
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombinatorial design of multimeric chelating peptoids for selective metal coordination†
Abel
Ricano‡
a,
Ilya
Captain‡
a,
Korey P.
Carter
 a,
Bryan P.
Nell
a,
Gauthier J.-P.
Deblonde
a and
Rebecca J.
Abergel
*ab
a,
Bryan P.
Nell
a,
Gauthier J.-P.
Deblonde
a and
Rebecca J.
Abergel
*ab
aChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: abergel@berkeley.edu
bDepartment of Nuclear Engineering, University of California, Berkeley, CA 94720, USA
First published on 12th June 2019
Abstract
Current methods for metal chelation are generally based on multidentate organic ligands, which are generated through cumbersome multistep synthetic processes that lack flexibility for systematically varying metal-binding motifs. Octadentate ligands incorporating hydroxypyridinone or catecholamide binding moieties onto a spermine scaffold are known to display some of the highest affinities towards f-elements. Enhancing binding affinity for specific lanthanide or actinide ions however, necessitates ligand architectures that allow for modular and high throughput synthesis. Here we introduce a high-throughput combinatorial library of 16 tetrameric N-substituted glycine oligomers (peptoids) containing hydroxypyridinone or catecholamide chelating units linked via an ethylenediamine bridge and, for comparison, we also synthesized the corresponding mixed ligands derived from the spermine scaffold: 3,4,3-LI(1,2-HOPO)2(CAM)2 and 3,4,3-LI(CAM)2(1,2-HOPO)2. Coordination-based luminescence studies were carried out with Eu3+ and Tb3+ to begin probing the properties of the new ligand architecture and revealed higher sensitization efficiency with the spermine scaffold as well as different spectroscopic features among the structural peptoid isomers. Solution thermodynamic properties of selected ligands revealed different coordination properties between the spermine and peptoid analogues with Eu3+ stability constants log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) β110 ranging from 28.88 ± 3.45 to 43.97 ± 0.49. The general synthetic strategy presented here paves the way for precision design of new specific and versatile ligands, with a variety of applications tailored towards the use of f-elements, including separations, optical device optimization, and pharmaceutical development.
β110 ranging from 28.88 ± 3.45 to 43.97 ± 0.49. The general synthetic strategy presented here paves the way for precision design of new specific and versatile ligands, with a variety of applications tailored towards the use of f-elements, including separations, optical device optimization, and pharmaceutical development.
Introduction
Interest in f-block elements has increased substantially in recent years as these elements have been used in a growing number of applications such as luminescent phosphors1,2 and diagnostic imaging and therapy.3–6 While the radioactive 5f actinides (An) have garnered significant attention due to the legacy of their use in energy and weapons,7–11 lanthanides (Ln), aside from promethium (Pm), consist wholly of stable isotopes. Many of the applications that utilize Ln metals are based on transitions within the 4f orbitals, which are shielded from external perturbations by filled 5s and 5p orbitals. This means that the 4f orbitals of Ln metal centers do not appreciably participate in bonding, thus local coordination environments do not overtly influence their respective solution or solid-state electronic and photophysical properties.Harnessing the unique capabilities of lanthanide metal cations so that one may selective tune the resulting properties involves exercising control over the first coordination sphere of Ln(III) ions. Using high-affinity, chelating ligands is one pathway for controlling Ln local coordination geometry, and currently the highest affinity f-block binding is achieved by organic, bio-inspired ligands.12–14 Raymond and coworkers15 have demonstrated the most striking example of f-block metal cation specificity by preparing a macrocyclic ligand that chelates Th4+ with a formation constant (Kf) of 1054, yet similar examples are limited as synthesis of such molecular structures is time consuming, expensive, and often not modular. As a result, new synthetic methods are needed to develop ligands in a high-throughput manner, which can also provide improved understanding of the subtleties of 4f-element coordination chemistry.
Most ligands with high-affinity for the f-block are multidentate and are composed of small metal-binding moieties placed on a molecular scaffold.7 Spermine (also called 3,4,3-LI) is described as a privileged scaffold due to the large number of ligands that have been derived from it and their relatively facile, low-cost, synthesis. Ligands of form X (Fig. 1) are typically synthesized by reacting spermine with activated esters of bidentate moieties and often display impressive binding characteristics. Mixed ligands of form Y (Fig. 1) that contain two different types of binding groups can be prepared by exploiting reactivity differences between the 3,4,3-LI primary and secondary amines, but are not very common due to low synthetic yields. Further variation of binding units along the 3,4,3-LI backbone has not been done due to impractical and low yielding syntheses of form Z ligands (Fig. 1). Access to ligands of the latter type is important, as it would allow for systematic binding unit variation along the backbone, leading to an enhanced understanding of Ln–ligand interactions.
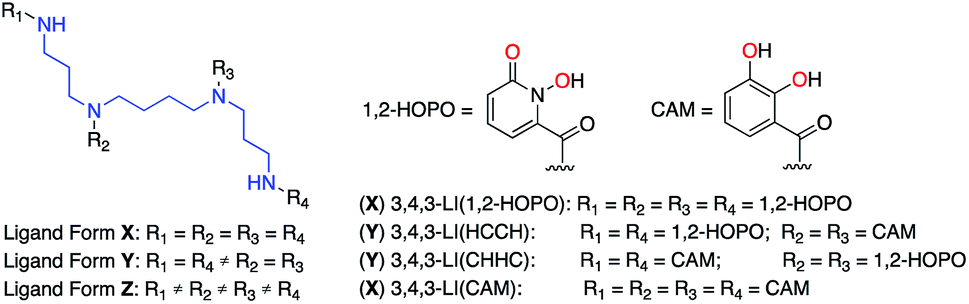 | ||
| Fig. 1 Spermine-based octadentate ligands. The 3,4,3-LI scaffold (blue) is connected to four metal-binding units (R1, R2, R3, and R4). With 1,2-HOPO and CAM (metal-binding atoms highlighted in red), only four ligand combinations are easily accessible through classical synthetic methods: 3,4,3-LI(1,2-HOPO), 3,4,3-LI(HCCH), 3,4,3-LI(CHHC), and 3,4,3-LI(CAM). | ||
To circumvent synthetic hurdles in the 3,4,3-LI system, we developed a new strategy by incorporating high-affinity metal-binding units onto peptoid scaffolds. Peptoids are structural analogs of peptides composed of N-substituted glycine oligomers. They are composed of primary amine sub-monomers, can be synthesized on solid support, and may be prepared using traditional automated peptide synthesizers.16 The combinatorial nature of peptoid synthesis and asymmetric scaffold provides a unique opportunity to control binding moiety sequence. This method can yield thousands of ligands in a matter of days, yet most studies have been limited to using commercially available amine sub-monomers. Peptoids have been examined as potential binders for a number of metals using combinatorial libraries,17–22 and here we aimed to improve upon the process of peptoid ligand synthesis by using custom-made sub-monomers known for their f-element binding properties. In this pilot study, we incorporated two well-characterized binding moieties into the peptoid scaffold using standard synthetic methods. The modularity of the scaffold allowed for the rapid preparation of all 16 possible tetrameric peptoid, octadentate ligands. In parallel, we also prepared two spermine-based ligands of form Y (Fig. 1) to serve as baseline comparisons. We then carried out luminescence-based coordination studies on all resulting ligands using Eu3+ and Tb3+, and stability constants for selected chelators with Eu3+ were determined. These systematic studies revealed that slight variations in ligand structure could result in significant differences in spectroscopic properties and complex stability, even for chemically similar Ln elements.
