
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
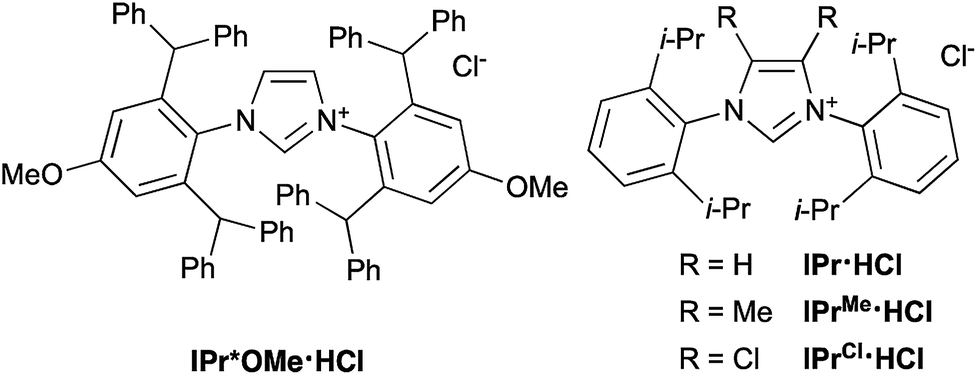
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic reduction of aryl trialkylammonium salts to aryl silanes and arenes†
Alexander W.
Rand
 and
John
Montgomery
*
and
John
Montgomery
*
Department of Chemistry, University of Michigan, 930 N. University Ave., Ann Arbor, MI 48109-1055, USA. E-mail: jmontg@umich.edu
First published on 26th April 2019
Abstract
A new approach for the reduction of aryl ammonium salts to arenes or aryl silanes using nickel catalysis is reported. This method displays excellent ligand-controlled selectivity based on the N-heterocyclic carbene (NHC) ligand employed. Utilizing a large NHC in non-polar solvents generates aryl silanes, while small NHCs in polar solvents promote reduction to arenes. Several classes of aryl silanes can be accessed from simple aniline building blocks, including those useful for cross-couplings, oxidations, and halogenations. The reaction conditions are mild, functional group tolerant, and provide efficient access to a variety of benzene derivatives.
Introduction
Aniline and its derivatives are available from a number of commercially available feedstocks and commodity chemicals, and are common structural motifs found in various materials and pharmaceuticals.1 Additionally, anilines are synthetically valuable due to their role as directing groups in electrophilic aromatic substitution and other arene functional group interconversions. While numerous types of aniline derivatives, such as diazoarenes, have been exploited in synthesis,2 aryl trialkylammonium salts are especially attractive given their ease of access, stability, safety in handling, and relatively weak C–N bond.3 Following the seminal work of Wenkert, in which aryl ammonium salts were utilized as coupling partners with Grignard reagents,4 numerous methods have been developed to create new C–C bonds via cross-couplings of aryl ammonium salts with organozinc,5 Grignard,6 and boron reagents.7 Likewise, the versatility of ammonium salts has been demonstrated in their conversion to heteroatoms via borylation,8 stannylation,9 phosphorylation,10 and transamination.11Despite these advances, the direct conversion of aryl trialkylammonium salts to aryl silanes has not been previously described. Aryl silanes are particularly useful in materials chemistry,12 as bioisosteres in medicinal chemistry,13 and are advantageous as nucleophilic coupling partners in Hiyama–Denmark couplings14 due to their high stability, low toxicity, and ability to be carried through multistep syntheses. Numerous methods for accessing aryl silanes have been developed through catalytic derivatization of aryl C–H15 and C–O16 bonds, but methods for the corresponding syntheses of aryl silanes from aryl C–N bonds are lacking. Notably, ligand control enables synthesis of either aryl silanes or simple arenes from trialkylammonium arenes and silanes utilizing Ni(0) catalysts with N-heterocyclic carbene (NHC) ligands. These findings provide simple and practical methods for the diversification of structural motifs easily accessed from widely available aniline feedstocks.
