
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-coordinated cobalt nanocrystals for oxidative dehydrogenation and hydrogenation of N-heterocycles†
Yue
Wu‡
a,
Zheng
Chen‡
a,
Weng-Chon
Cheong
a,
Chao
Zhang
a,
Lirong
Zheng
 b,
Wensheng
Yan
c,
Rong
Yu
d,
Chen
Chen
*a and
Yadong
Li
a
b,
Wensheng
Yan
c,
Rong
Yu
d,
Chen
Chen
*a and
Yadong
Li
a
aDepartment of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: cchen@mail.tsinghua.edu.cn
bBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
cNational Synchrotron Radiation Laboratory (NSRL), University of Science and Technology of China, Hefei, Anhui 230029, China
dNational Center for Electron Microscopy in Beijing, School of Materials Science and Engineering, Tsinghua University, 100084 Beijing, China
First published on 23rd April 2019
Abstract
To endow non-noble metals with the high catalytic activity that is typically exhibited by noble metals is the central yet challenging aim for substituting noble metals. In this regard, by exploiting the coordination effect of nitrogen, we prepared cobalt nanocrystals stabilized by nitrogen-doped graphitized carbon (Co NCs/N-C). The obtained Co NC/N-C catalyst showed extraordinary performances toward both oxidative dehydrogenation of N-heterocycles and its reverse hydrogenation process under extremely mild conditions. A nearly quantitative conversion could be achieved for oxidative dehydrogenation even at room temperature (25 °C), for which the coordination effect of nitrogen is responsible: the interaction of Co–N induces a partial positive charge on the Co surface, thereby promoting the reaction. In contrast, cobalt nanocrystals supported by pristine carbon (Co NCs/C) proved to be inactive for oxidative dehydrogenation, owing to the lack of nitrogen. Moreover, in Co NCs/N-C, the N-doped graphitized carbon formed a protective layer for Co NCs, which preserved the active valence of Co species and prevented the catalyst from leaching. It was found that the catalyst still retained its excellent catalytic activity after five regeneration cycles; in comparison, its cobaltous oxide counterpart (CoOx/N-C) was barely active. As for the mechanism, electron paramagnetic resonance (EPR) analysis revealed the formation of superoxide anion radicals during the dehydrogenation process. Interestingly, the pressure of feed hydrogen had little effect on the hydrogenation process. Our Co NC/N-C catalyst is capable of activating molecular oxygen and hydrogen as effectively as noble metals; the coordination effect of nitrogen and the protection by the carbon layer in combination confer tremendous potential on the Co NCs/N-C for substituting noble-metal-based catalysts and soluble catalysts for homogeneous reactions.
Introduction
With the rapid progress in the field of catalysis, noble metal catalysts have established dominance in both homogeneous and heterogeneous catalytic systems.1–3 However, the limited reserves and high cost of noble metals severely hinder their large-scale application and industrialization. To address these issues, extensive attention has been paid to exploiting non-noble metal catalysts4 as substitutes, by virtue of their relatively high abundance and low cost. The key objectives for substituting noble metals with non-noble metals are to achieve superior catalytic activity (generally exhibited by noble metals) under mild conditions and, in the meantime, to keep the non-noble metals intact during the reaction process, since non-noble metals are commonly susceptible to oxidation and leaching, particularly under harsh reaction conditions. To protect the relatively unstable non-noble metals, structural protection by inert layers and charge balance of non-noble metals through interaction with exotic atoms are two promising strategies. Since the first report of metal phthalocyanine complexes tested as cathode catalysts in fuel cells to replace expensive Pt,5 numerous transition metal catalysts have emerged in attempts to achieve high catalytic activity like that of noble metal ones. From the perspective of alternative energy technologies and hydrogen storage, catalytic oxidative dehydrogenation and hydrogenation have received significant attention.6 N-Heterocycles, a class of pharmacophores broadly present in numerous FDA-approved pharmaceuticals and other bioactive compounds,7 are an important research subject in this field. In particular, the catalytic oxidative dehydrogenation of saturated N-heterocycles provides one of the most atom-efficient routes8 towards yielding high-value products such as indoles, quinolones, and other N-heterocyclic aromatic compounds.To optimize the catalytic system for oxidative dehydrogenation of N-heterocycles and its reverse process, extensive research has been carried out on homogeneous and heterogeneous catalysis. In the most typical homogeneous catalytic system, Cp*Ir complexes9 gave almost quantitative yields for both oxidative dehydrogenation and hydrogenation. Inspired by this work, subsequent reports still focused on noble metals, such as Ir,6,10,11 Ru,12–14 Pt,15 Pd,15 and Rh.15 In spite of the development of noble metal catalytic systems, great efforts have been devoted to exploiting efficient catalysts derived from sustainable metals so as to circumvent the high cost, high toxicity and low abundance of noble metals. Iron-based7,16 and cobalt-based17 catalysts are two typical cases, performing at relatively high temperatures (100 °C or even higher), under pressurized conditions, and for a long reaction time. However, some of them still give barely satisfactory yields. Therefore, up to now it has seemed that non-noble metal catalysts typically require relatively harsh reaction conditions to deliver acceptable activities. In addition, homogeneous catalysis has an innate limitation in recycling,18 whereas for heterogeneous systems, there still exist plenty of challenges to develop inexpensive, earth-abundant metal catalysts instead of noble metal ones and, meanwhile, to retain superb performances with high activity (under reaction conditions which are as mild as possible) and robust stability for recycling.
The “heteroatom doping” method involves replacing certain carbon lattice atoms with heteroatoms.19 Nitrogen doping is the most common one, and has proved to be a powerful method to modify carbon materials for various applications.20 Due to the difference in electronegativity between carbon atoms and nitrogen, the nitrogen doping process can generally redistribute the charge density and spin density of carbon atoms in the lattice, thereby effectively regulating the work function and strengthening the adsorption of reactants at specific sites.19,21 In addition, the local geometry in the vicinity of nitrogen dopant atoms would be altered due to the difference in atomic sizes between C and the dopant N,19 as confirmed by density functional theory (DFT) calculations.22 N-Doped carbon nanomaterials are now one of the most important catalytic materials for a wide range of chemical and electrochemical catalytic reactions, such as selective oxidation of aromatic alkanes,23 reduction of nitrobenzene,24 hydration of different alkynes,25 the oxygen reduction reaction (ORR),26,27 the hydrogen evolution reaction (HER),28,29 and overall water splitting.30
Herein, cobalt nanocrystals stabilized by nitrogen-doped graphitized carbon (Co NCs/N-C) are developed for both oxidative dehydrogenation of N-heterocycles and its reverse process. This catalyst exhibits excellent catalytic activity for both oxidative dehydrogenation and hydrogenation. Remarkably, even at room temperature without extra O2 or additional pressure, the Co NCs/N-C can effectively facilitate oxidative dehydrogenation with a nearly complete conversion. The Co NC/N-C catalyst is readily recyclable by applying a magnetic field and can almost retain its excellent catalytic activity after 5 recycling rounds. It is the first catalytic system for the efficient oxidative dehydrogenation–hydrogenation reaction that proceeds under ambient conditions, which is a fundamental improvement from traditional catalytic methods for organic transformation of N-heterocycles (Fig. 1).