Results and discussion
Synthesis of peptoid library
Bidentate metal-binding units 1-hydroxy-pyridin-2-one (1,2-HOPO) and catecholamide (CAM) are well known moieties in f-element binding (Fig. 1).7 Extensive studies have established the octadentate 3,4,3-LI(1,2-HOPO) as a flagship chelator for An and Ln decorporation,13,23 and 3,4,3-LI(CAM) was recently evaluated for Eu3+, Th4+, and Zr4+ binding (Fig. 1).14 To accommodate the larger ionic radii of f-elements upon chelation, ethylenediamine bridges were inserted within the sub-monomer units to link 1,2-HOPO and CAM moieties to the peptoid scaffold (Scheme 1). Primary amine sub-monomers that we term H and C (corresponding to ethylenediamine-substituted 1,2-HOPO and CAM, respectively) were synthesized by reacting known precursor acyl chlorides with excess ethylenediamine to ensure single addition of chelating moieties (Scheme 1). The benzyl group was used to protect the N-hydroxyl of H while a diphenylacetal was used for the 1,2-diol on the C sub-monomer, and both sub-monomers were characterized by 1H and 13C NMR spectroscopy (ESI†). Acid labile protecting groups were chosen so they would be removed during cleavage of peptoids from solid support using trifluoroacetic acid (TFA).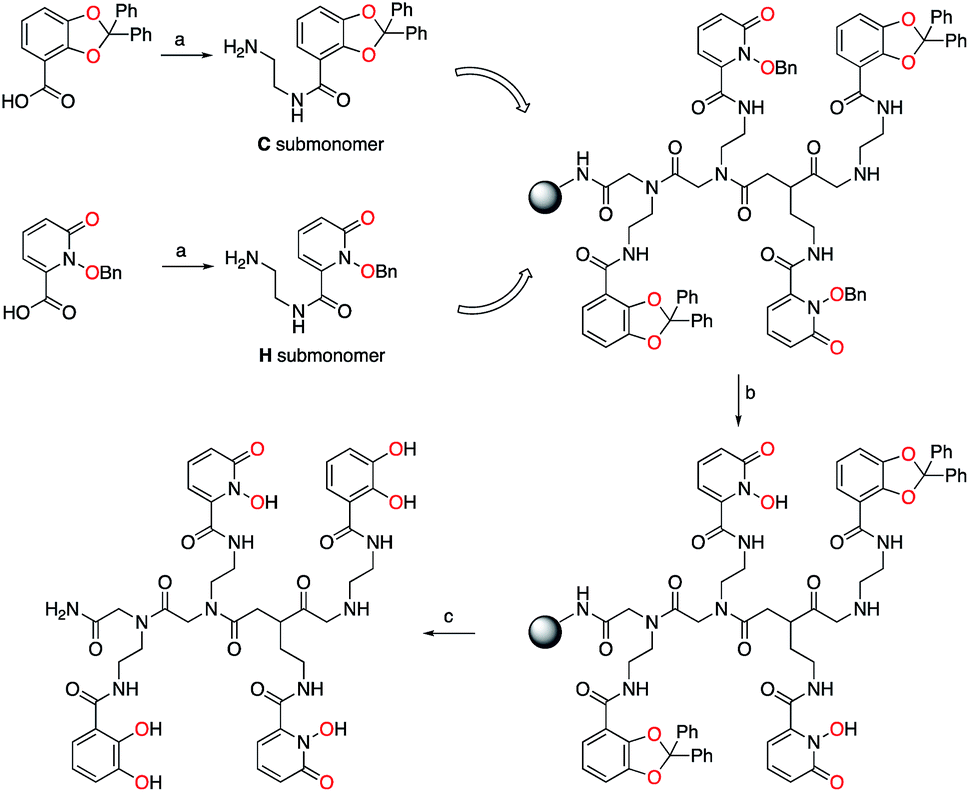 | ||
| Scheme 1 Synthesis of tetrameric peptoid ligands. (a) Oxalyl chloride, then ethylenediamine. (b) 0.1 M BBr3 in CH2Cl2. (c) 95/2.5/2.5 – TFA/water/triisopropylsilane. One peptoid structure, CHHC, shown, out of 16 possible tetrameric combinations with the two different types of sub-monomers C and H. Metal-binding oxygens are highlighted in red. | ||
H and C sub-monomers were then used to prepare the peptoids on Rink Amide resin employing standard coupling chemistry. In lieu of automated peptoids synthesis, we used fritted syringes as reaction vessels, which allowed for recovery of unreacted sub-monomers for future use. We were able to successfully reuse the solution of sub-monomer up to three times without any decrease in performance. Spent solutions were concentrated and re-purified by column chromatography.
Subsequent test cleavages on small samples of resin revealed that benzyl groups were not completely removed from H sub-monomers during TFA treatment, even with prolonged reaction times. After screening a number of conditions, we found that addition of 5% concentrated aqueous HCl to the TFA cleavage cocktail afforded the deprotected product but also led to significant peptoid backbone cleavage, which greatly complicated purification. Post-cleavage hydrogenolysis yielded the desired products but was impractical on milligram scales at which the peptoids were prepared. Alternatively, we found that the benzyl groups were removed by a BBr3 treatment without cleaving the peptoid from solid support. This allowed us to deprotect the peptoids and easily remove byproducts using a solvent wash. We finally settled on treating the resin with 0.1 M BBr3 in dichloromethane (CH2Cl2) followed by TFA cleavage to obtain the deprotected crude peptoid ligands. The ligands were purified using reverse phase preparative HPLC and were characterized by 1H and 13C NMR spectroscopy as well as high-resolution mass spectrometry, with TOF-MS/MS used to verify the peptoid sequence (ESI, Fig. S1–S23†). Ligands containing H moieties were generally more difficult to work with and to purify due to their propensity to bind iron and other trace metals present in instrument components even in acidic media. Due to the asymmetric nature of the peptoid scaffold (Scheme 1), the formation of 2n ligands is possible, where 2 denotes the number of sub-monomers (H and C) used and “n” the oligomer length. Sufficient amounts of all 16 possible tetrameric peptoid ligands were obtained in pure form. The synthesized peptoid chelators are listed in Table 1.
| a Synthesized tetrameric peptoids with H and C binding moieties. The leftmost designation in the ligand name denotes the first added moiety on the peptoid scaffold (i.e. nearest the terminal amide). | |||
|---|---|---|---|
| (1) HHHC | (2) CHHH | (3) HCHH | (4) HHCH |
| (5) CHHC | (6) HHCC | (7) CCHH | (8) HCHC |
| (9) HCCH | (10) CHCH | (11) HCCC | (12) CHCC |
| (13) CCHC | (14) CCCH | (15) CCCC | (16) HHHH |
Synthesis of 3,4,3-LI mixed ligands
In order to compare our new peptoid scaffold to the 3,4,3-LI backbone, we prepared two mixed derivatives, each either bearing two 1,2-HOPO units on the two internal tertiary amines and two CAM subunits on the terminal secondary amines of the spermine backbone (3,4,3-LI(CAM)2(1,2-HOPO)2, adopting the same binding group sequence as peptoid CHHC), or vice versa (3,4,3-LI(1,2-HOPO)2(CAM)2, adopting the same binding group sequence as peptoid HCCH). This synthesis takes advantage of the higher reactivity of the primary amines of spermine. One of the acid derivatives of 1,2-HOPO or CAM (with hydroxyl functionalities protected by benzyl or methyl groups, respectively) is first added onto the terminal amines through reaction with spermine in a controlled, dilute manner, and in the presence of amide coupling reagent carbonyldiimidazole (CDI). The acid chloride of the other subunit is then made in situ and directly added to the internal amines. Both methyl and benzyl protecting groups could be removed by BBr3 treatment, providing the crude ligands, which were purified using reverse phase preparative HPLC and characterized by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (ESI†).UV-visible spectroscopic properties