Results and discussion
While silanes are common reductants in metal-catalyzed processes, the vast majority of catalytic methods that utilize silanes result in formal transfer of a hydride unit to the substrate being reduced. The formation of C–Si bonds rather than C–H bonds using silane reductants is, in particular, a rare outcome. Recent work from our laboratory in the reaction between silyloxyarenes and silanes revealed either hydride or silyl transfer to the arene substrate could be favored depending on ligand choice on the nickel catalyst.17 Based on this precedent, we sought to extend this finding to aryl trialkylammonium salts in order to address the existing limitations in the reduction and silylation of this versatile substrate class. Furthermore, methods for the simple reduction of the C–N bond of anilines to generate Ar–H products have been described,18 but advances in scope and efficiency would further increase the utility of this process. In this study, we demonstrate methods for the conversion of aryl trialkylammonium ammonium salts to Ar–H and Ar–SiR3 products utilizing a nickel catalyst.Optimization studies
Initial efforts focused on utilizing ammonium salt 1, triethylsilane, and Ni(COD)2 with an NHC ligand in order to evaluate the feasibility of accessing either of the possible reductive outcomes (i.e. installation of –H or –SiEt3 to the starting aryl ammonium salt). Screening a variety of common NHCs uncovered a dramatic influence of ligand size on product formation, with IMes resulting in low yields of biphenyl 3a (Table 1, entry 1), while a more sterically encumbered NHC (entry 2) afforded small amounts of the aryl silane product 2a (more complete optimization in ESI†). More electron rich ligands led to higher yields compared to electron deficient ones, though low conversions were observed in each case (entries 2–4). However, turning to a more sterically demanding, electron-rich NHC, previously shown by our lab to promote C(sp2)–O bond activation in silyloxyarenes, increased the yield of the desired product to 89% (entry 5), with biphenyl (3a) as the remaining mass-balance. A strong dependence on the counter ion of the base was observed, with lower yields and conversions being observed with LiO-t-Bu and KO-t-Bu (entries 6 and 7). A similar dependence on counter ion has been noted in other nickel-catalyzed ammonium salt functionalizations.8a,11,17,19 Other ethereal solvents such as diethyl ether and THF provided inferior yields of the desired product with higher ratios of arene 3a (entries 8 and 9). Lastly, lowering the catalyst loading to 5 mol% resulted in a lower yield (entry 10) and nickel, base, and ligand were all necessary for the reaction (see ESI†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Base | Yielda2a (%) | Yielda3a (%) |
| a Determined by GCFID analysis of the crude mixture using tridecane as an internal standard. b 5 mol% Ni(COD)2 and 5 mol% IPr*OMe·HCl used. | |||||
| 1 | IMes·HCl | Dioxane | NaO-t-Bu | <1 | 6 |
| 2 | IPrCl·HCl | Dioxane | NaO-t-Bu | 12 | 8 |
| 3 | IPr·HCl | Dioxane | NaO-t-Bu | 14 | 7 |