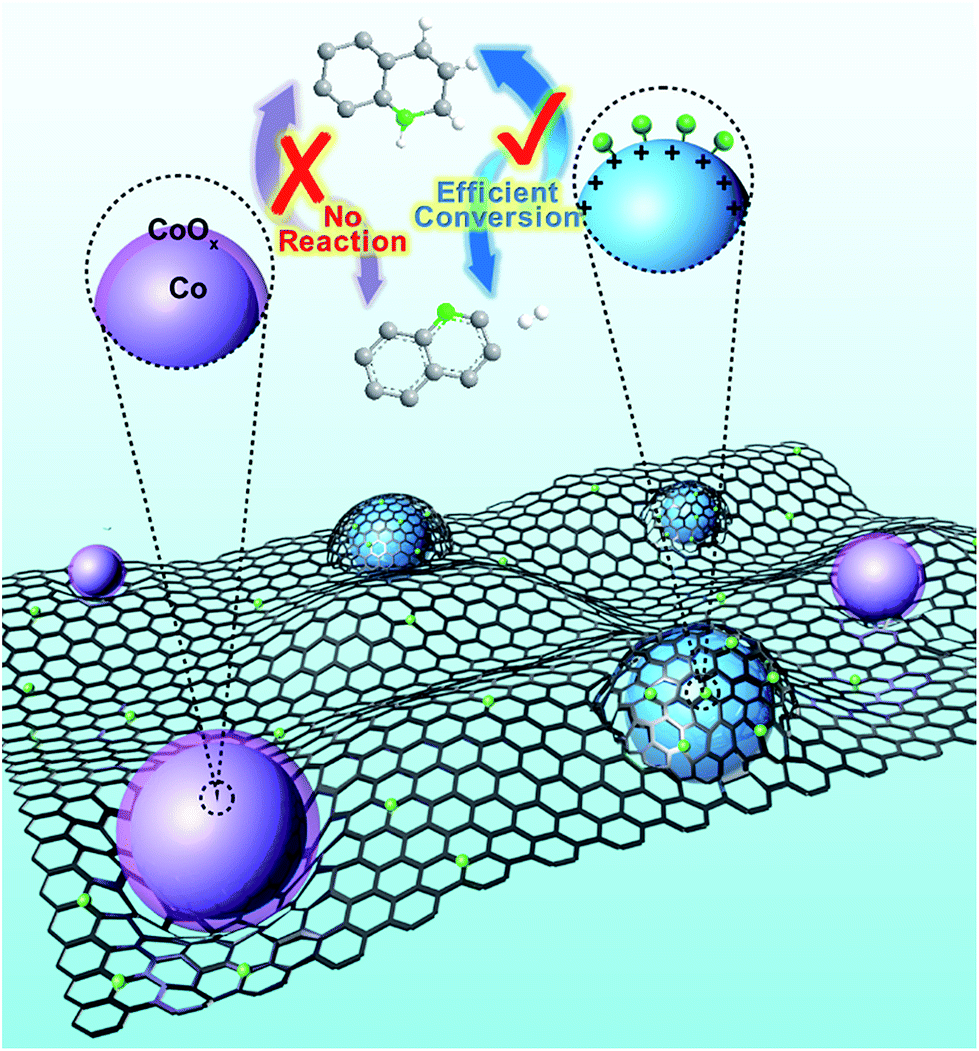 | ||
| Fig. 1 The scheme of Co NCs/N-C obtained by the pyrolysis of ZIF-67. | ||
Results and discussion
The pyrolysis of metal–organic frameworks (MOFs) at high temperatures is an effective and popular method for the synthesis of metal/nitrogen-doped carbon composites. Uniform Co-based metal–organic frameworks, ZIF-67 nanopolyhedra, can be synthesized from cobalt(II) nitrate (Co(NO3)2) and 2-methylimidazole linkers,31 and particles with average sizes of 200–300 nm could be obtained based on reported procedures.32 Then, ZIF-67 was carbonized at a high temperature above 700 °C for 3 h in a temperature-programmed furnace under a flow of argon. Subsequent treatment with HCl under ultrasonication selectively removed the unprotected cobalt particles generated during the pyrolysis, affording the resultant catalyst denoted as Co NCs (cobalt nanocrystals)/N-C (nitrogen-doped carbon). The entire synthetic strategy of Co NCs/N-C is shown in Fig. S1.† The transmission electron microscopy (TEM) images showed that ZIF-67 nanocrystals were all of a uniform rhombododecahedral shape (Fig. 2a), and the corresponding pyrolysis product well inherited the overall polyhedron-like morphology (Fig. 2b), with many Co nanocrystals formed on the carbonaceous support. Elemental mapping results (Fig. 2c) indicated that the Co particles were evenly dispersed in/on the support. Subsequent HCl treatment rendered the support highly porous, with the metallic Co nanocrystals distributed within, as confirmed from the TEM and the corresponding magnified images of a single rhombododecahedron (Fig. 2d–f). It is worth mentioning that the Co species are encapsulated in the porous carbon framework (Fig. 2g), and the interplanar spacing of Co nanocrystals wrapped in porous carbon is 0.214 nm, corresponding to the Co (111) lattice fringe. With aberration-corrected HRTEM, the surface of encapsulated Co NCs could be clearly observed at atomic-level resolution (Fig. 2h). The porous layers having lattice fringes with an interplanar distance of 0.340 nm are identified as graphitic carbon (Fig. 2i). | ||
| Fig. 2 TEM image of ZIF-67 (a) and Co NCs/N-C (b); (c) HAADF-STEM image and corresponding element maps showing the distribution of C (red), N (yellow) and Co (green); (d–f) TEM image and the magnified images of Co NCs/N-C after acid treatment; (g) magnified HAADF-STEM image of the Co NCs/N-C. (h) Atomic resolution STEM image of the Co NCs encapsulated in nitrogen-doped carbon; (i) magnified HAADF-STEM image of the nitrogen-doped graphitized carbon substrate. | ||
In the X-ray diffraction (XRD) patterns (Fig. S2†), the diffraction peaks at 44.2°, 51.5° and 75.8° were respectively ascribed to the (111), (200) and (220) reflections of face-centered cubic cobalt. It is noteworthy that after acid treatment the peak at 26.1° became quite prominent for all three samples (carbonized at 700 °C, 800 °C and 900 °C, respectively), and was indexed to graphitic carbon. This result is also consistent with the HAADF-STEM image (Fig. 2i). It is therefore reasonable to infer that acid washing removed not only Co nanocrystals on the surface but also some amorphous carbon, yielding sharper graphitic peaks in the XRD pattern. N-doped graphitic carbon is of vital importance for the catalytic performance of the whole material, because N-doping has been demonstrated to produce defects in the carbon support, which result in the formation of active sites for O2 adsorption.16,33–35 Meanwhile, N-doped graphene-like carbon materials also has the ability to reductively adsorb O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 and has been employed in the catalytic reduction of nitroarenes37 and for reductive amination.38
36 and has been employed in the catalytic reduction of nitroarenes37 and for reductive amination.38
X-ray photoelectron spectroscopy (XPS) was used to further identify the Co valence state in Co NC/N-C products carbonized at different temperatures (Fig. S3†); the survey spectrum revealed the presence of Co, C, N, and O. The C 1s peak can be deconvoluted into two peaks located at 284.1 eV (for the typical C–C bond of sp3 carbon) and 284.9 eV (with respect to C–N species), and the one centered at 285.5 eV is the typical graphitic carbon peak. Deconvolution of the N 1s peak indicates that there exist three kinds of nitrogen including pyridinic nitrogen (398.8 eV), pyrrolic nitrogen (400.0 eV), and graphitic nitrogen (401.2 eV) in Co NCs/N-C.39 As for the Co 2p spectrum which can be fitted into two major peaks (Co 2p3/2 and Co 2p1/2), the peaks around 778.3 eV and 793.3 eV correspond to Co0, and the two peaks at 780.6 eV and 803.5 eV can be ascribed to Co3+, whereas the peaks around 780.1 eV and 796.4 eV are assigned to Co2+. It is the two oxidized states not shown in XRD that are the manifestation of the nitrogen-coordination effect. The coordination interaction between the nitrogen atom and Co leads to the electron drifting from Co toward the nitrogen atom, resulting in a slightly positive charge on the metallic surface. Thus, cobalt existing in multiple valence states was revealed in XPS data, but only Co0 was found by XRD.