The electronic absorption spectra (ESI, Fig. S24†) of all sixteen peptoid ligands revealed the characteristic π–π* transitions of each aromatic system. (16) HHHH (λmax = 348 nm) and (15) CCCC (λmax = 320 nm) served as upper and lower bounds, respectively, for the maximum absorption of the peptoid ligands. Absorption maxima of peptoid ligands with more H units were red-shifted when compared with the multi-C unit peptoids (Fig. 2A), but featured similar extinction coefficients (Fig. 2B). Both 3,4,3-LI(CAM) and 3,4,3-LI(HCCH) free ligands contained a small shoulder around 285 nm in addition to their main absorption band centered at 320 nm and 350 nm, respectively. The absorption band for free 3,4,3-LI(CHHC) was centered at approximately 322 nm and changed in intensity and width upon complexation. All the complexes (of both Eu3+ and Tb3+) exhibited similar absorbance maxima at ca. 330 nm, with Ln–peptoid complexes with multiple H units blue-shifted with respect to the free ligands and vice versa for those with multiple C moieties (Fig. 2C and E for Eu3+ and Tb3+ complexes, respectively). Extinction coefficients decreased significantly upon Eu or Tb complexation for peptoid ligands containing more than 2 C sub-monomers, whereas the opposite trend was observed for the spermine-based ligands (Fig. 2D and F for Eu3+ and Tb3+ complexes, respectively).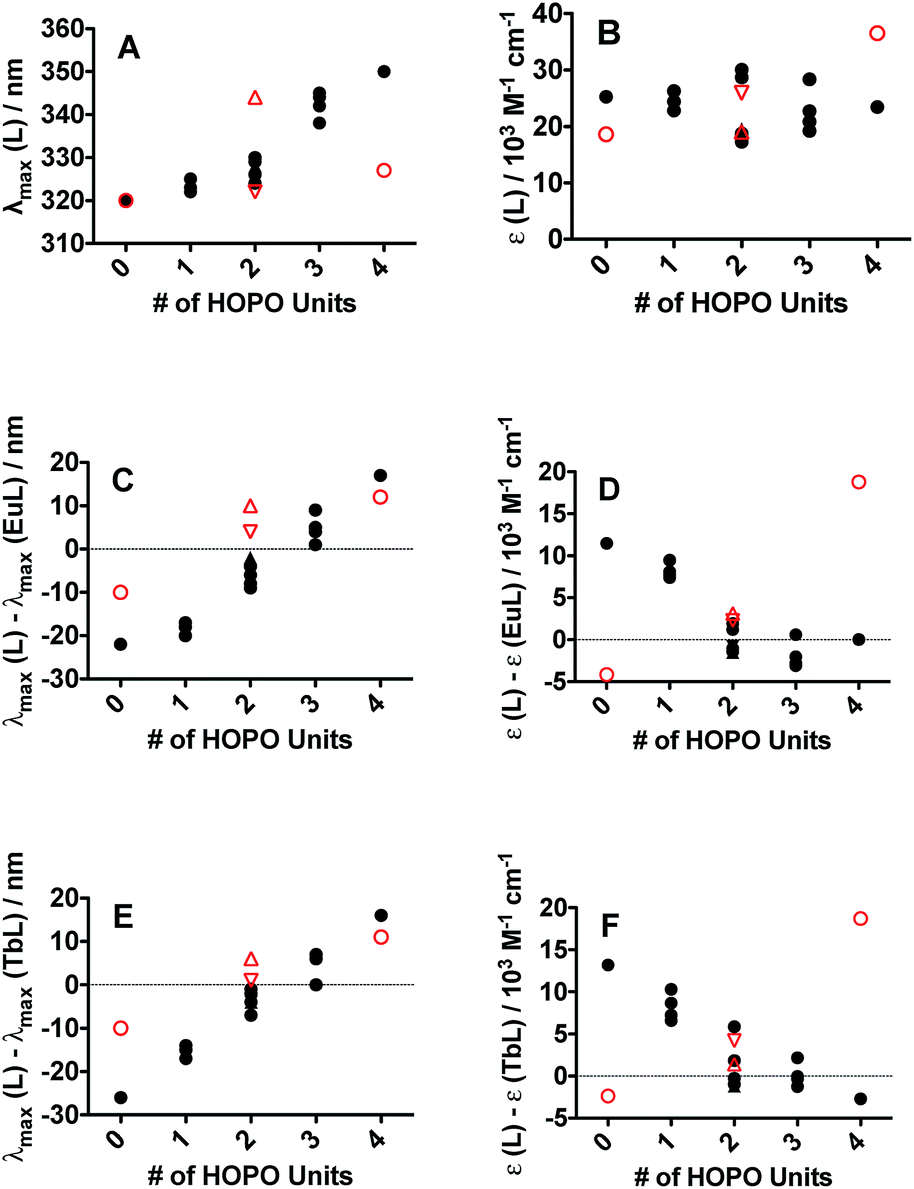 | ||
| Fig. 2 Ligand and complex UV-vis spectroscopic parameters dependence as a function of the number of 1,2-HOPO metal-binding units (0 for CCCC and 3,4,3-LI(CAM) to 4 for HHHH and 3,4,3-LI(1,2-HOPO)) on the peptoid (black full circles) and spermine-based scaffolds (red open symbols; circles for 3,4,3-LI(CAM) and 3,4,3-LI(1,2-HOPO); upward and downward triangles for 3,4,3-LI(HCCH) and 3,4,3-LI(CHHC), respectively). (A) Ligand maximum absorption wavelength. (B) Ligand extinction coefficient. (C) Wavelength shift upon Eu complexation. (D) Change in extinction coefficient upon Eu complexation. (E) Wavelength shift upon Tb complexation. (F) Change in extinction coefficient upon Tb complexation. | ||
Ligand-sensitization of Eu3+ and Tb3+ luminescence
The ligand of form X (Fig. 1) that bears 1,2-HOPO units (i.e. 3,4,3-LI(1,2-HOPO)) is known to sensitize the luminescence emission of selected Ln(III) cations through the formation of stable stoichiometric complexes and the so-called “antenna effect”,23,24 as depicted through a simplified Jablonski diagram in Fig. 3. To this end, we sought to determine if the peptoid ligands could also sensitize Ln(III) ions in a similar fashion, using Eu3+ and Tb3+ as representative systems. The emission spectra of the Eu3+ and Tb3+ peptoid complexes revealed relatively weak signals (ESI, Fig. S25–S30†). Quantum yields of Eu:HHCH (4) (ϕ = 0.0043 ± 0.0002) and Tb:3,4,3-LI(CHHC) (ϕ = 0.0011 ± 0.0001) were determined by the optical dilution method. These values were then used as benchmarks to estimate the values for the remaining complexes (ESI, Table S1†) by taking the ratio of the integrated emission spectra to that of the corresponding metal complex, assuming identical extinction coefficients. Here we can use Eu:3,4,3-LI(1,2-HOPO) as a reference with an estimated value of 0.14, consistent with the published quantum yield values of 15.6 ± 0.6% (isolated complex) and 14.0 ± 0.3% (complex formed in situ).25 This comparative method is not meant to be interpreted as rigorous for quantum yield determination, rather we aimed to probe relevant variations within the large number of ligands and the small scale of the pilot study. The largest quantum yields for Eu3+ and Tb3+, not including 3,4,3-LI(1,2-HOPO), are complexes formed with (16) HHHH and 3,4,3-LI(CHHC), respectively. Nonetheless, these observed weak emission properties provide evidence of poor energy transfer to the metal ([a] in Fig. 3), significant non-radiative de-excitation ([b] in Fig. 3), or a combination thereof.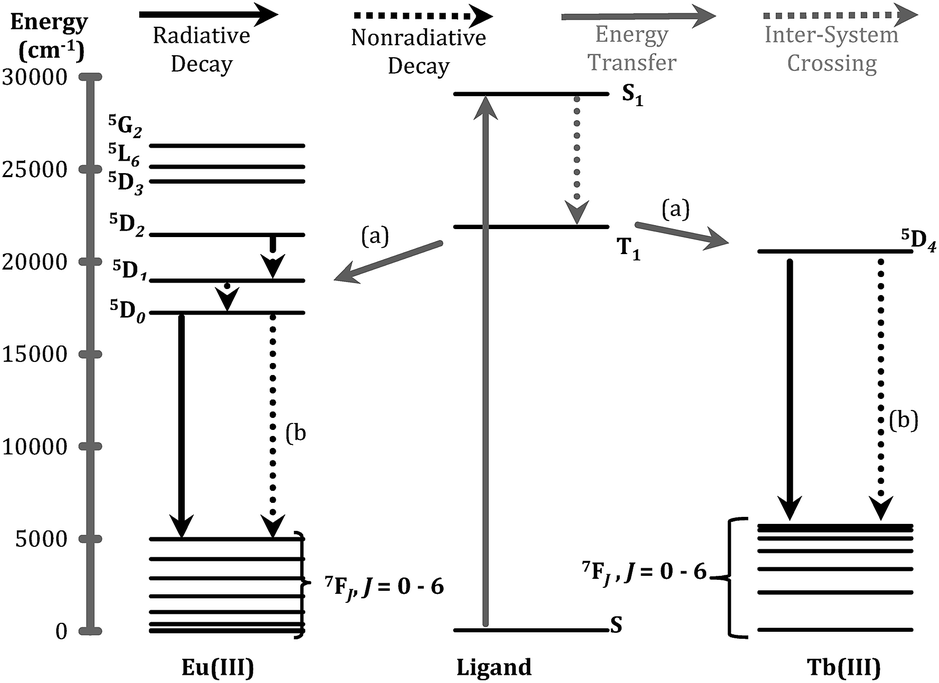 | ||
| Fig. 3 Jablonski diagram representing the major energy transfer pathways of sensitization. (a) Represents energy transfer from the ligand to the metal; (b) indicates non-radiative de-excitation. Energy levels for free ions were adapted from previous publications.26,27 | ||