| 4 | IPrMe·HCl | Dioxane | NaO-t-Bu | 21 | 5 |
| 5 | IPr*OMe·HCl | Dioxane | NaO-t-Bu | 89 | 11 |
| 6 | IPr*OMe·HCl | Dioxane | LiO-t-Bu | 41 | 5 |
| 7 | IPr*OMe·HCl | Dioxane | KO-t-Bu | 48 | 7 |
| 8b | IPr*OMe·HCl | Et2O | NaO-t-Bu | 5 | 6 |
| 9 | IPr*OMe·HCl | THF | NaO-t-Bu | 65 | 37 |
| 10b | IPr*OMe·HCl | Dioxane | NaO-t-Bu | 63 | 12 |
|
|||||
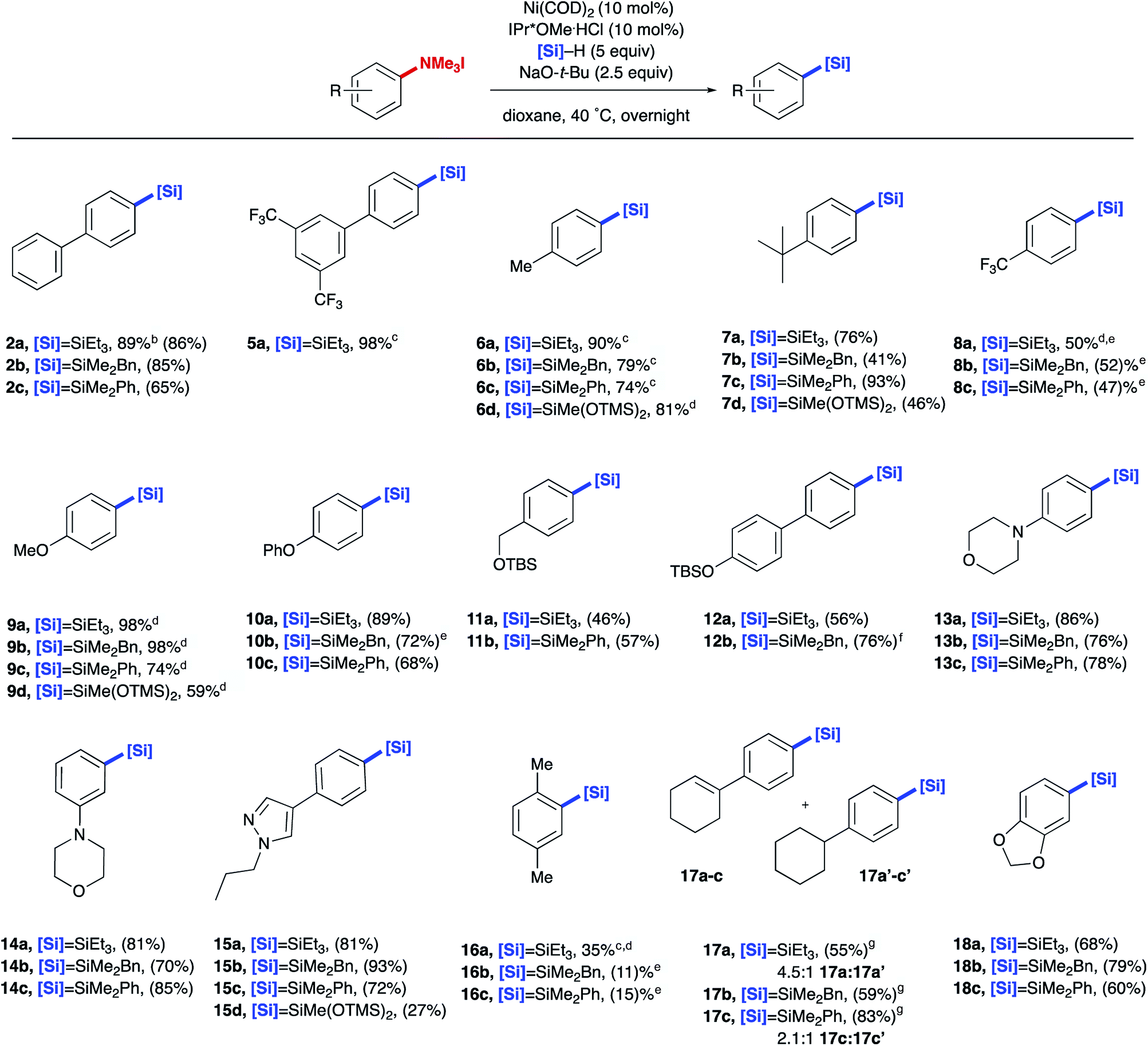
With methods in hand for the installation of –SiR3 to the aryl trialkylammonium salt, the scope of aryl silane production was explored (Table 2). Both biphenyl systems (2a–c and 5a) and alkyl substituted ammonium salts (6a–d and 7a–d) provided good yields of the desired product. Generally, electron deficient systems provided moderate yields (8a–c), whereas electron rich systems provided the corresponding aryl silane in high yield and tolerated a number of aryl or alkyl ethers (9a–d, 10a–c, and 11a, 11b). Additionally, aryl trialkylammonium salts display orthogonal reactivity to less reactive C(sp2)–O bonds (12a, 12b), allowing for further couplings in downstream functionalization using existing methods.17,20 A variety of heterocycles including morpholine and pyrazole, often found in medicinally relevant compounds, were transformed into their corresponding aryl silane derivatives in good yields (13a–c, 14a–c, and 15a–d). Ortho substitution on the aryl ammonium salts resulted in drastically lower yields (16a–c). Sensitive functional groups such as alkenes and acetals could be tolerated, although partial hydrogenation of alkenes was observed under the reaction conditions (17a–c and 18a–c). In addition to providing aryl triethylsilanes, this methodology was extended to other silanes including H–SiMe2Bn, H–SiMe2Ph, and H–Si(OTMS)2Me, which are suitable for further functional manipulation though oxidations, halogenation, and cross-couplings.21
| a Yields in parentheses are isolated yields. b Yield was determined by GCMS analysis using tridecane as an internal standard. c 3 equiv. of silane used. d Yield was determined by NMR using CH2Br2 as an internal standard. e 2 equiv. of silane used. f Run on a 0.52 mmol scale. g Reaction run under a stream of nitrogen. |
|---|
|
In our initial optimization of the synthesis of arylsilanes, some solvents resulted in significant amounts of biphenyl (3a) (Table 1, entry 9), prompting an investigation of this transformation. Typical methods to access C–H bonds from aniline derivatives rely on strong acids,18b elevated temperatures,18a or strong reductants.9,18c A more recently developed method utilized sodium isopropoxide as the terminal reductant,18a although we envisioned that improvements in scope and lower reaction temperatures could be achieved using a silane-mediated protocol. Despite the observance of appreciable yields of 3a when using a small NHC for the model system (1) developed in the silylation of aryl trialkylammonium salts, electron rich systems such as 4-phenoxybenzene trialkylammonium iodide 4 were less effective in the formation of the Ar–H reduction product 4a. Trialkylammonium salt 4 