We used the synchrotron radiation technique to further investigate the local atomic and electronic structures of Co NCs/N-C. In the NEXAFS spectra resulting from 2p → 3d dipole transitions, the Co L3 and L2 absorption lines are located around 773 eV and 787 eV, respectively. As shown in Fig. 3a, the Co L-edge NEXAFS spectra of Co NCs/N-C and Co NCs/C exhibit a peak at the L3-edge, which is characteristic of 3d-electrons, and no well-defined multiple structures are observed; the spectral profiles bear a greater similarity to those of Co rather than Co-based oxides,40,41 and the decrease in intensity after nitrogen hybridization implies higher Co 3d electron occupation in Co NCs/N-C, mainly due to the stronger electronic interactions introduced by nitrogen into the Co NCs.42 The X-ray absorption near-edge structure (XANES) curves of the Co K-edge (Fig. 3b) show that the near-edge absorption energy of Co NCs/N-C is stronger than that of Co NCs/C and Co foil (showed by the peak denoted by A in Fig. 3), indicating that the valence state of Co in Co NCs/N-C is slightly higher than that in Co NCs/C and Co foil.43 There is a strong peak located between 2.0 and 2.4 Å, which is related to Co–Co single scattering shown in extended X-ray absorption fine structure (EXAFS) spectra (Fig. 3c). In the locally magnified graph (inset in Fig. 3c), it is clear that the main peak of Co NCs/N-C is of the lowest intensity, suggesting that the Co–Co bond shrinks with the interaction with nitrogen.44 In the N K-edge spectrum (Fig. 3d), the peak at 396.3 eV and the peak at 398.8 eV mainly originated from pyridinic π* and graphitic π* transitions, respectively. The peak at 404.6 eV suggested the formation of the σ* C–N–C or C–N bond. The C K-edge spectrum displayed in Fig. 3e shows three peaks: peak A at 283.9 eV derived from the C–C π* (ring) excitations, peak B at 287.0 eV assigned to C–O–C or C–N–C and peak C at 290.7 eV originating from the C–C σ* (ring) transitions. Peaks A and C suggested that graphitization occurred during the pyrolysis process, while peak B indicated that defect sites existed in the carbon lattice.45 Therefore, the Co NC/N-C sample obtained after acid treatment show a higher degree of graphitization and more defects in the N-C substrate. Based on the above characterization, it can be inferred that the Co nanocrystals are stabilized by the coordination with nitrogen and the coating with graphitized carbon, as illustrated in Fig. 3f. To assess the activity of Co NCs/N-C for N-heterocycle oxidative dehydrogenation, we tested the catalyst obtained at 800 °C (denoted as Co NCs/N-C-800 °C) with 1,2,3,4-tetrahydroquinoline as a model substrate. Different solvents were screened, and we found that the prepared catalyst