One of the critical components in Ln(III) sensitization is the energy transfer efficiency between the first triplet-state of the ligand (T1) and the accepting state of the metal (Fig. 3).28 Since Gd3+ exhibits a size and atomic weight similar to Eu3+ but lacks an appropriately positioned electronic acceptor level, the triplet-state of the ligand can be estimated by measuring the phosphorescence spectra of the respective Gd(III) complex. Energy transfer is most efficient when the respective resonance levels are similar enough in energy such that thermal back-transfer can be minimized. For Eu3+ and Tb3+, this occurs when the triplet-state donor level is 2500–3500 cm−1 and 2500–4000 cm−1, respectively, higher than the metal accepting state.29 Triplet state energies were determined for all investigated compounds, according to described methods.30,31 The T1 energies were found close to the Eu3+ 5D2 level, similar to what has been reported with polyaminocarboxylates28 but contrasting with ligands such as Schiff bases32 (5D0) or β-diketonates33 (5D1). Structures with triplet state energies values nearest to the 5D2 level resulted, overall, in higher Eu emission intensities (Fig. 4A). To further probe the role of each HOPO or CAM binding unit on luminescence sensitization, phosphorescence spectral shapes were examined in more details for each Gd complex. Deconvolution of the main triplet-state peak of (15) CCCC and (16) HHHH with two Gaussian curves each revealed bands centered around 20000 cm−1 (∼498 nm) for both, 18100 cm−1 (∼552 nm) for (16) HHHH, and a higher energy contribution from (15) CCCC centered around 21400 cm−1 (∼467 nm). There was an additional small band in the (16) HHHH phosphorescence spectra centered around 25000 cm−1 (∼395 nm) that was also observed for (5) CHHC (∼391 nm). The extent of 1,2-HOPO and CAM triplet-state character in the rest of the peptoid ligands was gauged by the relative contribution of the spectral bands centered at 18100 cm−1 and 21400 cm−1, respectively, since both (15) CCCC and (16) HHHH featured a peak centered at ca. 20000 cm−1 (Fig. 4B). Luminescence intensity was found to increase with the number of H moieties on the peptoid scaffold (Fig. 4, panels C and D, and ESI†), which was notable as the 1,2-HOPO triplet-state contribution is just under 1000 cm−1 above the 5D2 manifold of Eu(III), much lower than the 2500 cm−1 energy gap suggested by Latva et al.28 for optimal energy transfer.
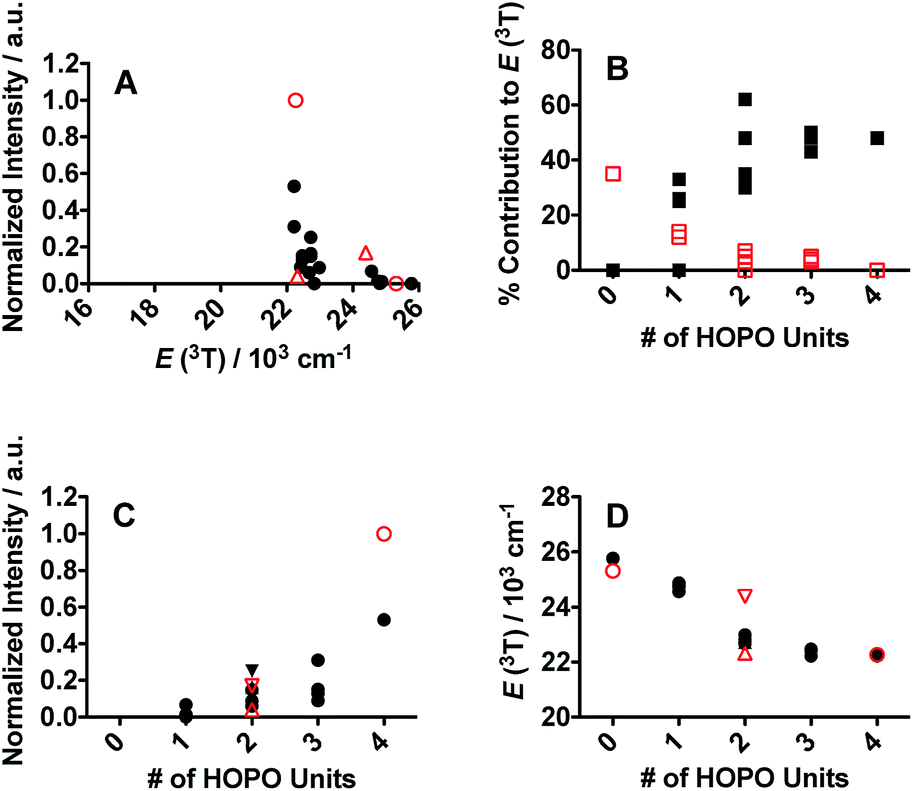 | ||
| Fig. 4 (A) Eu luminescence emission intensity as a function of ligand triplet-state energy for the peptoid (black full circles) and spermine-based scaffolds (red open symbols; circles for 3,4,3-LI(CAM) and 3,4,3-LI(1,2-HOPO); upward and downward triangles for 3,4,3-LI(HCCH) and 3,4,3-LI(CHHC), respectively). (B) % contribution of the 1,2-HOPO (full black squares) and CAM (open red squares) metal-binding units to the phosphorescence from the triplet excited state as a function of the number of 1,2-HOPO metal-binding units for peptoid ligands. (C) Eu luminescence emission intensity and (D) ligand triplet-state energy as a function of the number of 1,2-HOPO metal-binding units (0 for CCCC and 3,4,3-LI(CAM) to 4 for HHHH and 3,4,3-LI(1,2-HOPO)) on the peptoid and spermine-based scaffolds; symbols are as described for panel A. | ||
The triplet-state data of the spermine based ligands was similarly treated (Fig. 4). 3,4,3-LI(CAM) and 3,4,3-LI(1,2-HOPO) had deconvoluted bands centered around 22000 cm−1 (∼455 nm) and 18200 cm−1 (∼550 nm), respectively, and an additional component around 19800 cm−1 (∼506 nm). As in the peptoid analogue, 3,4,3-LI(1,2-HOPO) had an additional small peak centered around 24400 cm−1 (∼410 nm). A larger triplet-state character from 3,4,3-LI(CAM) was observed in 3,4,3-LI(CHHC) than in the 3,4,3-LI(HCCH). As with the peptoid scaffold ((15) CCCC), no fluorescence was observed for 3,4,3-LI(CAM) complexed with Eu3+. There was about a ∼30% weaker quantum yield for the Eu complex with 3,4,3-LI(HCCH) than 3,4,3-LI(CHHC), but this could very well be attributed to the low molar absorptivity (vide infra) of the 3,4,3-LI(HCCH) complex.
Further analysis of the Eu emission spectra revealed Stark splitting and small shifts in the 5D0 → 7F2 transition for most complexes (ESI, Fig. S25–S30†). Two comparisons are presented in Fig. 5. A 2 nm shift between the peaks of the Eu:1,2-HOPO complexes, 3,4,3-LI(1,2-HOPO) and (16) HHHH, as well as a shoulder peak at 618.5 nm for Eu:3,4,3-LI(1,2-HOPO) was observed (Fig. 5A). Panel B of Fig. 5 shows the directional isomers (3) HCHH and (4) HHCH and highlights the substantial differences that can result from small changes in the placement of H and C binding moieties. We note an approximate 4-fold intensity increase when comparing the Eu complexes of 3 and 4, along with some Stark splitting of the 7F2 transition for 4 that is not observed with the analogous Eu:HCHH (3) complex.