was therefore examined in optimization studies towards this product class. Low yields of 4a were observed in ethereal solvents such as dioxane and THF (Table 3, entries 1 and 2) using IMes as a ligand. To our delight, the yield of 4a was increased to 56% when switching to DMF (entry 3). By switching to DMF as the solvent the yield was greatly improved regardless of the NHC used, and only trace silylated or demethylated product could typically be detected. It is important to note, in the case of using DMF without a ligand, the reaction gave full conversion, but lower ratios of arene to arly silane were observed (see ESI†). For this reason we opted to conduct our substrate scope using IMes over ligand-free conditions. Contrary to what was observed in the silylation reaction, the size and electronics of the NHC had a less pronounced role in the reduction (entries 4 and 5). Substituting the silane source provided suitable yields of the desired product, though it was noted that increasing the size of the reductant resulted in decreased yield (entries 6–9). Lastly, although the reaction could be run at room temperature, slightly elevated temperatures were found to be optimal (entries 10 and 11).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Reductant | Yielda4a (%) |
| a Determined by GCFID analysis of the crude mixture using tridecane as an internal standard. b Reaction run at room temperature. c Reaction run at 40 °C. | ||||
| 1 | IMes·HCl | Dioxane | Et3Si–H | 29 |
| 2 | IMes·HCl | THF | Et3Si–H | 27 |
| 3 | IMes·HCl | DMF | Et 3 Si–H | 56 |
| 4 | SIMes·HCl | DMF | Et3Si–H | 47 |
| 5 | IPr*OMe·HCl | DMF | Et3Si–H | 49 |
| 6 | IMes·HCl | DMF | Et2HSi–H | 54 |
| 7 | IMes·HCl | DMF | Ph3Si–H | 41 |
| 8 | IMes·HCl | DMF | iPr3Si–H | 24 |
| 9 | IMes·HCl | DMF | Me(OTMS)2Si–H | 18 |
| 10b | IMes·HCl | DMF | Et3Si–H | 60 |
| 11 | IMes·HCl | DMF | Et 3 Si–H | 64 |
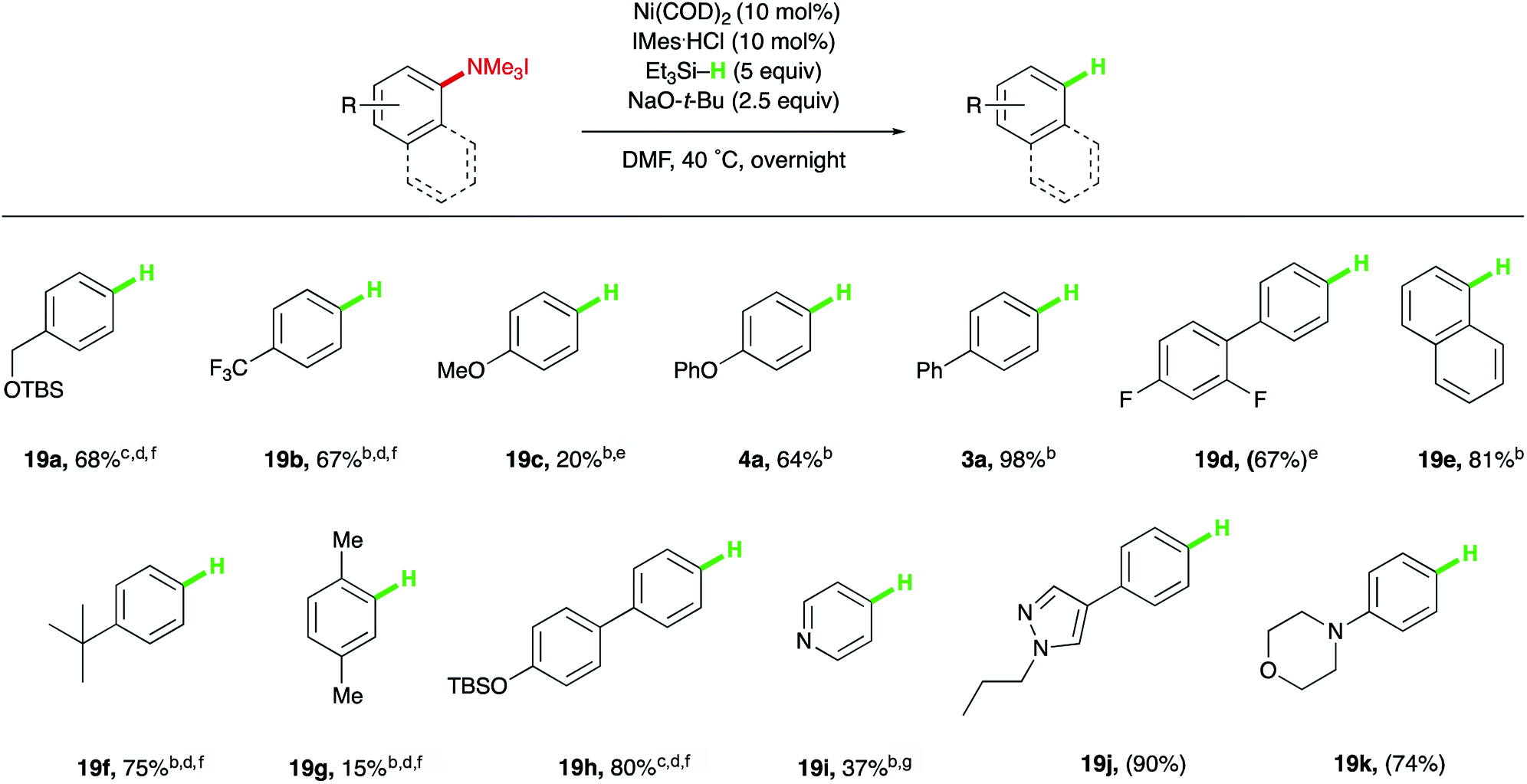
Having identified a mild method for reducing the C–N bond of aryl trialkylammonium salts to produce Ar–H bonds, the scope of this reduction was explored next, which revealed a trend opposite that seen for the electronic biases of the silylation chemistry (Table 4). Electron-deficient or electron-neutral systems, such as protected benzyl alcohol 19a and para-trifluoromethyl substrate 19b provided higher yields than electron-rich methoxy and phenoxy ethers (19c and 4a). Both isolated and extended π-systems provided reduced arenes in high yields (3a, 19d, and 19e). Alkyl substituents were tolerated (19f), with the substitution pattern greatly influencing reactivity, as ortho-methyl substitution resulted in low yields of the desired product (19g), just as observed with the developed silylation reaction. Additionally, other aryl electrophiles that can be utilized in orthogonal functionalizations, such as aryl silyl ethers, were suitable substrates (19h). Finally, common heterocyclic substrates, such as pyridines, pyrazoles, and morpholines could also be reduced to their benzene derivatives in moderate to excellent yields (19i, 19j, and 19k).