in general performed better in polar solvents. When the model reaction was tested at 80 °C in several nonpolar solvents (Table S1†) such as p-xylene, 1,4-dioxane, etc., the highest yield obtained was 24.9% after 4 h using p-xylene as the solvent. In contrast, polar solvents (Table S2†) performed better than all the tested nonpolar solvents. With methanol as the solvent, the reaction achieved approximately complete conversion with 100% selectivity at a very low temperature (50 °C) after only 4 h (Fig. 4b). Even more surprisingly, a conversion of 97.2% could be achieved with a longer reaction time (24 h) at room temperature (Table S3†).
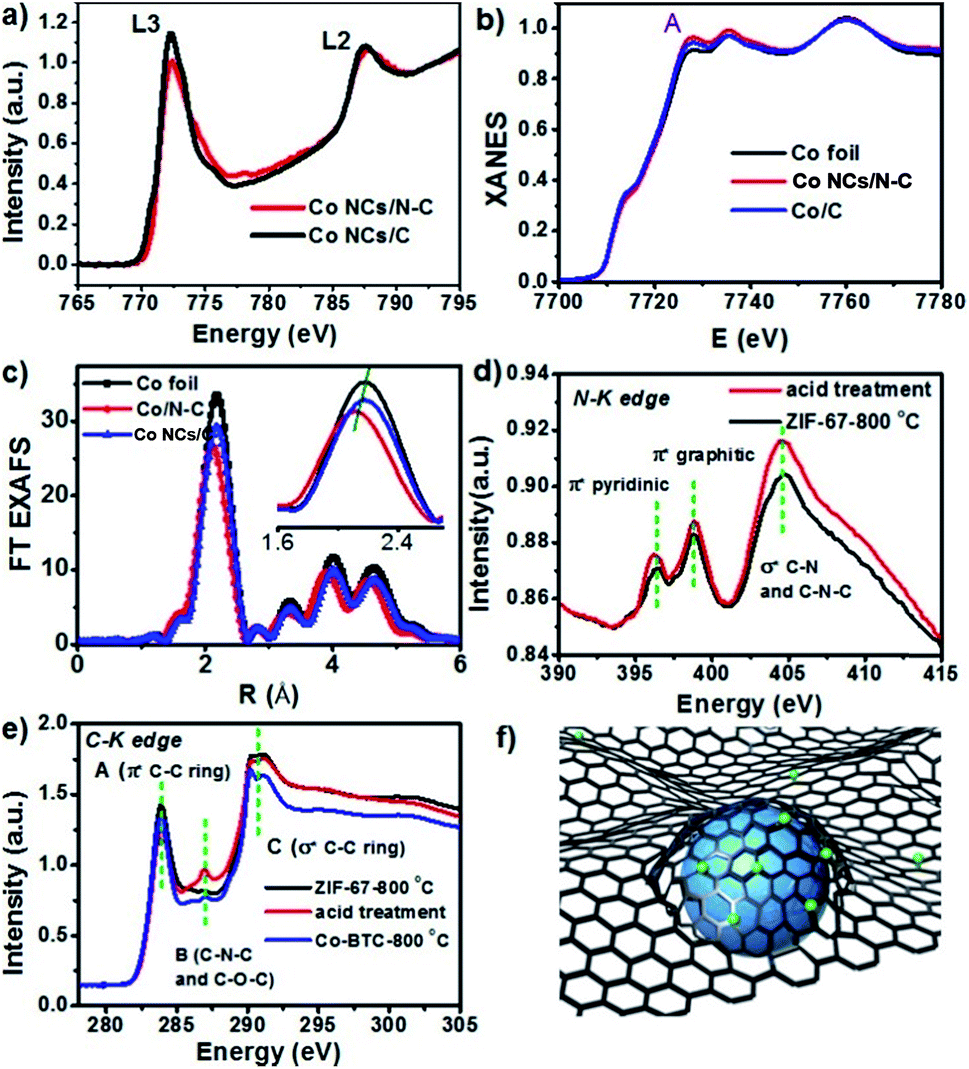 | ||
| Fig. 3 (a) NEXAFS spectra of Co NCs/N-C, (b) XANES spectra (the blue area highlights the near-edge absorption energy) and (c) Fourier transform (FT) spectra of ZIF-67, Co NCs/N-C, catalysts treated in 500 °C H2 and 300 °C air, and Co–BTC; (d) the N K-edge and (e) C K-edge of Co NCs/N-C; (f) the diagram of Co NCs encapsulated in N-doped carbon. | ||
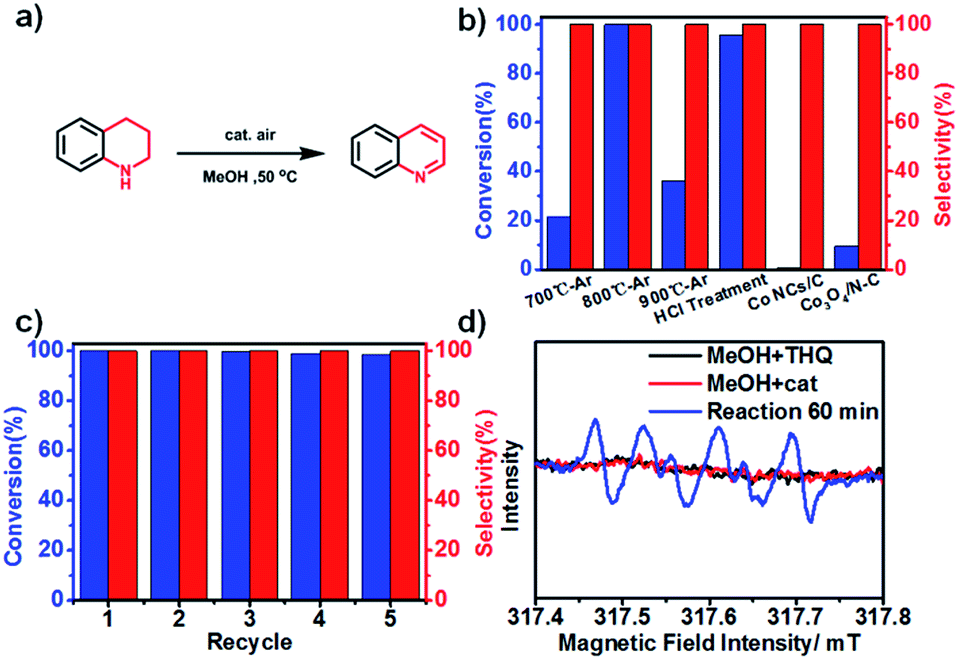 | ||
| Fig. 4 (a) Reaction for oxidative dehydrogenation of 1,2,3,4-tetrahydroquinoline; (b) comparison of conversion and selectivity for different catalysts towards oxidative dehydrogenation; (c) recycling test of the Co NC/N-C catalyst; (d) the DMPO spin-trapping ESR spectra for ˙O2− in this catalytic system. | ||
Next, we compared catalysts carbonized at different pyrolysis temperatures under optimized reaction conditions. After 4 h, Co NCs/N-C-700 °C and Co NCs/N-C-900 °C gave significantly lower conversions (21.4% and 36.1%, respectively) (Fig. 4b). We then assessed the recycling stability of the Co NC/N-C catalyst. As a magnetic catalyst, Co NCs/N-C has the advantage of easy separation by applying a magnetic field. The Co NCs/N-C could retain its catalytic performance after five-cycle measurements (corresponding morphology shown in Fig. S4c,† and XRD pattern shown in Fig. S5†), and no obvious decline in activity was found (Fig. 4c). In addition, there were no other by-products observed.
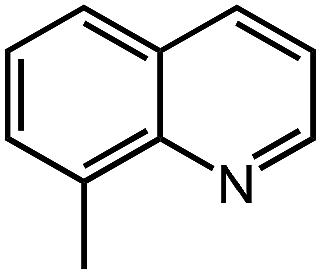
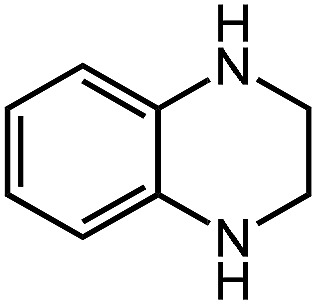
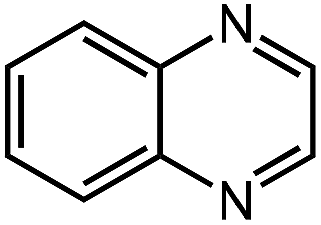
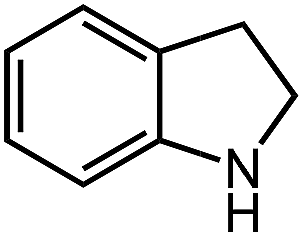
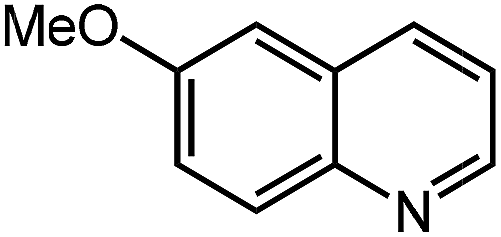
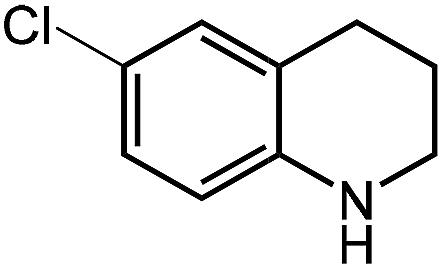
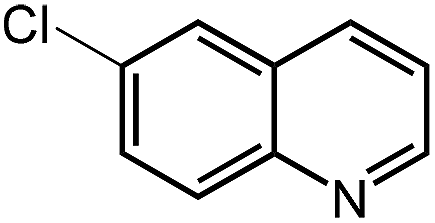
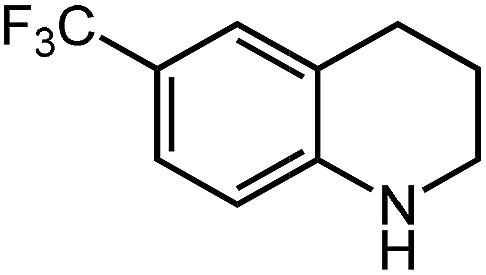
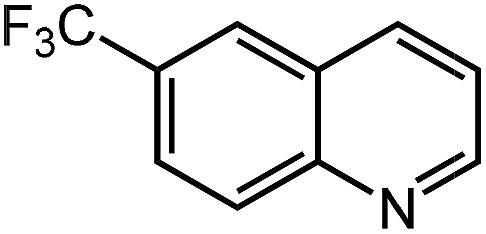
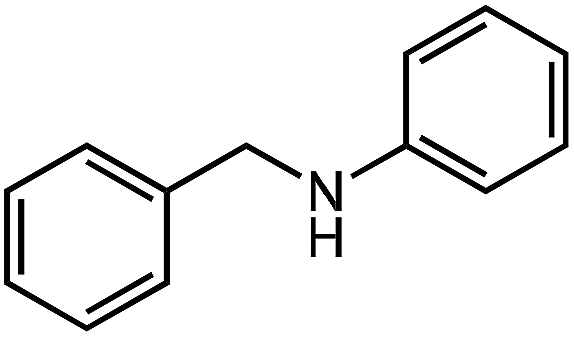
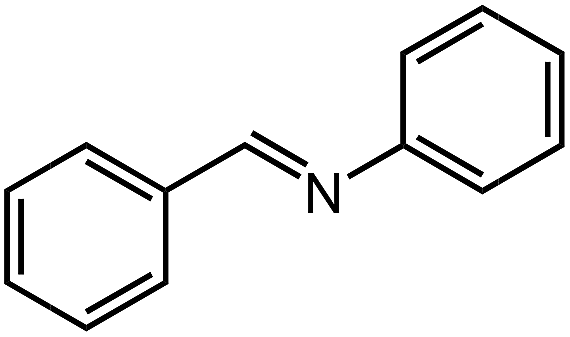
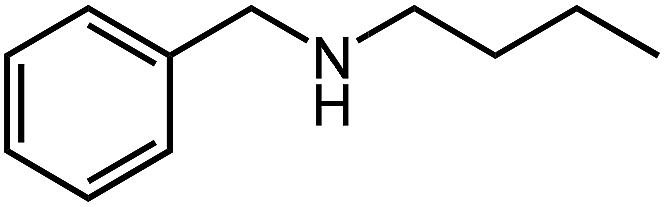