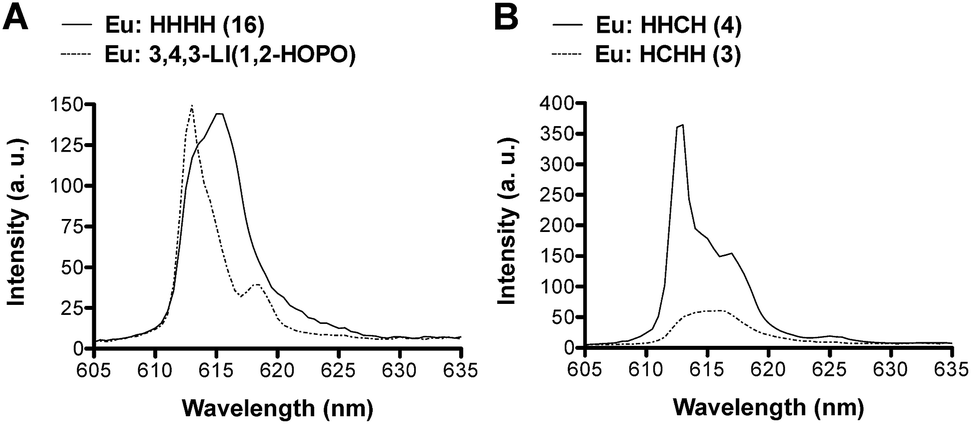 | ||
| Fig. 5 The 5D0 → 7F2 emissive transition of the Eu complexes formed with (A) 3,4,3-LI(1,2-HOPO) (dotted line) and (16) HHHH (solid line); (B) (3) HCHH (dotted line) and (4) HHCH (solid line). | ||
The Tb3+ spectra revealed much less efficient sensitization than with Eu3+ congeners as all spectra had significant ligand luminescence leakage around 450 nm. None of the triplet-states for any of the investigated ligands had optimum energy to efficiently populate the Tb3+ accepting level (∼20500 cm−1) and there was a significant difference in the sensitization efficiencies between the Eu(III) and Tb(III) systems. For Eu3+ the most luminescent peptoid was (16) HHHH, followed by (4) HHCH, whereas with Tb3+ it was (2) CHHH.
Luminescence lifetimes of Eu:peptoid complexes as well as 3,4,3-LI mixed ligand complexes with Eu3+ and Tb3+ revealed bi-exponential decays with the exception of Eu:HHCH (4), Eu:CHHC (5), both mono-exponential decays, and Eu:HCCC (11) and Tb:3,4,3-LI(CHHC), which were fit as tri-exponential. The longest lifetimes came from Eu complexes of (16) HHHH (827 μs) and 3,4,3-LI(CHHC) (733 μs), both corresponding to one coordinated water molecule according to Kimura's empirical equation (ESI, Table S2†).34 Comparing (16) HHHH (827 μs) with 3,4,3-LI(1,2-HOPO) (805 μs),24 reveals that when all binding units are 1,2-HOPO moieties there is little variation between 3,4,3-LI and peptoid scaffolds. With peptoid ((5) CHHC and (9) HCCH) and 3,4,3-LI mixed ligands each featuring two C and two H units the results get more interesting as we note a small (64 μs) increase in Eu(III) lifetime when going from (9) to 3,4,3-LI(HCCH) and a more substantial change (180 μs) when going from (5) to 3,4,3-LI(CHHC) (ESI, Table S2†).
The structural peptoid isomers bearing three H and one C units have lifetimes ranging from 432–679 μs (ESI, Table S2†) with the most noteworthy difference between (3) HCHH and (4) HHCH. By exchanging C units in the 2nd and 3rd positions we observe a ca. 250 μs change in Eu3+ lifetime, which corresponds to one and two bounds water molecules, respectively.34 This is indicative of a possible change in coordination of Eu(III) by (3) and (4) as a result of the difference in the order of H and C binding units, which suggests the location of H and C moieties may enhance/decrease recognition of Eu3+. The structural peptoid isomers bearing two H and two C units have lifetimes ranging from 425–553 μs (ESI, Table S2†) and an analogous comparison of Eu:peptoid complexes where C units are exchanged in the 2nd and 3rd positions ((7) CCHH and (10) CHCH) yields a difference of only ca. 25 μs, which is a much smaller change than what was noted above. For peptoid isomers with two H and two C units the more interesting results come from comparing complexes with H units in the 3rd and 4th positions. We observe decreases of ca. 75–125 μs in Eu(III) lifetimes when H moieties are switched from the 3rd position to the 4th position (comparing (5) CHHC with (10) CHCH and (8) HCHC with (9) HCCH), which is also possibly indicative of changes in ligand conformation about the Eu(III) metal center as a result of changing the order of binding moieties. The structural peptoid isomers bearing one H and three C units have lifetimes ranging from 569–682 μs (ESI, Table S2†) and display the least amount of variance (in lifetime values) with changes in the order of the binding units, suggesting that once a ligand includes a certain number of C units (three) only one binding conformation, about the Eu3+ metal center, is observed. Noteworthy, Gd coordination properties will be investigated in the future for those four ligand structures with which Eu3+ luminescence lifetimes suggested two inner-sphere water molecules, (3) HCHH, (7) CCHH, (9) HCCH, and (10) CHCH. The ability to form Gd:peptoid complexes with q values of 2 may result in enhanced relaxivity properties, which could prove attractive for magnetic resonance imaging applications when associated with high thermodynamic stability constants, as detailed below.
Solution thermodynamics
Inspired by previous efforts of our group to characterize 3,4,3-LI(CAM),14 we sought to investigate the solution thermodynamic properties of the peptoid analogue (15) CCCC. The last 3 pKa's of (15) CCCC were not resolvable by spectrophotometric titrations and were held constant at values comparable to the spermine analogue. The proton-independent stability constant (logβ110) of the (15) CCCC complex with Eu3+ was refined to 28.88 ± 3.45 (ESI, Table S3†) and this result is quite similar to Eu3+ complexed with 3,4,3-LI(CAM) (logβ110 = 29.65).14 Speciation modeling for equal concentrations of 3,4,3-LI(CAM) and (15) CCCC in the presence of Eu3+ reveals that 3,4,3-LI(CAM) predominately binds Eu3+ above a pH of 6 and further, the speciation diagram demonstrates that (15) CCCC is also fully binding Eu3+ under the conditions employed for fluorescence measurements (ESI†). Incorporating a mixture of CAM and HOPO groups onto a scaffold was expected to increase the affinity of the chelator to the metal while increasing the pH range at which it binds, in comparison to the parent ligands. The stability constant (logβ110) of 3,4,3-LI(CHHC) complexed with Eu3+ is 35.97 ± 0.06 (ESI, Table S3†). Thermodynamic modeling (Fig. 6) of 3,4,3-LI(1,2-HOPO), 3,4,3-LI(CAM), and 3,4,3-LI(CHHC) in the presence of Eu3+ at equal concentrations revealed that the predominant complexing ligand above a pH of 6 is the mixed 343-LI species 3,4,3-LI(CHHC). At lower pH values, 3,4,3-LI(1,2-HOPO) dominated while a small fraction of 3,4,3-LI(CAM) is bound around pH 11. The protonation constants for 3,4,3-LI(HCCH) were also investigated via spectrophotometric titrations, but the similarity of the Eu complex absorption signal with the signal from the free ligand (3,4,3-LI(HCCH)) prevented refinement with HypSpec. Therefore, competition batch titrations were employed against 3,4,3-LI(1,2-HOPO) (ESI†). The logβ111 value obtained for 3,4,3-LI(HCCH) was 43.34 ± 0.49. For comparison, we selected a peptoid with two H and two C moieties and the Eu complex formed with (7) CCHH displayed a stability constant (logβ110) of 36.01 ± 4.13, and is also fully formed at pH 7.4. As previously mentioned, having access to ligands with HOPO and CAM units opens up avenues for manipulating the properties of the chelator, as seen with the mixed ligands and when comparing the CAM analogues.