The reductive conversion of aryl trialkylammonium salts into aryl silanes or simple arenes presents interesting opportunities in multistep functionalization of simple aromatics. For example, the incorporation of strategies that utilize nitroarene-directed or aniline-directed synthetic steps prior to installation of the trialkylammonium functionality will allow a versatile approach for the conversion of simple arenes to highly functionalized aromatics. Nitrobenzenes, which are readily available by nitration of feedstock aromatics, are versatile intermediates due to their meta-directing influence in electrophilic aromatic substitution processes or through the promotion of nucleophilic aromatic substitutions of fluoro substituents. Following nitroarene derivatization, reduction to an aniline derivative enables ortho/para-directed aromatic substitutions or metallations directed by the amino group. The following syntheses are representative of the potential utility of this type of strategy (Scheme 1). The ability of anilines to direct Friedel–Crafts acylations was utilized starting from N,N-dimethylaniline (20), followed by subsequent Wolff–Kishner reduction and N-alkylation to access 21. The desired aryl silane (22) could be easily accessed in 66% yield, further highlighting the synthetic utility of this reaction. Additionally, beginning from 4-nitrofluorobenzene (23) allows for nucleophilic aromatic substitution with (−) menthol. Upon reduction of the nitro group with palladium on carbon, followed by reductive amination and methylation of the resulting N,N-dimethyl aniline, compound 24 was prepared in 21% yield over 4 steps. Next, using the developed silylation conditions, the desired aryl silane (25) was accessed in 58% yield. A similar approach using 4-nitrophenol (26) allows for the rapid build-up of complexity using a three-step allylation, Claisen rearrangement, and alkylation procedure to give 27. Nitro compound 27 could then be converted to the aryl trialkylammonium salt, providing product 28 in 33% yield over 6 steps, requiring only one silica gel column purification. Finally, 28 was silylated in 77% yield to give 29. The feasibility