Under optimized conditions, we tested the catalyst with other substrates. As shown in Table 1, the catalyst was also efficient for oxidative dehydrogenation of similar substrates at 50 °C in methanol. As expected, substrates with an electron-donating group were active, such as 8-methyl-1,2,3,4-tetrahydroquinoline (entry 1). In addition, 1,2,3,4-tetrahydroquinoxaline (entry 2) and indoline (entry 3) were also quantitatively dehydrogenated under model reaction conditions. Moreover, in spite of the passivation effect of electron-withdrawing groups on aromatic rings, substrates with an electron-withdrawing group at the 6th position were well tolerated. 6-Bromo-1,2,3,4-tetrahydroquinoline (entry 4) and 6-methoxy-1,2,3,4-tetrahydroquinoline (entry 5) both gave yields above 97%, while 6-chloroquinoline (entry 6) was produced with a yield of 93.8% under the conditions of prolonged reaction time. Surprisingly, 6-(trifluoromethyl)quinoline (entry 7), a substrate with a strong electron-withdrawing group, was obtained in a yield of up to 99.9%. Finally, we tested two benzylamines. Remarkably, N-benzylaniline transformed into N-(phenylmethylene)-benzenamine with 96.8% yield (entry 8). Besides, N-benzylbutan-1-amine gave a yield around 20% (entry 9). 1,2,3,4-Tetrahydroquinoline substituted with ester and nitro groups, and 1,2,3,4-tetrahydroisoquinoline gave moderate conversions towards corresponding oxidative dehydrogenation products (entry 10–12).
| Substrate | Product | Yieldb (%) | |
|---|---|---|---|
|
a Reaction conditions: 0.5 mmol substrate, 50 mg catalyst (the molar ratio of Co:substrate in this work is 7.6%), 3 mL MeOH, air, 50 °C, 12 h.
b The yields are determined by GC-MS and GC analysis.
c Reaction conditions: 0.5 mmol substrate, 50 mg catalyst, 3 mL MeOH, air, 50 °C, 24 h.
|
|||
| 1 |
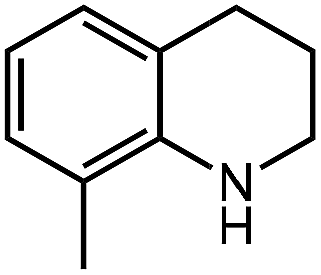
|
|
99.9 |
| 2 |
|
|
99.3 |
| 3 |
|
|
100.0 |
| 4 |
|
|
97.8 |
| 5 |
|
|
97.5 |
| 6 |
|
|
74.3 (93.8)c |
| 7 |
|
|
99.9 |
| 8 |
|
|
96.8 |
| 9 |
|
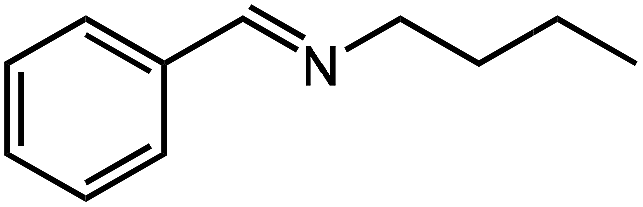
|
17.8 |
| 10 |
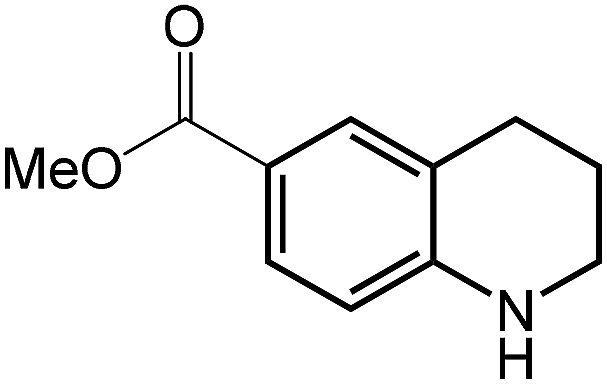
|
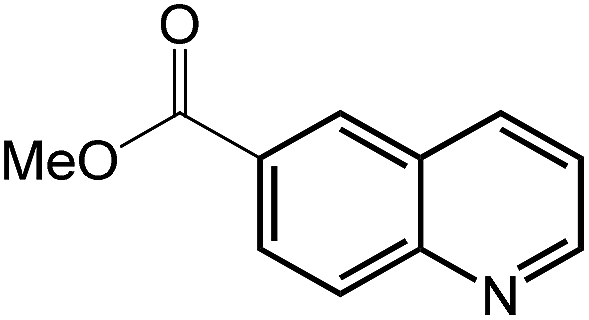
|
58.3 |
| 11 |
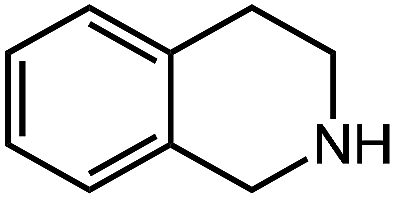
|
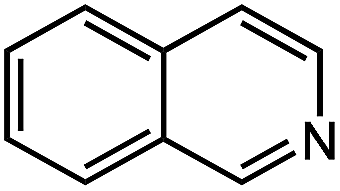
|
65.1 |
| 12 |
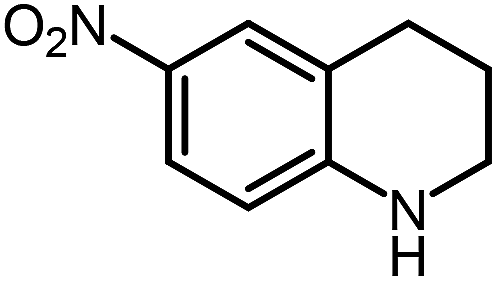
|
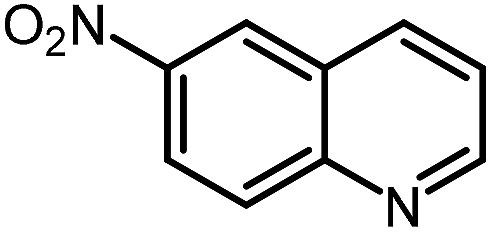
|
28.1 |
In light of the aforementioned excellent catalytic performance for the oxidative dehydrogenation of 1,2,3,4-tetrahydroquinoline, it was imperative to conduct further discussion on the catalytic mechanism. Herein, we investigate the mechanism from the following perspectives: (1) to identify the catalytically active species at the nanoscopic level; (2) to get insights into the chemical environment around the catalytically active species; (3) to explore their active valence states; and finally, (4) to infer the reaction pathway for the oxidative dehydrogenation process.