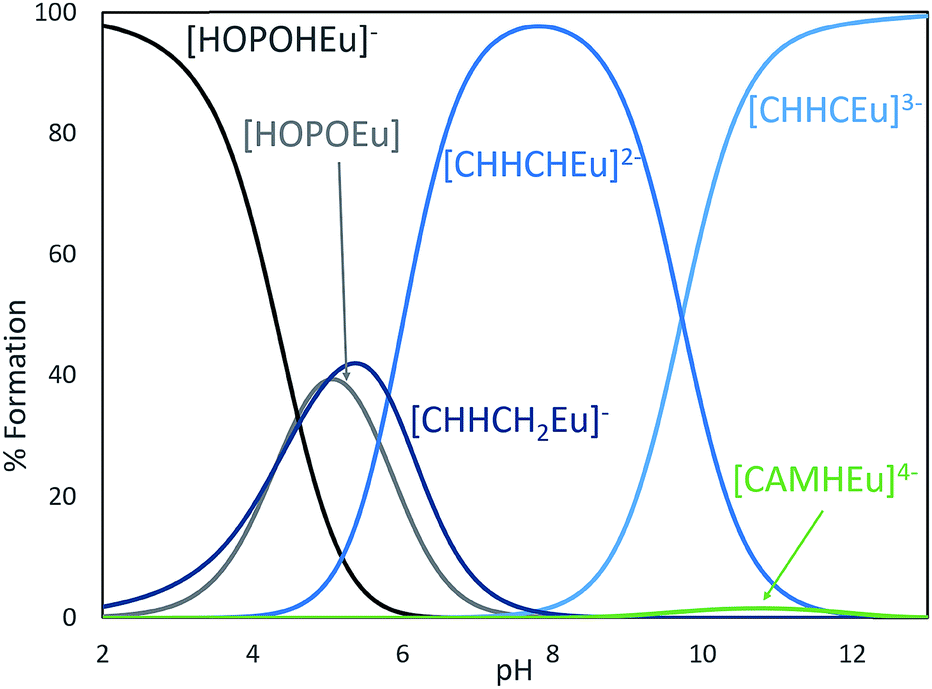 | ||
| Fig. 6 Speciation diagram for a competition between 3,4,3-LI(1,2-HOPO), 3,4,3-LI(CAM), and 3,4,3-LI(CHHC) abbreviated HOPO (gray), CAM (green), and CHHC (blue), respectively, in the presence of Eu3+. [HOPO] = [CAM] = [CHHC] = [metal] = 25 μM, I = 0.1 M, T = 25 °C. Only the major species were labeled on the plots for clarity. | ||
Conclusions
We report herein the synthesis of a new library of tetrameric peptoid ligands that can be used for chelation of f-block metals. We prepared and characterized a library of octadentate peptoids based upon 1,2-HOPO- and CAM-based sub-monomers, investigated their thermodynamic and photophysical properties with Eu3+ and Tb3+, and compared them with their spermine analogues. Both ligand systems show high affinity for Ln(III) metals with stability constants greater than 1029 for Eu3+ with the investigated chelators. Overall, the peptoid scaffold provides a platform to conduct a more comprehensive, systematic approach to study Ln–ligand interactions, and one can envision fine-tuning the peptoid manifold to incorporate the desired cavity size, binding moieties, and energetics of the chelator. As synthesis is done on solid-support, creativity and modularity can be imparted onto the ligand, depending on the desired function. We are now working on acquiring structural parameters of metal complexation, and further characterizing peptoid complexes with other f-block metals, by probing their ability to efficiently sensitize near-IR emitters as well as exploring the fundamental differences in energy transfer processes between 4f and 5f species. On-going work also focuses on expanding synthetic modifications of the scaffold for the development of targeted therapeutic applications.Materials and methods
Chemicals were obtained from commercial suppliers and were used as received unless stated otherwise. The ligands 3,4,3-LI(1,2-HOPO) and 3,4,3-LI(CAM) were prepared and characterized as previously described.14,35 Synthesis precursors 2,2-diphenylbenzo[d][1,3]dioxole-4-carboxylic acid and 1,6-carboxy-1-(benzyl)hydroxy-2-pyridinone were prepared according to literature procedures.14,36 While the mixed ligand 3,4,3-LI(CHHC) has been made before,7,37 herein we followed a slightly different approach and expanded efforts beyond what had been previously reported to produce both 3,4,3-LI(CHHC) and 3,4,3-LI(HCCH). Detailed syntheses and characterization of the two compounds can be found in the ESI.† All NMR spectra were recorded at ambient temperature on Bruker FT-NMR spectrometers, using tetramethylsilane as an internal reference. SilicaFlash G60 (particle size 60–200 μm) was used for flash column chromatography. LC-MS was performed on an Agilent LC/MS system consisting of an Agilent 1200 binary LC pump, a temperature-controlled autosampler, a PDA UV detector, and a 6530 Accurate Mass Q-TOF mass spectrometer (Wilmington, DE, USA). The mass spectrometer was equipped with a JetStream® ESI probe operating at atmospheric pressure. The ESI source parameter settings were: mass range m/z 100–1200, gas temperature 350 °C, gas flow 10 L min−1, nebulizer 50 psi, sheath gas temperature 400 °C, sheath gas flow 12 L min−1, capillary voltage (Vcap) 3500 V, nozzle voltage 500 V, fragmentor 200 V, skimmer 65 V, octopole RF (OCT 1 RF Vpp) 750 V. Reverse phase preparatory HPLC was performed on a Varian ProStar system with a Vydac C18 column. HRMS and MS-MS were obtained on a Waters Xevo G2 Qtof mass spectrometer, and leucine encephalin lockspray with mass correction was used for HR-MS.Ethylenediamine substituted CAM submonomer, “C”
2,2-Diphenylbenzo[d][1,3]dioxole-4-carboxylic acid (3.84 g, 12.06 mmol) was suspended in 30 ml of toluene under an argon atmosphere. Oxalyl chloride (1.14 ml, 13.3 mmol) was then added followed by a catalytic amount of N,N-dimethylformamide. The suspension was heated to 40 °C and stirred until it became clear and the evolution of gas had ceased (∼1 hour). The volatiles were then removed using the manifold vacuum and the resulting white solid was dissolved in dry CH2Cl2. A separate 1 L roundbottom flask outfitted with an addition funnel was charged with ethylenediamine (8 ml, 120 mmol) and 50 ml dry CH2Cl2; the resulting solution was cooled to 0 °C using an ice bath. The aforementioned solution of acyl chloride was transferred into the addition funnel and was diluted with CH2Cl2 to a total volume of 700 ml. The acyl chloride solution was then added into the vigorously stirred ethylenediamine over 1.5 hours at 0 °C. Following the addition, the reaction solution was transferred into a separatory funnel and was washed with 0.5 M NaOH in 50% saturated aq. NaCl (50 ml × 2). The organic phase was dried over MgSO4 and was concentrated on a rotary evaporator yielding the crude. The crude was purified using silica column chromatography (5 → 10% MeOH in CH2Cl2 with 1% Et3N, Rf = 0.35 in 10% MeOH in CH2Cl2). The desired fractions were combined, concentrated under reduced pressure, and dried under vacuum yielding the CAM submonomer as a sticky yellow oil (3.49 g, 9.68 mmol, 80% yield). 1H NMR (300 MHz, CDCl3) δ 7.64 (1H, br s, NH), 7.56–7.61 (5H, m, ArH), 7.37–7.42 (6H, m, ArH), 7.01 (1H, dd, J = 7.7, 1.4 Hz, ArH), 6.94 (1H, t, J = 7.9 Hz, ArH), 3.56 (2H, q, J = 6.0 Hz, NHC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 2.97 (2H, t, J = 6.0 Hz, NH2C2), 2.75 (2H, s, NH2). 13C NMR (75 MHz, CDCl3) δ 163.7, 147.2, 144.7, 139.4, 129.5, 128.4, 126.2, 122.4, 122.0, 118.0, 116.0, 111.6, 42.3, 41.5. HRMS-ESI (m/z) [M + H] calcd For C22H20N2O3 + H, 361.1563; found, 361.1581.
2), 2.97 (2H, t, J = 6.0 Hz, NH2C2), 2.75 (2H, s, NH2). 13C NMR (75 MHz, CDCl3) δ 163.7, 147.2, 144.7, 139.4, 129.5, 128.4, 126.2, 122.4, 122.0, 118.0, 116.0, 111.6, 42.3, 41.5. HRMS-ESI (m/z) [M + H] calcd For C22H20N2O3 + H, 361.1563; found, 361.1581.
Ethylenediamine substituted 1,2-HOPO submonomer, “H”
1,6-Carboxy-1-(benzyl)hydroxy-2-pyridinone36 (3.84 g, 12.06 mmol) was suspended in 30 ml of toluene under an argon atmosphere. Oxalyl chloride (1.14 ml, 13.3 mmol) was then added followed by a catalytic amount of N,N-dimethylformamide. The suspension was heated to 40 °C and was stirred until it became clear and the evolution of gas has ceased (∼1 hour). The volatiles were then removed using the manifold vacuum and the resulting white solid was dissolved in dry dichloromethane. A separate 1 L roundbottom flask outfitted with an addition funnel was charged with ethylenediamine (8 ml, 120 mmol) and 50 ml dry dichloromethane; the resulting solution was cooled to 0 °C using an ice bath. The aforementioned solution of acyl chloride was transferred into the addition funnel and was diluted with dichloromethane to a total volume of 700 ml. The acyl chloride solution was then added into the vigorously stirred ethylenediamine over 1.5 hours at 0 °C. Following the addition, the reaction solution was transferred into a separatory funnel and was washed with 0.5 M NaOH in 50% saturated aq. NaCl (50 ml × 2). The organic phase was dried over MgSO4 and was concentrated on a rotary evaporator yielding the crude. The crude was purified using silica column chromatography (5 → 10% MeOH in DCM with 1% Et3N, Rf = 0.35 in 10% MeOH in DCM). The desired fractions were combined, concentrated under reduced pressure, and dried under vacuum yielding the HOPO submonomer as a yellow oil (3.49 g, 9.68 mmol, 80% yield). 1H NMR (300 MHz, CDCl3) δ 7.42–7.49 (3H, m, ArH and NH), 7.31–7.40 (3H, m, ArH), 7.26 (1H, dd, J = 9.2, 6.8 Hz, CHCCH), 6.67 (1H, dd, J = 9.2, 1.6 Hz, CHCHC), 6.44 (1H, dd, J = 6.7, 1.6 Hz, CCHCH), 5.28 (2H, s, CH2Ph), 3.36 (2H, t, J = 6.0 Hz, NHC2), 3.80 (2H, q, J = 6.1 Hz, NH2C2). 13C NMR (75 MHz, CDCl3) δ 160.3, 158.5, 142.5, 138.0, 133.2, 130.1, 129.4, 128.6, 124.0, 106.4, 79.3, 42.7, 40.7. Calcd for C15H17N3O3 + H, 288.1328; found, 288.1361.
Synthesis of peptoids
Unless noted otherwise, all steps were carried out in fritted polypropylene syringes, which allowed for recovery of submonomer for re-use. Automatic peptoid synthesis was not an option for this work due to difficulty of sub-monomer preparation. 100–150 mg of Rink amide resin was added to a fritted syringe. The resin was swollen by adding 2 ml of DMF and rocking for 30 minutes, and then solution was ejected to isolate the swollen resin. 1 ml of 20% 4-methylpiperidine in DMF (v/v) was added to deprotect the Fmoc group followed by agitation for two minutes, draining of the solution, and repeating for 12 minutes in total. Immediately following, the resin was rinsed with DMF (2 ml, 5 times for 1 minute). To bromoacetylate, premix 0.8 M bromoacetic acid in DMF with 0.8 M N,N-diisopropylcarbodiimide (DIC) (2 ml total solution with 0.4 M of each reagent). Draw the solution into the syringe, agitate for five minutes, and rinse again with 2 ml DMF (5 × 1 minute). In order for displacement to occur, draw in 1.5 ml of submonomer solution (0.2 M in DMF), agitate for one1 hour at 45 °C, and then rinse with DMF (5 × 1 minute). Repeat bromoacetylation and displacement steps until synthesis is finished and then wash with DCM (3 × 1 minute) after the last DMF wash. Finally, dry resin by pulling the plunger out and applying a gentle vacuum onto the syringe needle.Test cleavage
A small amount of resin was placed into an LC-MS vial and was treated with ∼200 μl of cleavage cocktail (95/2.5/2.5 TFA/water/triisopropylsilane). The suspension was agitated for one hour at room temperature, after which it was diluted to 1 ml with methanol and the beads were filtered off. The solution was analyzed by LC-MS for the desired mass. This procedure was only used analytically as benzyls from 1,2-HOPO containing peptoids are not cleaved under these conditions (keep this in mind when looking for the desired mass).Deprotection, cleavage, and purification
Dry resin (100–150 mg) was placed into a scintillation vial and was swollen in 9 ml of DCM by shaking for 30 minutes. 1 ml of 1.0 M BBr3 in hexanes was added via a syringe and the vial was capped and shaken for 60 minutes ensuring that all of the resin was thoroughly submerged, which removes benzyl protecting groups from HOPO units. The solvent was carefully removed with a glass pipette and the resin was washed with DCM (2 ml) and methanol (2 ml × 2), followed by DCM (2 ml × 2). The peptoid was then cleaved from resin by treatment with cleavage cocktail for 60 minutes (the treatment also deprotects CAM units). The cleavage cocktail was filtered from resin and a small aliquot was removed and diluted with methanol for LC-MS analysis (1 → 30% MeCN in H2O over 20 minutes, both with 0.1% formic acid) while the resin was washed of TFA traces and discarded. Most LC-MS analyses showed a relatively clean desired compound. Iron complexes were sometimes observed, which we believe came from stainless steel components of the instrument. Volatiles were then removed from the filtrate using a vacuum pump. The resulting residue was dissolved in 90/10 acetic acid/water (0.5–1 ml) and the resulting clear solution was stirred at 42 °C and treated with water in 0.5 ml increments. The solution turned turbid upon addition of water and slowly clarified with continued stirring (5–15 min between additions). A total of ∼2.5 ml of water was added, at which point the solution remained turbid even with prolonged stirring. The turbid solution was taken up into a syringe and injected onto reverse-phase prep-HPLC through a 0.45 μm filter in no more than 2.0 ml batches (∼2 injections per peptoid).Photophysical measurements
UV-visible absorption spectra were recorded on a Varian Cary 5G double-beam absorption spectrophotometer, using either 1.00 cm or 2.0 mm path length quartz cells. Each of the measured peptoid solutions was 45.4 μM (100 μM for 3,4,3-LI ligands) with 1.1 equivalents of metal (M = Eu3+ or Tb3+), to ensure the absence of free ligand, and buffered in 50 mM HEPES at pH 7.4. Fluorescence and time-resolved measurements were acquired on a HORIBA Jobin Yvon IBH Fluorolog-3 spectrophotometer. The excitation source was a xenon flash lamp. A 400 nm long-pass filter was used when the second harmonic of the excitation beam overlapped with the emission spectra. For luminescence measurements, the ligands (5.0 μM) were mixed with 0.9 equivalents of metal (M = Eu3+ or Tb3+) to ensure full metal complexation in 50 mM HEPES buffer (pH 7.4), and excited at the absorption maximum for each of the complex. Quantum yield measurements were obtained using the optical dilution method with quinine sulfate as a reference. 38 Samples of quinine sulfate at various absorbance (0.02–0.1) were prepared in 50 mM sulfuric acid. Ln complexes (complexes = Tb:3,4,3-LI(CHHC) or Eu:HHCH (4)) at various absorbance (0.02–1) were prepared in 0.1 M KCl, buffered in 50 mM HEPES at pH 7.4. All samples were background corrected for solvent auto fluorescence. The ligand luminescence leakage for Tb3+ spectra was background corrected with QTIPlot. At least five samples were used per trial for both quinine sulfate and metal complexes. The reported quantum yields and uncertainty were based off three independent trials. Additionally, the triplet-state energies of the ligands were determined by preparing Gd3+ complexes in situ (91% EtOH, 4.5% DMSO, and 4.5% H2O) and measuring the phosphorescence of the ligand under cryogenic conditions. The main peak in 3,4,3-LI(CAM), 3,4,3-LI(1,2-HOPO), CCCC (15), and HHHH (16) phosphorescence spectra was modeled by two Gaussian curves each and further used to model the triplet-state for the mixed peptoid and spermine ligands. It was assumed that the triple-state character of the mixed ligands was a linear combination of the pure CAM and 1,2-HOPO ligands.Solution thermodynamics
Absorbance spectra for spectrophotometric titrations were recorded on an Ocean Optics USB 4000 spectrophotometer (slit 50 nm, grating 600 grooves per mm, blaze 400 nm) equipped with a PX-3 pulsed xenon or HPX-2000 high-powered xenon light source. Spectral data were acquired with a 1 cm path length dip probe (Ocean Optics, Inc.). A microCombi Electrode (Metrohm) H+-response electrode was calibrated prior to each titration and used in conjunction with a Metrohm Titrando 907 to measure the pH of the experimental solutions. Absorption and pH measurements were taken after each incremental addition of carbonate-free 0.1 M KOH (prepared from Baker Dilut-It concentrate) by the Metrohm autoburet. The instrument set-up was completely automated by in-house titration software. All spectrophotometric titration samples were prepared under acidic conditions (∼pH 1) with 25 μM ligand buffered in HEPES, CHES, MES, and acetic acid (3 mM each) in 0.1 M ionic strength (KCl supporting electrolyte) at 25° under positive argon pressure. Solutions used for stability constants had 1 equivalent of metal (M = Eu3+). Data from each titration were imported and refined by the nonlinear least squares fitting program HypSpec (see ESI†). The stability constant of Eu:3,4,3-LI(HCCH) complex was determined by fluorimetric batch titrations. Solutions of 100 nM Eu:3,4,3-LI(HCCH), with varying equivalents of 3,4,3-LI(1,2-HOPO) (∼0.6–32) buffered in 50 mM HEPES at pH 7.4 at 0.1 M ionic strength (KCl supporting electrolyte) were prepared, allowed to equilibrate at 25° for at least 1 day (until no spectral change could be noted) and emission spectra recorded. The analytical signal used was from the Eu:3,4,3-LI(1,2-HOPO) complex, as previously described.24Author contributions
IC, AR, and RJA designed the research. IC developed the synthetic pathway for peptoid ligands and IC, AR, and BN prepared all ligands. AR and KPC performed the spectroscopic and thermodynamic analyses of metal complexes. GJPD assisted with experimental design of solution thermodynamic measurements and interpretation of thermodynamic and spectroscopic data. All authors discussed the experimental results and contributed to the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
IC and RJA are listed as inventors on a patent application filed by the Lawrence Berkeley National Laboratory and describing inventions related to the research results presented here. The authors declare no other competing financial interest.Acknowledgements
Early peptoid library development was supported by the Fuel Cycle Research and Development Campaign (FCRD)/Fuel Resources Program, Office of Nuclear Energy, the U.S. Department of Energy (DOE) under Contract No. DE-AC02-05CH11231 at the Lawrence Berkeley National Laboratory (LBNL). Spermine-based ligand development and all spectroscopic characterization were supported by the DOE, Office of Science Early Career Research Program and Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division at LBNL under Contract DE-AC02-05CH11231. Peptoid ligand purification and analysis were performed at the Molecular Foundry, a User Facility supported by the Office of Science, Office of Basic Energy Sciences, of the DOE under Contract No. DE-AC02-05CH11231. The authors thank Drs Ronald Zuckermann and Michael Connolly from the LBNL Molecular Foundry for invaluable help with peptoid preparation.References
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2016, ch. 287, vol. 50, pp. 141–176 Search PubMed.