of using commercially available, air-tolerant IPr*OMe-nickel(0) acrylate catalysts to form 2a was also demonstrated, albeit in slightly lower yield than the in situ generated catalyst (Scheme 2). Lastly, the silylation reaction was scaled to 3.27 mmol to give the desired product (9a) in 78% yield after isolation, using 5 mol% Ni(COD)2 and IPr*OMe·HCl (Scheme 3).
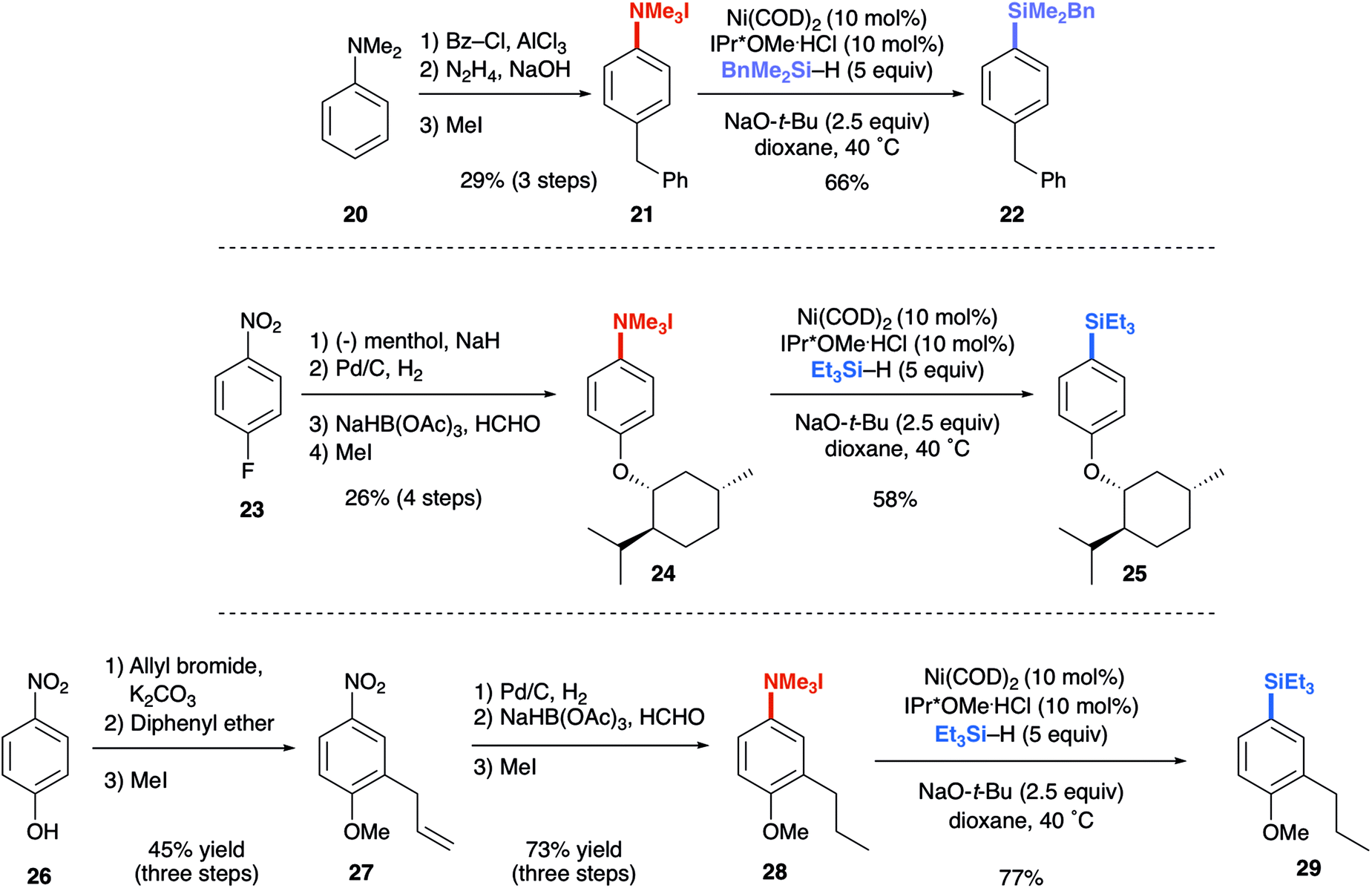 | ||
| Scheme 1 Synthetic application of aryl trialkylammonium salt silylation. | ||
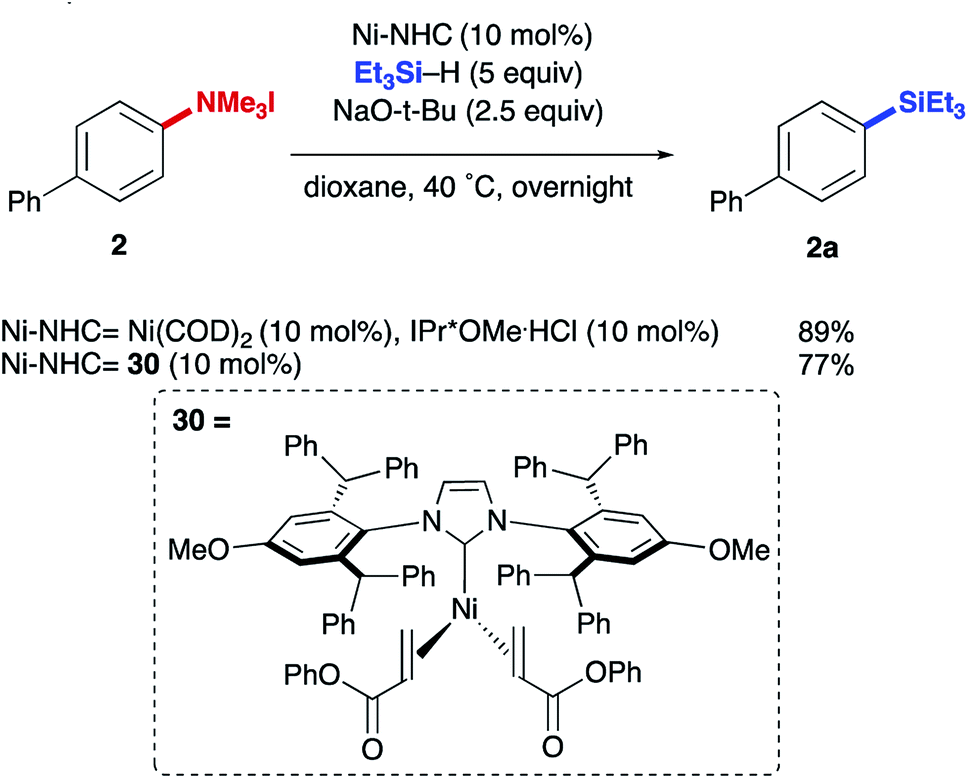 | ||
| Scheme 2 Comparison an in situ formed and air-tolerant nickel complex. | ||
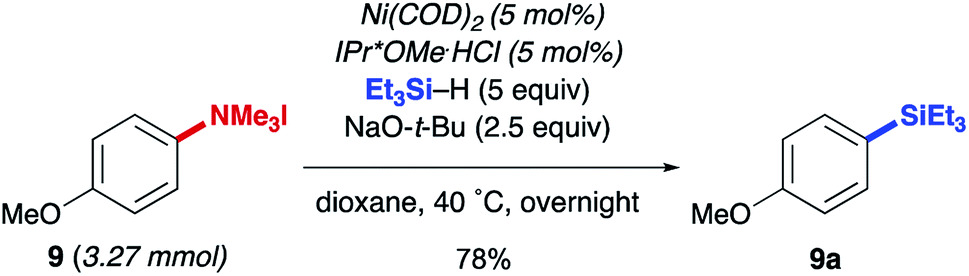 | ||
| Scheme 3 Large-scale aryl trialkylammonium salt silylation. | ||
Conclusions
Complementary methods to form aryl silanes from aryl trialkylammonium salts have been developed using several different classes of silanes, allowing for their use in subsequent oxidations, halogenations, and cross-couplings. Having garnered a better understanding of aryl trialkylammonium salt silylation, reduction to benzene derivatives was accomplished under ambient conditions and in synthetically useful yields. The silylation/reduction selectivity of aryl trialkylammonium salts was controlled though the size of NHC ligand and judicious choice of solvent. Several classes of silanes were utilized under mild reaction conditions, providing a new method to access synthetically useful aryl silanes. This new method to access high-value aryl silanes offers a streamlined approach to a range of functionalities, starting from commercially available nitro- and aniline-containing compounds. Moreover, the methods developed above offer a new strategy to functionalize C(sp2)–N bonds, a common motif found in biologically relevant molecules and commercial feedstocks, using economical, earth abundant nickel catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1565837) for support of this research. Alex J. Nett is thanked for helpful discussions.Notes and references
- B. R. Smith and J. T. Njardarson, J. Chem. Educ., 2012, 87, 1348–1359 Search PubMed.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC.