As shown in the TEM images above, there were two kinds of Co NCs in the catalysts, some partially embedded on the surface and some encapsulated in the porous N-doped carbon framework. After treatment with HCl, the Co nanocrystals on the surface were removed, and the wrapped ones were acid-resistant and therefore remained intact. The resulting catalyst (denoted as Co NCs/N-C-HCl) had a cobalt content of about 4.5%, as determined by ICP-OES measurement (Inductively Coupled Plasma Optical Emission Spectrometer), in contrast to the untreated catalyst Co NCs/N-C (with 30.4% cobalt content) (Table S4†). The activity test with 50 mg Co NCs/N-C-HCl in the model reaction gave a reaction yield as high as 95.5% after 12 h, indicating that Co NCs/N-C-HCl retained almost all the original catalytic activity, even with only one sixth of the initial Co content. Therefore, it is the Co NCs encapsulated in the N-doped carbon framework that essentially drive the reaction during the oxidative dehydrogenation of tetrahydroquinoline coordination.
To determine the relationship between the N-coordination environment and the catalytic activity, we changed the ligand for MOFs from 2-methylimidazole to 1,3,5-benzenetricarboxylic acid (BTC) (Fig. S4d†). As a control experiment, we adopted the synthesis method of Co NCs/N-C, and the resulting catalyst was denoted as Co NCs/C-BTC. As confirmed by XPS data (Fig. S3†), Co NCs/C-BTC contained C, O, and Co, while N could barely be detected. As for the activity test, the Co NCs/C-BTC with 60.0% Co content (measured by ICP-OES) (Table S4†) gave a marginal yield of 0.86%. Therefore, the N-coordination environment not only protected the Co NCs from oxidation and deactivation, but also induced electrons to deviate from Co, leading to a slightly positive surface charge on the metallic surface, as confirmed by XPS data. This coordination effect further offered an excitation mechanism for the catalytic centers to activate O2, by which the encapsulated Co NCs delivered a remarkable catalytic efficiency.
From the XRD and XPS results, it was determined that Co species in the catalyst was mostly in the zero valence state, while a local charge existed at the interface between Co NCs and the N ligand. For more proof of the catalytic activity of Co0, we thermally treated the catalyst (previously carbonized at 800 °C) in air at 300 °C and in H2 at 500 °C, respectively; the products are denoted as Co NCs/N-C-300Air (Fig. S4a and S5†) and Co NCs/N-C-500H2 (Fig. S4b and S5†). After oxidation in air, Co NCs/N-C-300Air only gave a sharply decreased yield of 9.47% after 4 h (Fig. S6†). The valence state of catalytic species could explain this significant deterioration in performance. After thermal treatment in air, Co NCs were transformed into Co3O4, as confirmed by XRD (Fig. S5†) (so it is also denoted as Co3O4/N-C in Fig. 4b). The activities of Co3+ and Co2+ were far below that of Co0. As for Co NCs/N-C-500H2, a yield of 68.3% was achieved (Fig. S6†). The catalysts gave almost identical XRD patterns (Fig. S5†) before and after H2 reduction, and no Co3+ peak was found in the Co spectrum. We inferred that the H2 atmosphere at 500 °C damaged the coordination interaction between N and Co, which caused the decrease in activity. To verify this inference, Co NCs/N-C-400H2 was prepared and then tested for comparison. Treated at a lower temperature (400 °C), the catalyst retained most of the catalytic activity with the yield reaching 93.0%, which confirmed the importance of N-coordination for the catalytic process.
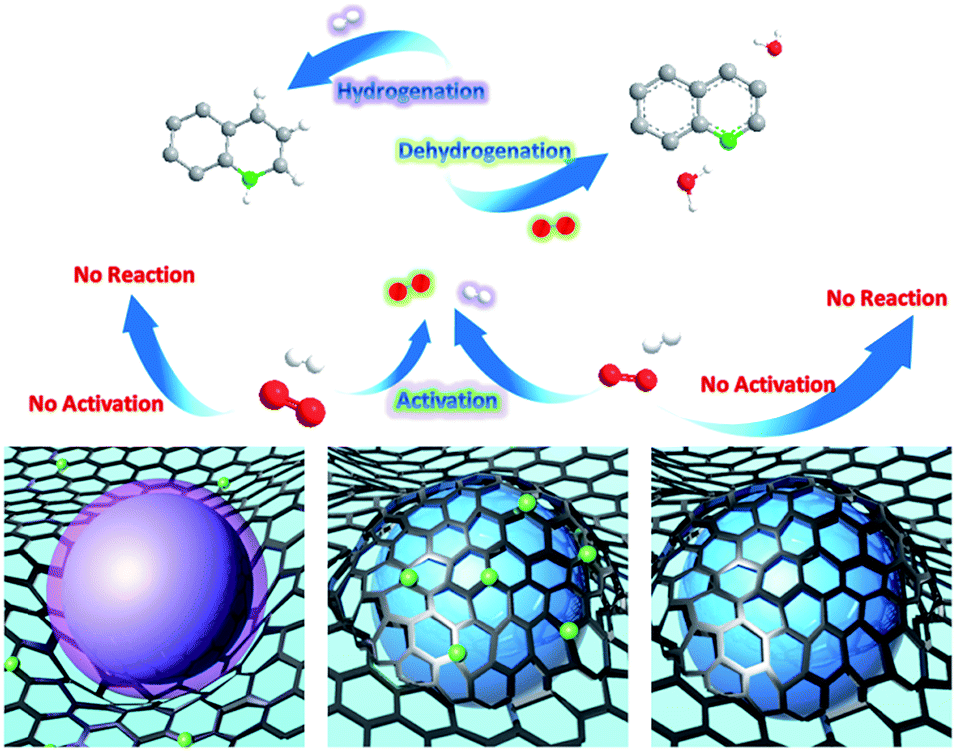 | ||
| Fig. 5 The mechanism of oxidative dehydrogenation and hydrogenation on three types of Co NCs: Co NCs exposed on the nitrogen-doped carbon substrate (left), Co NCs encapsulated within nitrogen-doped carbon (middle), and Co NCs encapsulated within carbon (right). | ||
There has been speculation that this reaction progresses via a radical-mediated pathway, which involves the superoxide anion radical (˙O2−). The subsequently generated H2O2 is quickly consumed, with the formation of quinoline and water.16 Mechanistic studies on the role of ˙O2− in oxidative stress processes are particularly difficult owing to its low steady-state concentration and the lack of highly specific probes that allow its unequivocal identification and quantification.46 Herein, we detected ˙O2− by means of electron paramagnetic resonance (EPR). Fortunately, after significantly elevating the concentration of tetrahydroquinoline in the catalytic system, we captured the sextet signal (Fig. 4d) which is characteristic of ˙O2−. As mentioned above, in N-doped graphene-like carbon materials, nitrogen atom doping leads to defects, resulting in the formation of adsorption active sites for O2 adsorption and reduction. Therefore, we speculate that the catalytically active species, namely, Co NCs encapsulated by N-doped carbon, could adsorb O2 from the surrounding air, and donate electrons to reduce O2 to ˙O2−, which drives the oxidative dehydrogenation of tetrahydroquinoline. In a N-coordination environment, the electron “gas” hanging over the surface of Co NCs shifted toward nitrogen atoms, facilitating electron transfer for ˙O2− production. As a protic solvent, methanol could serve as an outstanding proton donor and transform ˙O2− into HO2−, the effective oxidizing species. The stronger the protic solvent, the more significantly the oxidative dehydrogenation was facilitated, which explains the experimental fact that methanol performed best compared with aprotic solvents and other protic solvents tested in this work. Based on the experimental results and our discussion, a plausible reaction mechanism (Fig. S7†) is proposed. Meanwhile, it was compared the mechanism of oxidative dehydrogenation and hydrogenation on three types of Co NCs (Fig. 5).