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858–883 CrossRef CAS.
- R. D. Teo, J. Termini and H. B. Gray, J. Med. Chem., 2016, 59, 6012–6024 CrossRef CAS PubMed.
- A. J. Amoroso, I. A. Fallis and S. J. A. Pope, Coord. Chem. Rev., 2017, 340, 198–219 CrossRef CAS.
- J. A. Rees, G. J. P. Deblonde, D. D. An, C. Ansoborlo, S. S. Gauny and R. J. Abergel, Sci. Rep., 2018, 8, 4419 CrossRef PubMed.
- A. E. V. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Coord. Chem. Rev., 2014, 266–267, 171–193 CrossRef CAS.
- G. J. P. Deblonde, M. Sturzbecher-Hoehne, P. B. Rupert, D. D. An, M.-C. Illy, C. Y. Ralston, J. Brabec, W. A. de Jong, R. K. Strong and R. J. Abergel, Nat. Chem., 2017, 9, 843–849 CrossRef CAS PubMed.
- G. J.-P. Deblonde, M. P. Kelley, J. Su, E. R. Batista, P. Yang, C. H. Booth and R. J. Abergel, Angew. Chem., Int. Ed., 2018, 57, 4521–4526 CrossRef CAS PubMed.
- K. P. Carter, R. G. Surbella III, M. Kalaj and C. L. Cahill, Chem.–Eur. J., 2018, 24, 12747–12756 CrossRef CAS PubMed.
- G. J. P. Deblonde, M. Sturzbecher-Hoehne and R. J. Abergel, Inorg. Chem., 2013, 52, 8805–8811 CrossRef CAS PubMed.
- M. Sturzbecher-Hoehne, T. A. Choi and R. J. Abergel, Inorg. Chem., 2015, 54, 3462–3468 CrossRef CAS PubMed.
- I. Captain, G. J. P. Deblonde, P. B. Rupert, D. D. An, M.-C. Illy, E. Rostan, C. Y. Ralston, R. K. Strong and R. J. Abergel, Inorg. Chem., 2016, 55, 11930–11936 CrossRef CAS PubMed.
- T. A. Pham, A. B. Altman, S. C. E. Stieber, C. H. Booth, S. A. Kozimor, W. W. Lukens, D. T. Olive, T. Tyliszczak, J. Wang, S. G. Minasian and K. N. Raymond, Inorg. Chem., 2016, 55, 9989–10002 CrossRef CAS.
- G. M. Figliozzi, R. Goldsmith, S. C. Ng, S. C. Banville and R. N. Zuckermann, in Methods in Enzymology, Academic Press, 1996, vol. 267, pp. 437–447 Search PubMed.
- A. S. Knight, E. Y. Zhou, J. G. Pelton and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17488–17493 CrossRef CAS PubMed.
- A. S. Knight, E. Y. Zhou and M. B. Francis, Chem. Sci., 2015, 6, 4042–4048 RSC.
- M. Baskin and G. Maayan, Chem. Sci., 2016, 7, 2809–2820 RSC.
- B. F. Parker, A. S. Knight, S. Vukovic, J. Arnold and M. B. Francis, Ind. Eng. Chem. Res., 2016, 55, 4187–4194 CrossRef CAS.
- A. S. Knight, R. U. Kulkarni, E. Y. Zhou, J. M. Franke, E. W. Miller and M. B. Francis, Chem. Commun., 2017, 53, 3477–3480 RSC.
- M. Baskin and G. Maayan, Dalton Trans., 2018, 47, 10767–10774 RSC.
- M. Sturzbecher-Hoehne, C. Ng Pak Leung, A. D'Aleo, B. Kullgren, A.-L. Prigent, D. K. Shuh, K. N. Raymond and R. J. Abergel, Dalton Trans., 2011, 40, 8340–8346 RSC.
- R. J. Abergel, A. D'Aléo, C. Ng Pak Leung, D. K. Shuh and K. N. Raymond, Inorg. Chem., 2009, 48, 10868–10870 CrossRef CAS PubMed.
- L. J. Daumann, D. S. Tatum, B. E. R. Snyder, C. Ni, G.-l. Law, E. I. Solomon and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 2816–2819 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- V. V. Utochnikova and N. P. Kuzmina, Russ. J. Coord. Chem., 2016, 42, 679–694 CrossRef CAS.
- M. Congiu, M. Alamiry, O. Moudam, S. Ciorba, P. R. Richardson, L. Maron, A. C. Jones, B. S. Richards and N. Robertson, Dalton Trans., 2013, 42, 13537–13545 RSC.
- X. Q. Song, Q. F. Zheng, L. Wang and W. S. Liu, Luminescence, 2012, 27, 459–465 CrossRef CAS PubMed.
- R. D. Archer, H. Chen and L. C. Thompson, Inorg. Chem., 1998, 37, 2089–2095 CrossRef CAS.
- S. Sato and M. Wada, Bull. Chem. Soc. Jpn., 1970, 43, 1955–1962 CrossRef CAS.
- T. Kimura, R. Nagaishi, Y. Kato and Z. Yoshida, J. Alloys Compd., 2001, 323–324, 164–168 CrossRef CAS.
- R. J. Abergel, P. W. Durbin, B. Kullgren, S. N. Ebbe, J. Xu, P. Y. Chang, D. I. Bunin, E. A. Blakely, K. A. Bjornstad, C. J. Rosen, D. K. Shuh and K. N. Raymond, Health Phys., 2010, 99, 401–407 CrossRef CAS PubMed.
- M. A. Deri, S. Ponnala, P. Kozlowski, B. P. Burton-Pye, H. T. Cicek, C. Hu, J. S. Lewis and L. C. Francesconi, Bioconjugate Chem., 2015, 26, 2579–2591 CrossRef CAS PubMed.
- L. C. Uhlir, P. W. Durbin, N. Jeung and K. N. Raymond, J. Med. Chem., 1993, 36, 504–509 CrossRef CAS PubMed.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, LC-MS, high resolution MS, TOF-MS/MS, and NMR spectra for peptoid ligands, UV-vis and luminescence spectra for Eu and Tb peptoid and 343-LI complexes, luminescence lifetime values, example data sets from solution thermodynamic titrations, and quantum yield data are provided. See DOI: 10.1039/c9sc01068h |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2019 |