- (a) K. Ouyang, W. Hao, W. X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS PubMed; (b) J. B. Bonanno, T. P. Henry, D. R. Neithamer, P. T. Wolczanski and E. B. Lobkovsky, J. Am. Chem. Soc., 1996, 118, 5132–5133 CrossRef CAS; (c) X. Cong, F. Fan, P. Ma, M. Luo, H. Chen and X. Zeng, J. Am. Chem. Soc., 2017, 139, 15182–15190 CrossRef CAS PubMed; (d) Z. C. Cao, X. L. Li, Q. Y. Luo, H. Fang and Z. J. Shi, Org. Lett., 2018, 20, 1995–1998 CrossRef CAS PubMed; (e) Z.-C. Cao, S.-J. Xie, H. Fang and Z.-J. Shi, J. Am. Chem. Soc., 2018, 140, 13575–13579 CrossRef CAS PubMed.
- (a) A.-L. H. Ernest Wenkert and C.-J. Jenny, J. Chem. Soc., Chem. Commun., 1988, 975–976 RSC; (b) E. L. M. Ernest Wenkert, C. S. Swindell and M. Tingoli, J. Org. Chem., 1984, 49, 4894–4899 CrossRef.
- (a) L. G. Xie and Z. X. Wang, Angew. Chem., Int. Ed. Engl., 2011, 50, 4901–4904 CrossRef CAS PubMed; (b) X. Q. Zhang and Z. X. Wang, J. Org. Chem., 2012, 77, 3658–3663 CrossRef CAS PubMed.
- (a) W.-J. Guo and Z.-X. Wang, Tetrahedron, 2013, 69, 9580–9585 CrossRef CAS; (b) J. T. Reeves, D. R. Fandrick, Z. Tan, J. J. Song, H. Lee, N. K. Yee and C. H. Senanayake, Org. Lett., 2010, 12, 4388–4391 CrossRef CAS PubMed.
- (a) S. B. Blakey and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 6046–6047 CrossRef CAS PubMed; (b) T. K. Tetsuro Koreeda and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 7236–7239 CrossRef; (c) N. C. Satoshi Ueno and F. Kakiuchi, J. Am. Chem. Soc., 2007, 129, 6098–6099 CrossRef PubMed; (d) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313–5316 CrossRef CAS PubMed; (e) P. Maity, D. M. Shacklady-McAtee, G. P. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280–285 CrossRef CAS PubMed.
- (a) J. Hu, H. Sun, W. Cai, X. Pu, Y. Zhang and Z. Shi, J. Org. Chem., 2016, 81, 14–24 CrossRef CAS PubMed; (b) H. Zhang, S. Hagihara and K. Itami, Chemistry, 2015, 21, 16796–16800 CrossRef CAS PubMed; (c) C. H. Basch, K. M. Cobb and M. P. Watson, Org. Lett., 2016, 18, 136–139 CrossRef CAS PubMed.
- A. B. Chopa, M. T. Lockhart and G. Silbestri, Organometallics, 2001, 20, 3358–3360 CrossRef CAS.
- B. Yang and Z. X. Wang, J. Org. Chem., 2019, 84, 1500–1509 CrossRef CAS PubMed.
