista, K. S. Boland, J. A. Bradley, S. R. Daly, S. A. Kozimor, W. W. Lukens, R. L. Martin, D. Nordlund, G. T. Seidler, D. K. Shuh, D. Sokaras, T. Tyliszczak, G. L. Wagner, T. C. Weng and P. Yang, J. Am. Chem. Soc., 2013, 135, 1864–1871 CrossRef CAS PubMed.
- R. A. Penneman, R. G. Haire and M. H. Lloyd, presented in part at ACS Symposium Series, Washington, D.C., 1980 Search PubMed.
- E. C. Buck, D. J. Wronkiewicz, P. A. Finn and J. K. Bates, J. Nucl. Mater., 1997, 249, 70–76 CrossRef CAS.
- S. V. Krivovichev, P. C. Burns and I. G. Tananaev, in Structural Chemistry of Inorganic Actinides, ed. S. V. Krivovichev, P. C. Burns and I. G. Tananaev, Elsevier, Amsterdam, 1st edn, 2007, ch. 4, pp. 95–182 Search PubMed.
- N. D. Dahale, M. Keskar and K. D. S. Mudher, J. Alloys Compd., 2006, 415, 244–250 CrossRef CAS.
- A. Tabuteau, M. Pages and W. Freundlich, Mater. Res. Bull., 1972, 7, 691–697 CrossRef CAS.
- A. Tabuteau and M. Pages, J. Solid State Chem., 1978, 26, 153–158 CrossRef CAS.
- M. N. Sokolova, A. M. Fedosseev, G. B. Andreev, N. A. Budantseva, A. B. Yusov and P. Moisy, Inorg. Chem., 2009, 48, 9185–9190 CrossRef CAS PubMed.
- J. N. Cross, P. M. Duncan, E. M. Villa, M. J. Polinski, J.-M. Babo, E. V. Alekseev, C. H. Booth and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2013, 135, 2769–2775 CrossRef CAS PubMed.
- J. N. Cross, S. K. Cary, J. T. Stritzinger, M. J. Polinski and T. E. Albrecht-Schmitt, Inorg. Chem., 2014, 53, 3148–3152 CrossRef CAS PubMed.
- J. Xu, E. Radkov, M. Ziegler and K. N. Raymond, Inorg. Chem., 2000, 39, 4156–4164 CrossRef CAS PubMed.
- N. E. Brese and M. O'Keefe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189–1203 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. F. Klevtsova and S. V. Borisov, Sov. Phys. Cryst., 1970, 14, 776–778 Search PubMed.
- M. J. Polinski, S. Wang, E. V. Alekseev, J. N. Cross, W. Depmeier and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 11541–11548 CrossRef CAS PubMed.
- M. J. Polinski, S. Wang, J. N. Cross, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 7859–7866 CrossRef CAS PubMed.
- C. Apostolidis, B. Schimmelpfennig, N. Magnani, P. Lindqvist-Reis, O. Walter, R. Sykora, A. Morgenstern, E. Colineau, R. Caciuffo, R. Klenz, R. G. Haire, J. Rebizant, F. Bruchertseifer and T. Fanghänel, Angew. Chem., Int. Ed., 2010, 49, 6343–6347 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and R. G. Pappalardo, J. Chem. Phys., 1970, 53, 2922–2938 CrossRef CAS.
- N. L. Banik, B. Schimmelpfennig, C. M. Marquardt, B. Brendebach, A. Geista and M. A. Denecke, Dalton Trans., 2010, 39, 5117–5122 RSC.
- T. M. Eaton, J. Lin, J. N. Cross, J. T. Stritzinger and T. E. Albrecht-Schmitt, Chem. Commun., 2014, 50, 3668–3670 RSC.
- R. R. Jacobs, W. F. Krupke and M. J. Weber, Appl. Phys. Lett., 1978, 33, 410–412 CrossRef CAS.
- Y. Dong, G. Zhou, J. Xu, G. Zhao, F. Su, L. Su, G. Zhang, D. Zhang, H. Li and J.-L. Si, Mater. Res. Bull., 2006, 41, 1959–1963 CrossRef CAS.
- P. A. Tanner, Chem. Soc. Rev., 2013, 42, 5090–5101 RSC.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356–5360 CrossRef CAS PubMed.
- S. Dey, R. A. Ricciardo, H. L. Cuthbert and P. M. Woodward, Inorg. Chem., 2014, 53, 4394–4399 CrossRef CAS PubMed.
- X.-D. Wen, R. L. Martin, T. M. Henderson and G. E. Scuseria, Chem. Rev., 2013, 113, 1063–1096 CrossRef CAS PubMed.
- O. Gunnarsson and B. I. Lundqvist, Phys. Rev. B: Solid State, 1976, 13, 4274–4298 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kotliar, S. Y. Savrasov, K. Haule, V. S. Oudovenko, O. Parcollet and C. A. Marianetti, Rev. Mod. Phys., 2006, 78, 865–951 CrossRef CAS.
- A. Georges, G. Kotliar, W. Krauth and M. J. Rozenberg, Rev. Mod. Phys., 1996, 68, 13–125 CrossRef CAS.
- V. Anisimov and Y. Izyumov, in Electronic Structure of Strongly Correlated Materials, Springer-Verlag Berlin, Heidelberg, 1st edn, 2010, vol. 3, pp. 47–120 Search PubMed.
- M. C. Gutzwiller, Phys. Rev., 1965, 137, A1726–A1735 CrossRef.
- X.-Y. Deng, L. Wang, X. Dai and Z. Fang, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075114 CrossRef.
- K.-M. Ho, J. Schmalian and C.-Z. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 073101 CrossRef.
- N. Lanatà, Y. Yao, C.-Z. Wang, K.-M. Ho and G. Kotliar, Phys. Rev. X, 2015, 5, 011008 Search PubMed.
- N. Lanatà, Y. Yao, X. Deng, V. Dobrosavljević and G. Kotliar, Phys. Rev. Lett., 2017, 118, 126401 CrossRef PubMed.
- P. R. C. Kent and G. Kotliar, Science, 2018, 361, 348–354 CrossRef CAS PubMed.
- K. Haule, J. Phys. Soc. Jpn., 2018, 87, 041005 CrossRef.
- N. Lanatà, T. H. Lee, Y. X. Yao, V. Stevanović and V. Dobrosavljević, npj Comput. Mater., 2019, 5, 30 CrossRef.
- K. Schwarz and P. Blaha, Comput. Mater. Sci., 2003, 28, 259–273 CrossRef CAS.
- K. Haule, C.-H. Yee and K. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 195107 CrossRef.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767–808 CrossRef CAS.
- K. Haule, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 155113 CrossRef.
- A. Damascelli, Phys. Scr., 2004, T109, 61–74 CrossRef CAS.
- H. B. Gray and C. J. Ballhausen, J. Am. Chem. Soc., 1963, 85, 260–265 CrossRef CAS.
- J. Diwu, A.-G. D. Nelson, S. Wang, C. F. Campana and T. E. Albrecht-Schmitt, Inorg. Chem., 2010, 49, 3337–3342 CrossRef CAS PubMed.
- P. G. Laubereau and J. H. Burns, Inorg. Chem., 1970, 5, 1091–1095 CrossRef.
- S. K. Cary, S. S. Galley, M. L. Marsh, D. L. Hobart, R. E. Baumbach, J. N. Cross, J. T. Stritzinger, M. J. Polinski, L. Maron and T. E. Albrecht-Schmitt, Nat. Chem., 2017, 9, 856–861 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1902033–1902039. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01174a |
| This journal is © The Royal Society of Chemistry 2019 |