After achieving efficient catalytic oxidative dehydrogenation of N-heterocycles at relatively low temperatures, we became interested in whether the reverse process, namely, hydrogenation of quinolone to 1,2,3,4-tetrahydroquinoline, could be accomplished using the same catalyst. To evaluate the catalytic activity for hydrogenation, we chose external hydrogen as a hydrogen source, and separately explored the effects of temperature, time, and pressure. We first carried out the hydrogenation reaction in tetrahydrofuran (THF) under 2 MPa pressure for 6 h (Fig. 6b), and found that with the increase of temperature, the conversion was obviously elevated. At ambient temperature (25 °C), almost no transformation was detected. However, when the reaction was carried out at 60 °C, the conversion reached 80.0%, and almost quantitative transformation was achieved at 120 °C. Kinetic measurement showed that the reaction rate reached a maximum from 0.5 h to 1 h, and the conversion remained constant after 6 h (Fig. 6c). In addition, the catalytic efficiency is barely affected by the pressure of H2 (Fig. 6d), as confirmed by the performance test under different pressures in the range from 1 MPa to 5 MPa. Based on the above results, it can be concluded that Co NCs/N-C could serve as a bifunctional catalyst for both the oxidative dehydrogenation of N-heterocycles and its reverse process.
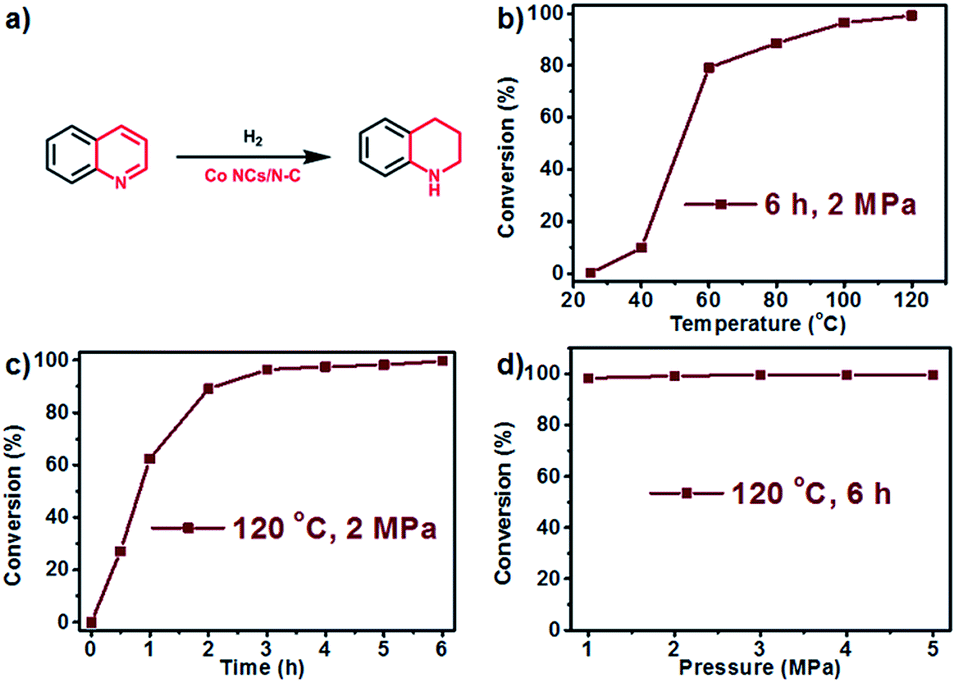 | ||
| Fig. 6 (a) Reaction for hydrogenation of quinoline; and the conversion rate vs. temperature (b), time (c), and pressure of H2 (d) for hydrogenation. | ||
Conclusions
In conclusion, we developed a bifunctional catalyst, namely, Co NCs/N-C, which is capable of catalyzing the oxidative dehydrogenation and hydrogenation of N-heterocycles with high efficiency. Particularly for oxidative dehydrogenation, the catalyst requires only an extremely mild and simple catalytic system, without extra O2, relatively high temperature or pressure. Prepared under optimal conditions, the catalyst can promote oxidative dehydrogenation with methanol as the solvent at 50 °C with exposure to air, with a nearly complete conversion within 4 h. The activity shows barely any loss after 5 rounds of magnetic regeneration. Through the Co–N coordination effect (nitrogen modulating the electronic structure of Co NCs) and the protection effect of the carbon layer, we successfully substituted noble metals with an inexpensive and readily recyclable transition metal, and significantly elevated the catalytic efficiency. As a result, the oxidative dehydrogenation of N-heterocycles can now be carried out under extremely mild conditions. Compared with conventional catalytic systems, our catalyst offers a highly effective alternative for N-heterocycle transformation, and stands as a new model for converting homogeneous catalytic systems into heterogeneous ones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (JQ18007), National Key R&D Program of China (2016YFA0202801 and 2017YFA0700101), National Natural Science Foundation of China (21872076, 21573119, 21590792, and 21890383), and China Postdoctoral Science Foundation (2018M631444). We acknowledge the Institute of High Energy Physics in Beijing and the National Synchrotron Radiation Laboratory in Hefei for XAFS measurement.Notes and references
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343(6177), 1339–1343 CrossRef CAS PubMed
.