- X. Q. Zhang and Z. X. Wang, Org. Biomol. Chem., 2014, 12, 1448–1453 RSC.
- N. Auner and J. Weis, Organosilicon chemistry VI: from molecules to materials, Wiley-VCH, 2005 Search PubMed.
- M. J. Barnes, R. Conroy, D. J. Miller, J. S. Mills, J. G. Montana, P. K. Pooni, G. A. Showell, L. M. Walsh and J. B. Warneck, Bioorg. Med. Chem. Lett., 2007, 17, 354–357 CrossRef CAS PubMed.
- (a) T. Komiyama, Y. Minami and T. Hiyama, Angew. Chem., Int. Ed. Engl., 2016, 55, 15787–15791 CrossRef CAS PubMed; (b) H.-o. S. Hajime Ito, K. Arimoto, K. Miura and A. Hosomi, Chem. Lett., 1997, 26, 639–640 CrossRef.
- (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed; (b) M. Murata, N. Fukuyama, J.-i. Wada, S. Watanabe and Y. Masuda, Chem. Lett., 2007, 36, 910–911 CrossRef CAS; (c) T. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 4879–4886 CrossRef CAS PubMed; (d) S. Toshiyasu, T. Yuko, S. Touru and T. Masato, Chem. Lett., 1987, 16, 2375–2378 CrossRef; (e) T. Ishiyama, T. Saiki, E. Kishida, I. Sasaki, H. Ito and N. Miyaura, Org. Biomol. Chem., 2013, 11, 8162–8165 RSC; (f) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592–595 CrossRef CAS PubMed; (g) E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef CAS PubMed; (h) K. Fumitoshi, I. Kimitaka, M. Mitsutaka, C. Naoto and M. Shinji, Chem. Lett., 2001, 30, 422–423 CrossRef; (i) K. Fumitoshi, I. Kimitaka, M. Mitsutaka, H. Tomoo, C. Naoto and M. Shinji, Chem. Lett., 2002, 31, 396–397 CrossRef; (j) F. Kakiuchi, M. Matsumoto, K. Tsuchiya, K. Igi, T. Hayamizu, N. Chatani and S. Murai, J. Organomet. Chem., 2003, 686, 134–144 CrossRef CAS; (k) H. Ihara and M. Suginome, J. Am. Chem. Soc., 2009, 131, 7502–7503 CrossRef CAS PubMed; (l) A. Modak, T. Patra, R. Chowdhury, S. Raul and D. Maiti, Organometallics, 2017, 36, 2418–2423 CrossRef CAS; (m) A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed. Engl., 2017, 56, 14903–14907 CrossRef CAS PubMed; (n) Z. Xu, L. Chai and Z. Q. Liu, Org. Lett., 2017, 19, 5573–5576 CrossRef CAS PubMed; (o) W. B. Liu, D. P. Schuman, Y. F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS PubMed; (p) A. A. Toutov, W. B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS PubMed; (q) K. Nozawa-Kumada, S. Osawa, M. Sasaki, I. Chataigner, M. Shigeno and Y. Kondo, J. Org. Chem., 2017, 82, 9487–9496 CrossRef CAS PubMed; (r) S. Banerjee, Y. F. Yang, I. D. Jenkins, Y. Liang, A. A. Toutov, W. B. Liu, D. P. Schuman, R. H. Grubbs, B. M. Stoltz, E. H. Krenske, K. N. Houk and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6880–6887 CrossRef CAS PubMed; (s) Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS; (t) S. Liu, P. Pan, H. Fan, H. Li, W. Wang and Y. Zhang, Chem. Sci., 2019, 10, 3817–3825 RSC.
- C. Zarate, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2017, 139, 1191–1197 CrossRef CAS PubMed.
- E. M. Wiensch, D. P. Todd and J. Montgomery, ACS Catal., 2017, 7, 5568–5571 CrossRef CAS.
- (a) Y. Q. Yi, W. C. Yang, D. D. Zhai, X. Y. Zhang, S. Q. Li and B. T. Guan, Chem. Commun., 2016, 52, 10894–10897 RSC; (b) B. Scholl, S. Jolidon and H.-J. Hansen, Helv. Chim. Acta, 1986, 69, 184–194 CrossRef CAS; (c) N. A. Paras, B. Simmons and D. W. C. MacMillan, Tetrahedron, 2009, 65, 3232–3238 CrossRef CAS.
- (a) Q. Zhou, H. D. Srinivas, S. Dasgupta and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 3307–3310 CrossRef CAS PubMed; (b) B. Wang, Q. Zhang, J. Jiang, H. Yu and Y. Fu, Chem.–Eur. J., 2017, 23, 17249–17256 CrossRef CAS.
- (a) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS; (b) E. M. Wiensch and J. Montgomery, Angew. Chem., Int. Ed., 2018, 57, 11045–11049 CrossRef CAS PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and copies of NMR spectra. See DOI: 10.1039/c9sc01083a |
| This journal is © The Royal Society of Chemistry 2019 |