- Z. Chen, Q. Zhang, W. Chen, J. Dong, H. Yao, X. Zhang, X. Tong, D. Wang, Q. Peng, C. Chen, W. He and Y. Li, Adv. Mater., 2018, 30(5), 1704720 CrossRef PubMed
- Y. Wu, D. Wang, G. Zhou, R. Yu, C. Chen and Y. Li, Sophisticated Construction of Au Islands on Pt–Ni: An Ideal Trimetallic Nanoframe Catalyst, J. Am. Chem. Soc., 2014, 136(33), 11594–11597 CrossRef CAS PubMed
- Y. Qian, P. Du, P. Wu, C. Cai and D. F. Gervasio, J. Phys. Chem. C, 2016, 120(18), 9884–9896 CrossRef CAS
- R. Jasinski, Nature, 1964, 201, 1212 CrossRef CAS
- Á. Vivancos, M. Beller and M. Albrecht, ACS Catal., 2018, 8(1), 17–21 CrossRef
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136(24), 8564–8567 CrossRef CAS PubMed
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57(24), 10257–10274 CrossRef CAS PubMed
- R. Yamaguchi, C. Ikeda, Y. Takahashi and K.-i. Fujita, J. Am. Chem. Soc., 2009, 131(24), 8410–8412 CrossRef CAS PubMed
- J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52(27), 6983–6987 CrossRef CAS
- W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53(5), 1390–1394 CrossRef CAS PubMed
- K.-N. T. Tseng, A. M. Rizzi and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135(44), 16352–16355 CrossRef CAS PubMed
- K. H. He, F. F. Tan, C. Z. Zhou, G. J. Zhou, X. L. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56(11), 3080–3084 CrossRef CAS PubMed
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42(13), 1480–1483 CrossRef CAS PubMed
- C. Deraedt, R. Ye, W. T. Ralston, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2017, 139(49), 18084–18092 CrossRef CAS PubMed
- X. Cui, Y. Li, S. Bachmann, M. Scalone, A.-E. Surkus, K. Junge, C. Topf and M. Beller, J. Am. Chem. Soc., 2015, 137(33), 10652–10658 CrossRef CAS PubMed
- R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5(11), 6350–6354 CrossRef CAS
- X.-L. Du, L. He, S. Zhao, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2011, 50(34), 7815–7819 CrossRef CAS PubMed
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31(13), 1805121 CrossRef PubMed
- Y. Gao, D. Ma, G. Hu, P. Zhai, X. Bao, B. Zhu, B. Zhang and D. S. Su, Angew. Chem., Int. Ed., 2011, 50(43), 10236–10240 CrossRef CAS PubMed
- P. Yan, J. Liu, S. Yuan, Y. Liu, W. Cen and Y. Chen, Appl. Surf. Sci., 2018, 445, 398–403 CrossRef CAS
- M. D. Esrafili, Comput. Theor. Chem., 2013, 1015, 1–7 CrossRef CAS
- Y. Zhu, W. Sun, W. Chen, T. Cao, Y. Xiong, J. Luo, J. Dong, L. Zheng, J. Zhang, X. Wang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Funct. Mater., 2018, 28(37), 1802167 CrossRef
- N. Liu, L. Ding, H. Li, M. Jia, W. Zhang, N. An and X. Yuan, J. Colloid Interface Sci., 2017, 490, 677–684 CrossRef CAS PubMed
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., 2010, 122(38), 6965–6968 CrossRef
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56(24), 6937–6941 CrossRef CAS PubMed
- Z.-Y. Wu, X.-X. Xu, B.-C. Hu, H.-W. Liang, Y. Lin, L.-F. Chen and S.-H. Yu, Angew. Chem., Int. Ed., 2015, 54(28), 8179–8183 CrossRef CAS PubMed
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W.-C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56(50), 16086–16090 CrossRef CAS PubMed
- W. Zhou, T. Xiong, C. Shi, J. Zhou, K. Zhou, N. Zhu, L. Li, Z. Tang and S. Chen, Angew. Chem., Int. Ed., 2016, 55(29), 8416–8420 CrossRef CAS PubMed
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W.-C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140(7), 2610–2618 CrossRef CAS PubMed
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319(5865), 939–943 CrossRef CAS PubMed
- A. Aijaz, J. Masa, C. Rösler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55(12), 4087–4091 CrossRef CAS PubMed
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108(6), 2064–2110 CrossRef CAS PubMed
- J.-L. Shui, N. K. Karan, M. Balasubramanian, S.-Y. Li and D.-J. Liu, J. Am. Chem. Soc., 2012, 134(40), 16654–16661 CrossRef CAS PubMed
- U. I. Kramm, M. Lefèvre, N. Larouche, D. Schmeisser and J.-P. Dodelet, J. Am. Chem. Soc., 2014, 136(3), 978–985 CrossRef CAS PubMed
- J. L. Li, G. L. Liu, X. D. Long, G. Gao, J. Wu and F. W. Li, J. Catal., 2017, 355, 53–62 CrossRef CAS
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342(6162), 1073–1076 CrossRef CAS PubMed
- S. Tobias, S. Annette-Enrika, P. Marga-Martina, J. Kathrin and B. Matthias, ChemSusChem, 2014, 7(11), 3012–3016 CrossRef PubMed
- Z. Yu, Y. Bai, S. Zhang, Y. Liu, N. Zhang, G. Wang, J. Wei, Q. Wu and K. Sun, ACS Appl. Mater. Interfaces, 2018, 10(7), 6245–6252 CrossRef CAS PubMed
- C. Pengzuo, Z. Tianpei, X. Lili, X. Kun, T. Yun, X. Hui, Z. Lidong, Y. Wensheng, C. Wangsheng, W. Changzheng and X. Yi, Angew. Chem., Int. Ed., 2017, 56(2), 610–614 CrossRef PubMed
- G. S. Chang, E. Z. Kurmaev, D. W. Boukhvalov, L. D. Finkelstein, S. Colis, T. M. Pedersen, A. Moewes and A. Dinia, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(19), 195215 CrossRef
- L. Chen, F. Hu, H. Duan, Q. Liu, H. Tan, W. Yan, T. Yao, Y. Jiang, Z. Sun and S. Wei, Appl. Phys. Lett., 2016, 108(25), 252402 CrossRef
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55(36), 10800–10805 CrossRef CAS PubMed
- M. Junjie, C. Wenxing, S. Wenming, C. Zheng, P. Jiajing, H. Dongsheng, L. Chunlin, W. Dingsheng and L. Yadong, Angew. Chem., Int. Ed., 2017, 56(39), 11971–11975 CrossRef PubMed
- C. Wenxing, P. Jiajing, H. Chun-Ting, W. Jiawei, R. Hanlin, Z. Youqi, W. Yu, D. Juncai, T. Shubo, C. Weng-Chon, L. Siqi, Z. Lirong, Z. Xusheng, Y. Wensheng, Z. Zhongbin, C. Chen, P. Qing, W. Dingsheng and L. Yadong, Angew. Chem., Int. Ed., 2017, 56(50), 16086–16090 CrossRef PubMed
- M. Hardy, F. Poulhés, E. Rizzato, A. Rockenbauer, K. Banaszak, H. Karoui, M. Lopez, J. Zielonka, J. Vasquez-Vivar, S. Sethumadhavan, B. Kalyanaraman, P. Tordo and O. Ouari, Chem. Res. Toxicol., 2014, 27(7), 1155–1165 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00475k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |