
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral diversification through the assembly of achiral phenylacetylene macrocycles with a two-fold bridge†
Ryo
Katoono
 *,
Keiichi
Kusaka
,
Yuki
Saito
,
Kazuki
Sakamoto
and
Takanori
Suzuki
*,
Keiichi
Kusaka
,
Yuki
Saito
,
Kazuki
Sakamoto
and
Takanori
Suzuki
Department of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: katoono@sci.hokudai.ac.jp; Fax: +81 11 706-2714; Tel: +81 11 706-3396
First published on 2nd April 2019
Abstract
We demonstrate so-called “chiral diversification”, which is a design strategy to create multiple chiral molecules through the assembly and double-bridging of achiral components. We used phenylacetylene macrocycles (PAMs) as an achiral element. In a molecule, two achiral rings of [6]PAM are stacked one above the other, or bound to each other mechanically. As an alternative, a single enlarged ring of [12]PAM was also assumed to be a doubled form of [6]PAM. In any case, one or two ring(s) are doubly-bridged by covalent bonds to exert chirality. Through intramolecular two-bond formation, these multiple chiral molecules were obtained as a set of products in one reaction. The dynamic chirality generated in molecules with either two helically-stacked rings of [6]PAM or a single helically-folded ring of [12]PAM was characterized by induced Cotton effects with the aid of an external chiral source. Thus, a chiral structure based on [12]PAM could be demonstrated as the first success. Alternatively, enantiomeric separation was achieved for molecules with two interlocked rings of [6]PAM to show remarkable chiroptical properties.
Introduction
Chirality is a central topic in chemistry. Once an asymmetric center is attached to a molecule, it can be chiral. On the other hand, considerable effort has been exerted to desymmetrize molecules without the use of an asymmetric center.1–8 In such molecules, chirality is achieved by the arrangement of achiral components. Especially, the assembly of cyclic components is interesting because multiple forms are possible. The best example would be helical stacking of achiral rings,9 where helical chirality is generated when one ring is stacked above the other and twisted in a clockwise or counterclockwise manner with respect to the other ring. Alternatively, mechanically linked achiral rings (catenanes) may be chiral,5,7,10–12 when the rings have directionality (topological chirality),5a,7,10 or simply one ring is relatively tilted with respect to the other ring (mechanical helical chirality).5a,12 For a case where two rings are further bridged in addition to the mechanical linkage, so called pretzelanes,13 the further bridging of the mechanically-interlocked rings plays a key role in the characterization of chirality.13aWe have been interested in a design strategy to provide multiple chiral structures through the assembly and double-bridging of achiral components.14–16 Here, we used phenylacetylene macrocycles (PAMs)17 as achiral components for the creation of multiple chiral molecules. The assembly of two rings provides multiple forms (Scheme 1). Site-specific modification of PAMs18a would be suitable for bridging of the two rings at arbitrary positions. In the case where two achiral rings of [6]PAM18 are stacked and doubly-bridged (Schemes 1 and 2a), helically-chiral structures would be created through the twisting of one ring with respect to the other ring in a clockwise or counterclockwise manner. These two enantiomeric structures would conformationally interconvert to each other. In the case where two achiral rings of [6]PAM are mechanically bound and doubly-bridged (Schemes 1 and 2b), the relative arrangement of the two rings would be defined, based on the shape-persistency of [6]PAM, and the resulting chirality would be configurationally stable. In the last case where a single larger ring of [12]PAM19 is generated (Schemes 1 and 2c), double-bridging would lead to a specifically-folded structure since such a larger ring would prefer nonplanar conformations rather than a planar conformation. To the best of our knowledge, there has been no previous report on any chiral structure based on [12]PAM, and nothing is known about its chiroptical properties.
 | ||
| Scheme 1 Generation of multiple forms through the assembly of achiral [6]PAMs. | ||
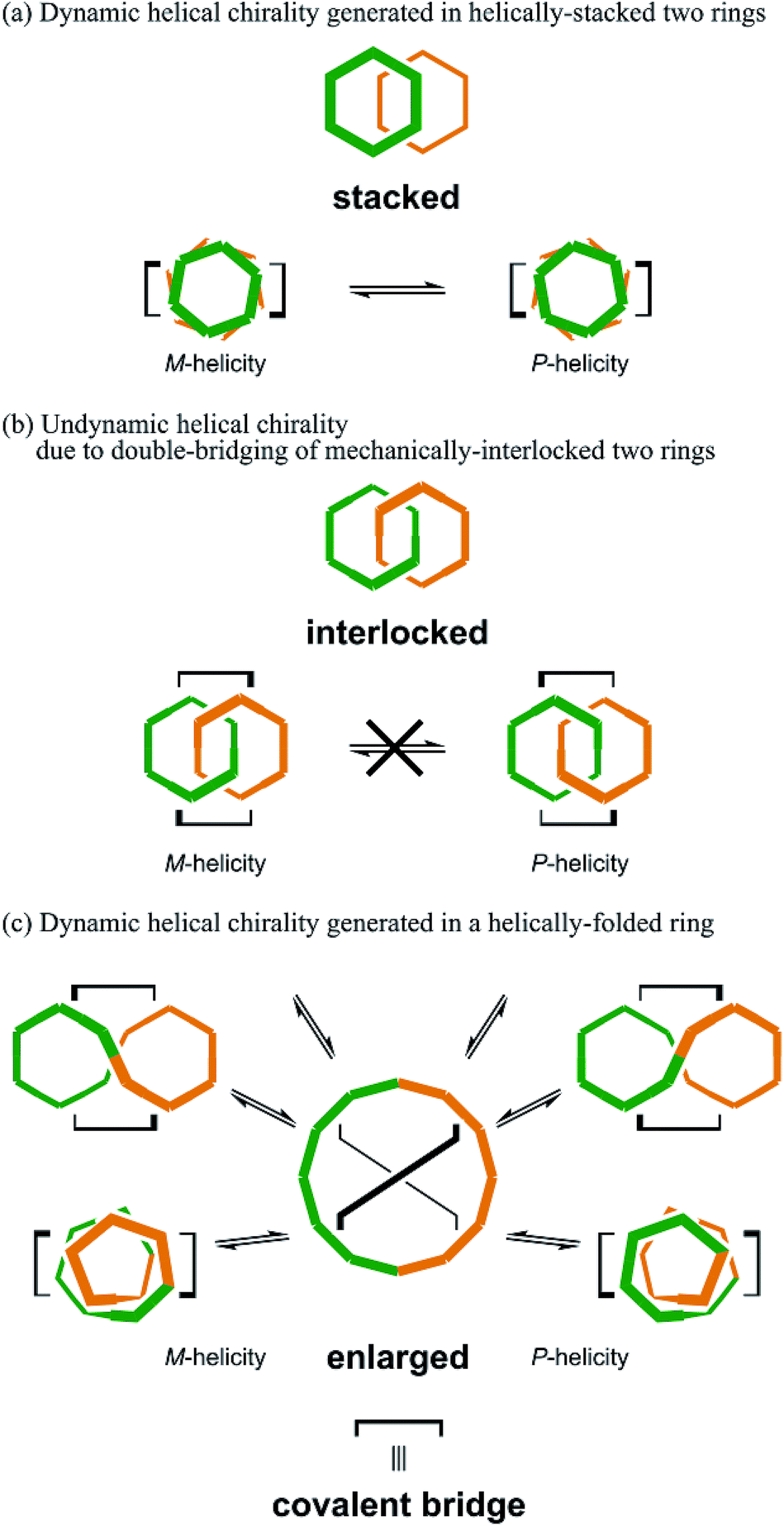 | ||
| Scheme 2 Dynamic or undynamic helical chirality generated in (a) doubly-bridged two rings of [6]PAM stacked one above the other, (b) doubly-bridged two rings of [6]PAMs mechanically-interlocked to each other, and (c) a doubly-bridged single ring of [12]PAM. | ||
Thus, we designed two series (1–5 and 6–8) of chiral molecules in which two rings of [6]PAM are stacked one above the other (1 and 6), two rings of [6]PAM are bound to each other mechanically (3–5 and 8), and a single larger ring of [12]PAM is helically-folded (2 and 7) (Fig. 1 and S1†). Double bridging not only assures the generated stereochemistry in 3–5 and 8, it is also the origin of diversification. For descriptive purposes, we numbered six benzene rings starting from either of the two benzene rings that is bridged to another [6]PAM. Double-bridging at the 1- and 3-positions led us to assume three different chiral molecules (3, 4 and 5). With double-bridging at the 1- and 4-positions, only a single chiral molecule could be designed (8). The type of chirality generated here should be classified as mechanical helical chirality due to the equivalence of the 1- and 3-, or 1- and 4-positions due to arbitrary numbering. Thus, the ring is not oriented to have any priority for the 1-position relative to the 3- or 4-position, which is necessary for topologically-chiral molecules.6,7
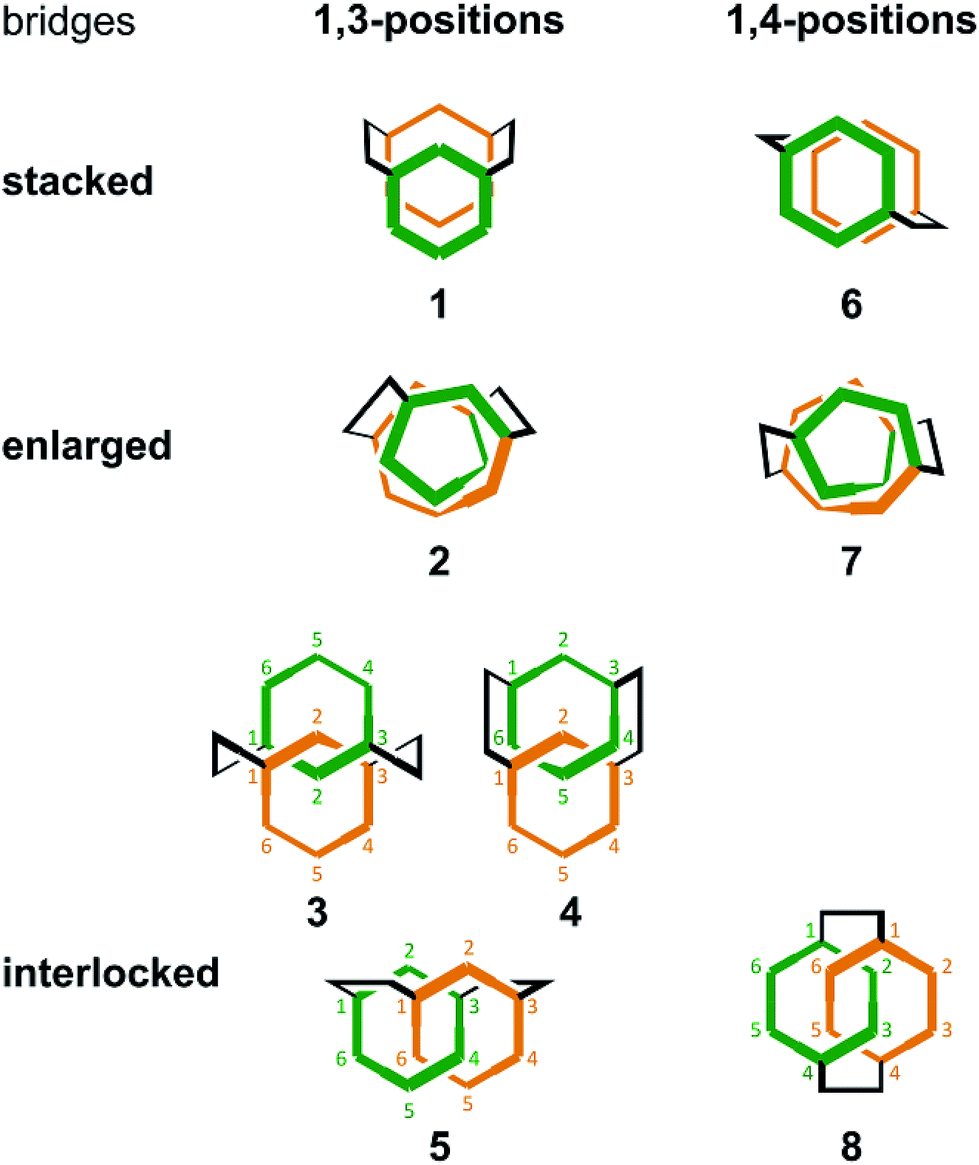 | ||
| Fig. 1 Schematic representation of 1 and 6 (stacked, [6]PAM ×2), 2 and 7 (helically-folded, [12]PAM ×1), and 3–5 and 8 (interlocked, [6]PAM ×2). Bridges were at the 1- and 3-positions of each PAM for 1–5 and at the 1- and 4-positions for 6–8. The position number is arbitrary. Only one of two enantiomeric conformers/isomers is depicted. Interlocked (±)-5 have not been synthesized. | ||
We should be able to observe the chiroptical properties of dynamic chiral molecules when they interact with an external chiral source, such as in diastereomeric complexation with a chiral guest. Through the supramolecular transmission of external chirality to dynamic chiral structures in a complex, one of two enantiomeric conformations would be preferred over the other. Based on the imbalance between their populations, chiroptical properties would be observable. Terephthalamide was used to serve as both the binding site for capturing a guest and the chirality-transmitting site in bridges. Alternatively, undynamic chiral molecules will show chiroptical properties after enantiomeric separation.
The results of chiral diversification through the assembly of achiral PAMs with a two-fold bridge, molecular structures and chiroptical properties are described below.
Results and discussion
Synthesis
Templated synthesis is one of the most important methods for producing sophisticated molecules.11,20 Also, a “covalent template-strategy” would be useful.21 By considering the case of an intramolecular two-fold ring-closure reaction,15 we assumed that there could be multiple products in which two bonds are formed: a pair of rings and a single enlarged ring (Scheme 3). | ||
| Scheme 3 Synthetic strategy based on intramolecular two-bond formation (“covalent template-strategy”). | ||
For intramolecular two-bond formation (covalent template-strategy), we designed achiral precursors 9 and 10 (Scheme 3 and Fig. 2). They are composed of two linear [6]phenylacetylene (PA) sequences that are covalently assembled to form a macrocyclic framework. Two-bond formation was implemented by a two-fold Sonogashira coupling reaction of α-iodo-ω-ethynyl sequences.18a Based on the difference in the relative direction of the paired sequences, two achiral isomers, a (C2h) and b (C2v), were possible for both 9 and 10. We can assume two reaction pathways I and II (Scheme 3 and Fig. 2). One will give two rings of [6]PAM and the other will give a single larger ring of [12]PAM with or without crossing. These two pathways would be competitive if the conformational flexibility of the linear sequences remained in the macrocyclic framework. Thus, we expected at most five helically-chiral molecules (1–5 with a two-fold bridge at the 1- and 3-positions) from 9, and three helically-chiral molecules (6–8 with a two-fold bridge at the 1- and 4-positions) from 10.
 | ||
| Fig. 2 Chemical structures of ring-closing precursors 9a/b and 10a/b. | ||
Achiral ring-closing precursors 9a and 9b were separable from each other.22 A set of three products (1, 3 and 4) were obtained from 9a (Scheme 4a). All of them were considered to generate via pathway I. This result indicated that pathway II was invalid, which could have led to the generation of a [12]PAM without crossing. Another set of products (2, 3 and 4) were obtained from 9b (Scheme 4a). Compound 5 was not obtained from either 9a or 9b.
 | ||
| Scheme 4 Synthetic results of the intramolecular ring-closure reactions with (a) 9a or 9b, or (b) a mixture of 10a and 10b. aPseudo-high-dilution conditions: Pd(PPh3)4, CuI, Et3N, THF, 80 °C. bFolded 2 and interlocked (±)-5 could not be found. cStacked 1 and interlocked (±)-5 could not be found. | ||
Achiral ring-closing precursors 10a and 10b were inseparable (approx. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture), and they were subjected to the final reaction together. A set of three products (6, 7 and 8) were obtained (Scheme 4b).
:1 mixture), and they were subjected to the final reaction together. A set of three products (6, 7 and 8) were obtained (Scheme 4b).
Products 1–4 each exhibited a specific retention time on silica gel, and could be successfully isolated through HPLC separation. Similar separation was applied to 6–8. The 1H NMR spectra of 1–4 and 6–8 showed that they were distinct from each other (Fig. 5, described later). Mass analyses showed that they each had a molecular weight of 1746. Based on these results, we could confirm that products 1–4 and 6–8 were all isomers with an identical composition (C128H88N4O4). Enantiomeric separation was possible only for (±)-3 and (±)-8. Unfortunately, racemates (±)-4 remained inseparable even with considerable effort.23
Model rings of [6]PAM 11 (ref. 16a) and 12 were prepared for comparison of NMR and UV spectra. A model macrocycle 13 is composed of two halves of a [6]PA sequence that are doubly-bridged, and was used for monitoring complexation by 1H NMR spectroscopy, since most protons were well-assigned. Chiral ditopic guests (R,R)-14 and (S,S)-14 were used as an external chiral source.16 They could be captured by the formation of hydrogen bonds at terephthalamide bridges (Fig. 3, Schemes S1, S3 and S4†).
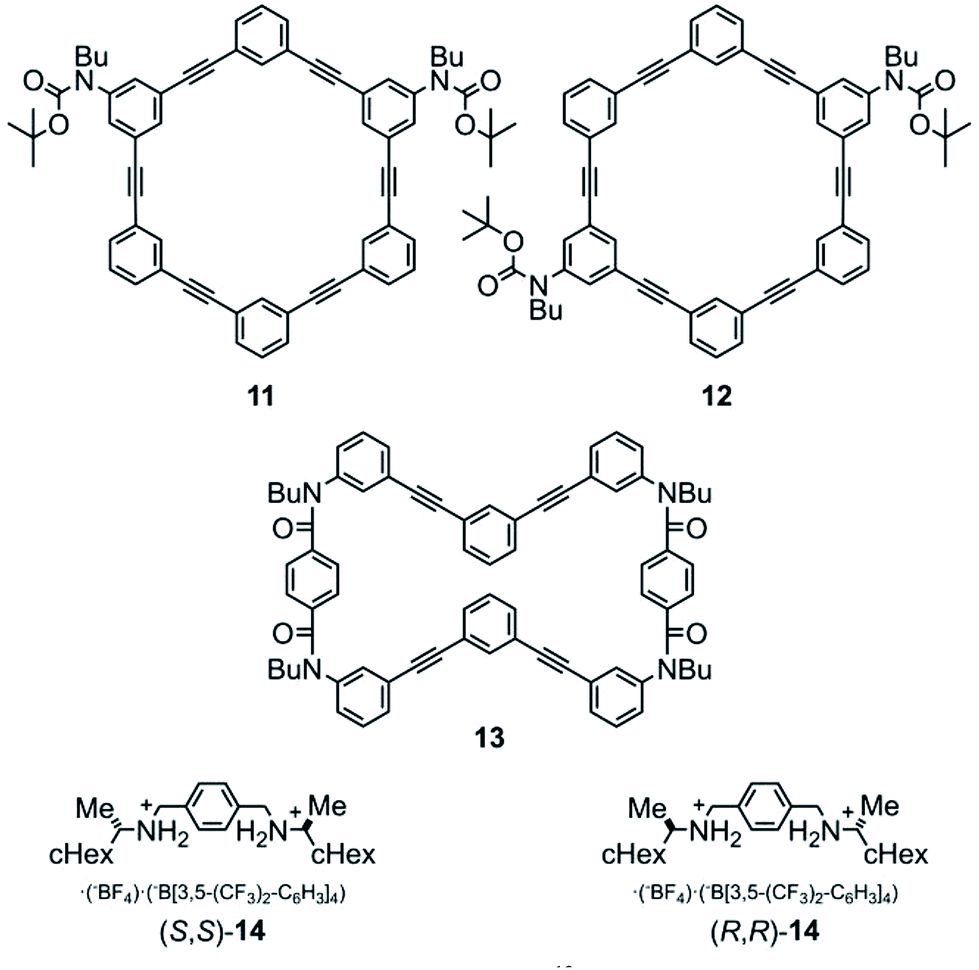 | ||
| Fig. 3 Chemical structures of model [6]PAMs 11 (ref. 16a) and 12, a model macrocycle 13, and chiral guests (R,R)-14 and (S,S)-14.16 | ||
Molecular structures
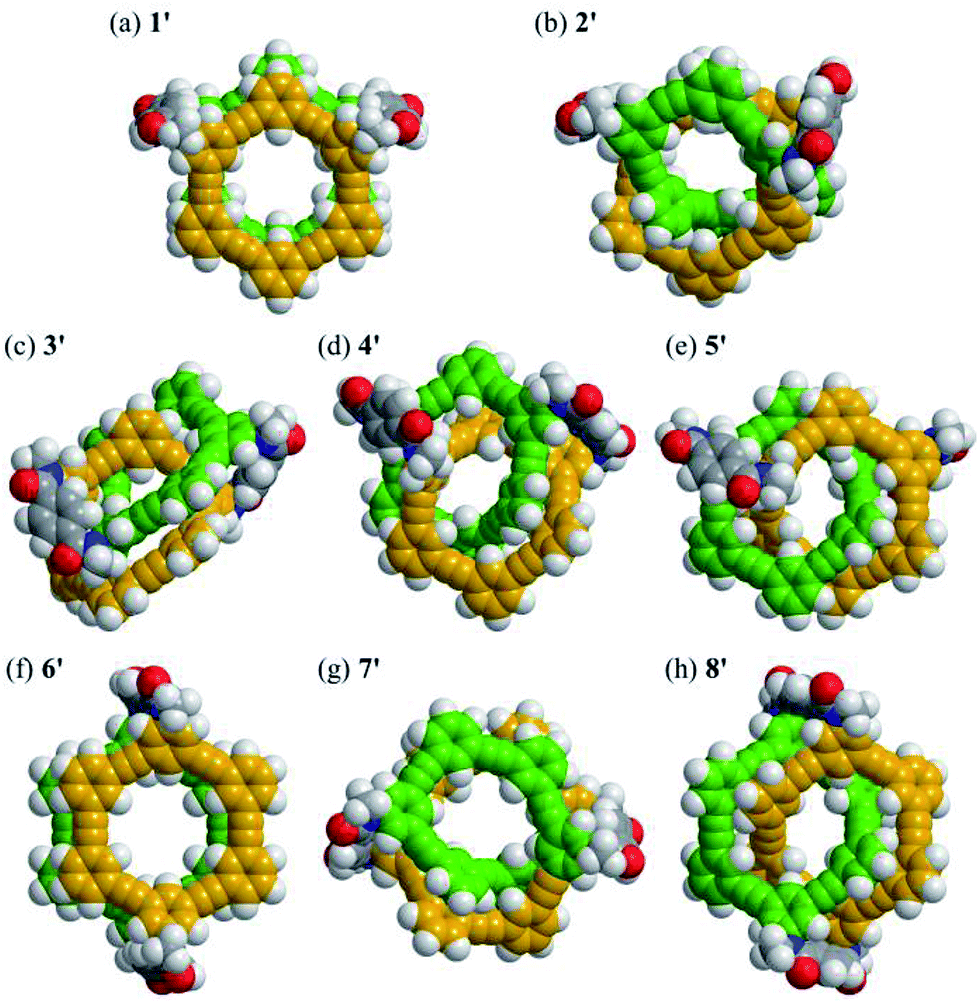 | ||
| Fig. 4 The lowest energy structures for models 1′–8′ (NMe, C116H64N4O4): (a) 1′ (+6.5 kJ mol−1), (b) 2′ (+30.6 kJ mol−1), (c) 3′ (+24.0 kJ mol−1), (d) 4′ (+5.0 kJ mol−1), (e) 5′ (+14.6 kJ mol−1), (f) 6′ (+5.4 kJ mol−1), (g) 7′ (+23.0 kJ mol−1) and (h) 8′ (rel. 0 kJ mol−1), obtained by conformational searches using MacroModel software (v11.8 OPLS3e, Monte Carlo Multiple Minimum method, non-solvated, 100000 steps). | ||
For models 1′ and 6′ with two rings of [6]PAM stacked one above the other, the lowest energy structures were found to be achiral (Fig. 4a and f). Alternatively, a helically-chiral structure was found at a higher energy level for both model isomers (Fig. S1† and S2B†). These achiral and helically-chiral structures would be conformationally accessible to each other.
For models 2′ and 7′ with a larger ring of [12]PAM, helically-folded structures were found. The global chirality seems to enable the bridges to adopt only one form with a particular sense of local helicity and/or a non-helical form (Fig. S2C†). If so, the inversion of local helicity in the bridge would accompany the inversion of global helicity, similar to 1′ and 6′.
For models 3′, 4′, 5′ and 8′ with two rings of [6]PAM bound to each other mechanically, chiral structures were definitely found. No rings adopted a planar conformation. While dynamic motion in the bridge would lead to a different structure, the stereochemistry of these molecules should be maintained (Fig. S1 and S2D†).
For model 13′, several structures were found within a very tight range of energy levels (Fig. S2A†). It seems that this would be desirable for a synthetic precursor to generate multiple structures in the current system. In fact, similar conformations were found in the structures of 1′ and 2′.
 | ||
| Fig. 5 Partial 1H NMR spectra (400 MHz) of (a) 1–4 and 11, and (b) 6–8 and 12, measured in chloroform-d at room temperature. | ||
Integral values for inner protons (HA, HB and HC) of 1 showed that the two rings of [6]PAM were equivalent. At lower temperatures, several species in different populations were observed. They were averaged with a rise in temperature through dynamic conformational interconversions. The two rings of [6]PAM in 6 were also equivalent. The different chemical shifts for HA and HA′ show a situation where one ring is stacked away from right above the other.
Although broadened resonances for 2 provided less information on the structure, the chemical shift values for most protons were maintained to some extent over the entire range of temperatures. This suggested the existence of a dominant conformation in solution. This suggestion also applied to 7.
At each temperature examined, we observed only one singlet peak for phenylene protons (HT) of terephthalamide in 3 and 8, while the corresponding protons in 4 were split to be nonequivalent (HT and HT′) below 283 K. This was used to determine 3 or 4.
Possible explanations for the identification of 1–4 were discussed based on not only less informative results of NMR spectroscopy, but also several preliminary synthetic results with the use of model intermediates with a single ring of [6]PAM (Scheme S2) in the ESI_1.†
For model macrocycle 13, we observed a single set of averaged resonances over the entire range of temperatures. Most protons were well-assigned (Fig. S5h and S6a†). The chemical shift of averaged resonances for some protons changed with temperature. This was attributed to dynamic interconversion among several conformations with different structures.
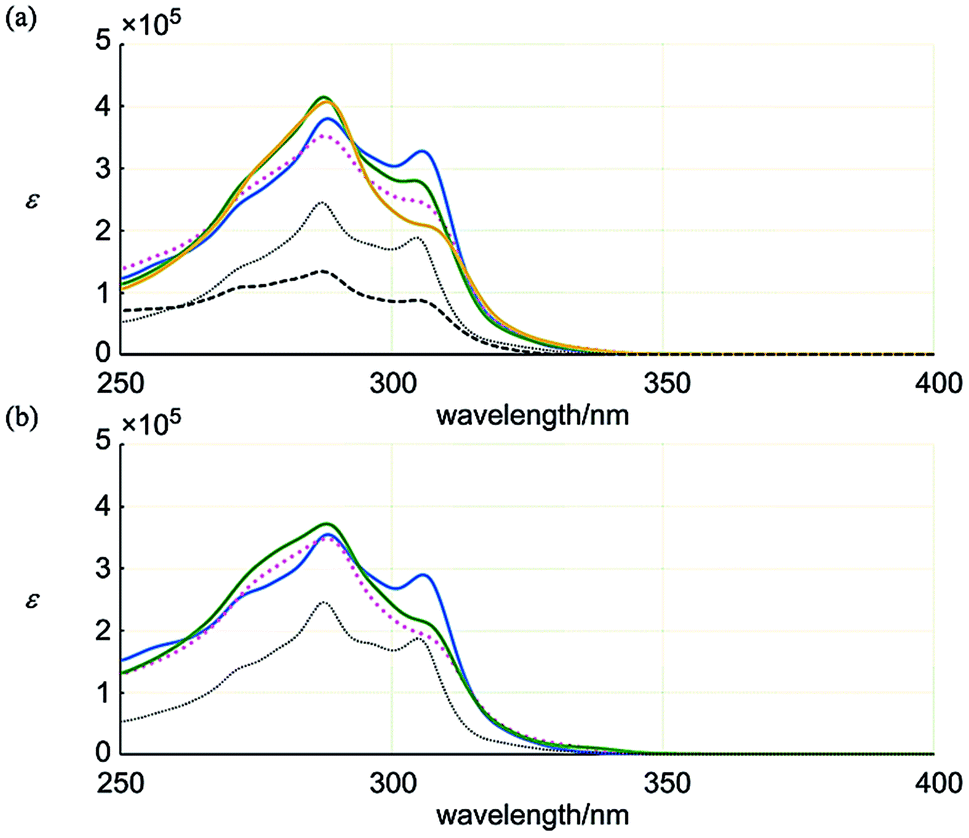 | ||
| Fig. 6 UV spectra of (a) 1 (blue solid line), 2 (pink dotted line), 3 (green solid line), 4 (yellow solid line), 11 (black thin dotted line) and 13 (black thick dashed line), and (b) 6 (blue solid line), 7 (pink dotted line), 8 (green solid line) and 12 (black thin dotted line). All spectra were measured in dichloromethane at room temperature. | ||
Next, we considered the absorption intensity based on the ratio of molar extinction coefficients ε1 and ε2 (Table 1). The value for 6 was close to that of 12 with a single ring of [6]PAM. This similarity indicated that the two rings of [6]PAM in 6 behaved electronically-independently to maintain the original electronic property in a stacked structure of 6. The close values for 7 and 8, which were distinct from that of 6, suggests that they share some common physical features.
| λ max/nm | ε 1/105 | λ max/nm | ε 2/105 | ε 1/ε2 | |
|---|---|---|---|---|---|
| 1 | 306 | 3.28 | 288 | 3.81 | 0.86 |
| 2 | 306 | 2.44 | 288 | 3.52 | 0.69 |
| 3 | 304 | 2.81 | 287 | 4.15 | 0.68 |
| 4 | 309 | 1.97 | 288 | 4.08 | 0.48 |
| 11 | 305 | 1.88 | 287 | 2.45 | 0.77 |
| 13 | 305 | 0.879 | 287 | 1.35 | 0.65 |
| 6 | 306 | 2.89 | 288 | 3.54 | 0.82 |
| 7 | 308 | 1.83 | 288 | 3.48 | 0.53 |
| 8 | 308 | 2.02 | 288 | 3.72 | 0.54 |
| 12 | 305 | 1.87 | 287 | 2.45 | 0.77 |
In the series 1–4, none of the structures had a value close to that of 11. These structures, especially 4, might undergo some physical perturbation that deviates from the single ring of [6]PAM itself.
Chiroptical properties
:97 (v/v) acetonitrile-d3/chloroform-d, upfield shifts were induced for both the central phenylene protons (HF) in the bridge of 13 and the central phenylene protons (Ha) in 14. This result showed that the ditopic guest was captured at the two amide groups in the bridge. For other protons far from the binding site, several complexation-induced shifts were observed. This result showed a change in populations of several conformations at equilibrium before and after complexation. Based on these complexation-induced changes in the chemical shift, Job plots were created to confirm that the stoichiometry of a complex was 1:2. Through a titration experiment, the binding constants were estimated (K1 = 5.8 × 102 M−1, K2 = 1.5 × 103 M−1) (Fig. S6a†).24 We considered that the sigmoidal curve was caused by a change in the conformation of 13 upon complexation.
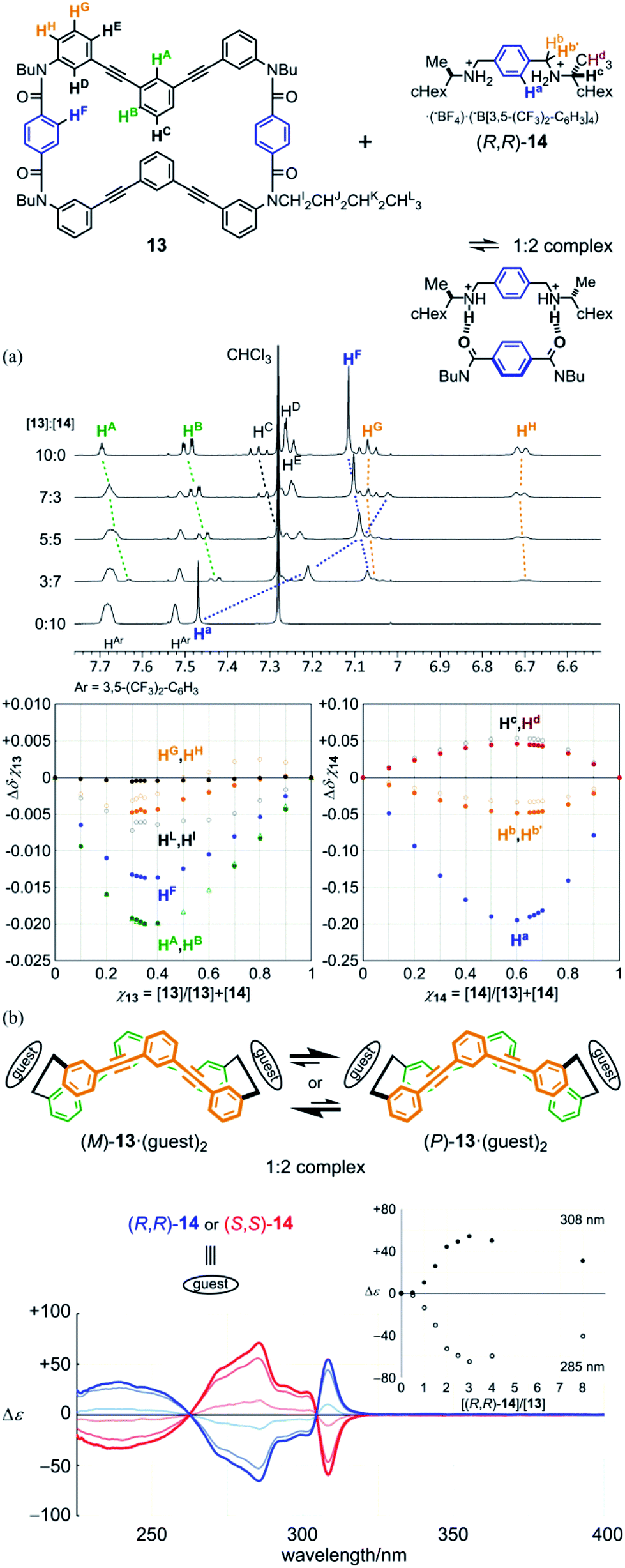 | ||
| Fig. 7 Complexation of model macrocycle 13 with chiral guests (R,R)-14 or (S,S)-14: (a) partial 1H NMR spectra of 13 in the presence of (R,R)-14 [[13]:[14] = 0:10 (14 only), 3:7, 5:5, 7:3 and 10:0 (13 only)], and Job plots ([13] + [(R,R)-14] = 2 mM, measured in 3:97 (v/v) acetonitrile-d3/chloroform-d at 303 K); (b) CD spectra of 13 (1.3 × 10−4 M) in the presence of (R,R)-14 (blue lines: 1, 2 and 3 equiv.) or (S,S)-14 (red lines), measured in dichloromethane at 293 K [inset: plots of induced molar CD versus equivalents of guest added]. | ||
Next, we monitored 1:2 complexation, at a lower concentration in dichloromethane, by UV and CD spectroscopy. When the guest (R,R)-14 was added to a solution of 13, we observed changes in the appearance of the UV spectrum of 13 (Fig. S6b†). This result showed that dynamic equilibrium among conformations was perturbed upon complexation, as shown by changes in the chemical shift in 1H NMR spectroscopy. In the CD spectrum of 13 (Fig. 7b), Cotton effects were induced in the absorption region of 13. During addition of the guest at up to 3 equivalents, while the appearance of the spectrum was maintained, the intensity increased with an increase in the concentration of the guest. If we consider that Cotton effects originated only from chiral species, any intermediary complex, e.g., a 1:1 complex, was negligible due to the positive allosterism. Mirror images were obtained by addition of the antipodal guest (S,S)-14. This result showed that model macrocycle 13 could adopt dynamic chiral conformations [(M)-13 and (P)-13]. When it formed diastereomeric complexes with a particular chiral guest [(M)-13·(R,R)-142 and (P)-13·(R,R)-142], one complex with a particular chiral sense was preferred over the other. There is no chiral-sense preference in 13 itself. In an earlier stage of the addition of the guest, Cotton effects were sigmoidally increased (Fig. 7b, inset), as in the NMR titration experiment. The induced Cotton effects decreased to some extent in the presence of abundant equivalents. This decrease in intensity was attributed to the emergence of higher-ordered complexes [1:n (n > 2)], in which some ditopic guest was forced to be bound in a monotopic manner, and was not enabled to transfer its chirality. At room temperature or above,25 induced Cotton effects were attenuated with an increase in temperature (Fig. S6c†), as often seen in most systems based on dynamic equilibrium.
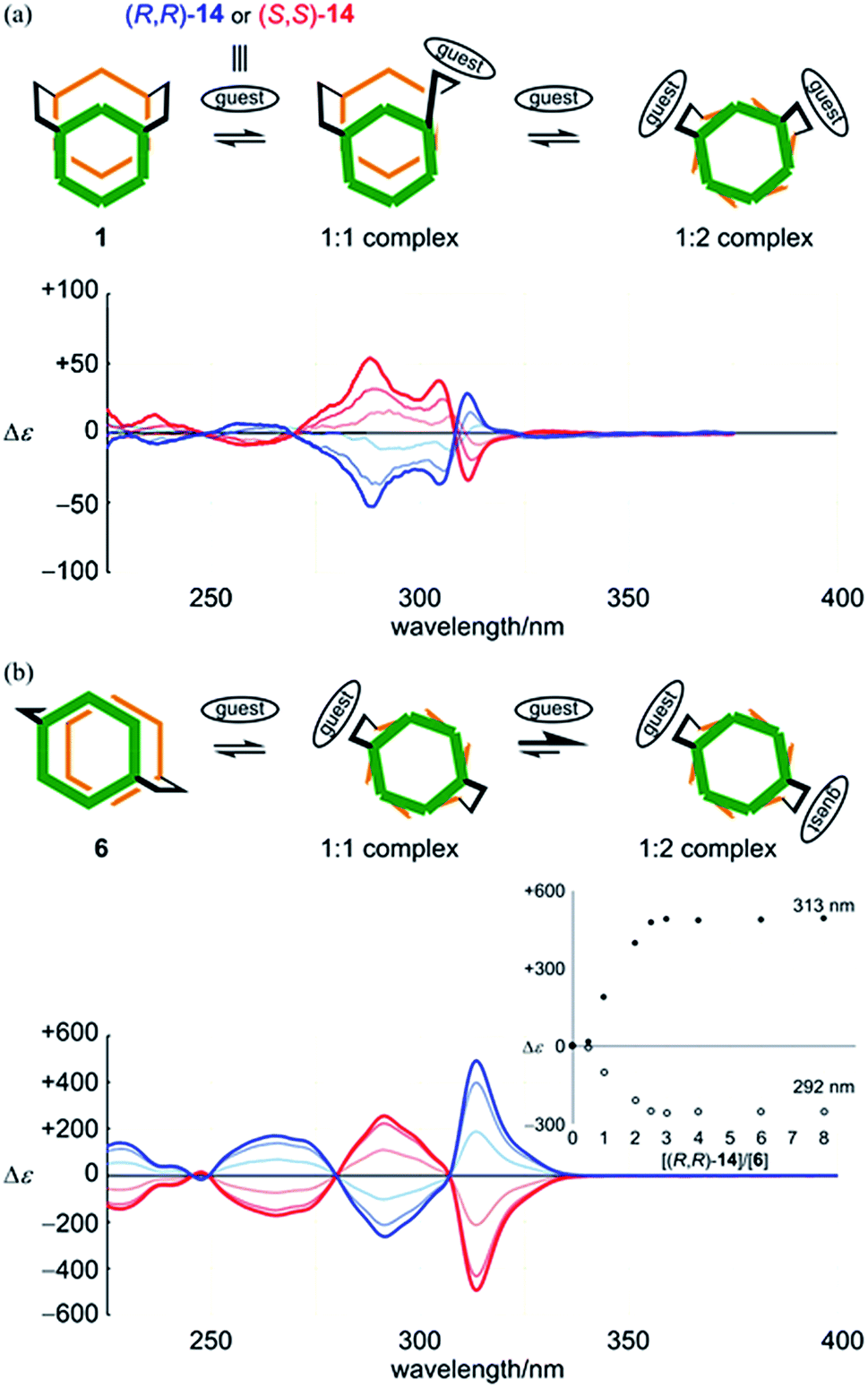 | ||
| Fig. 8 CD spectra of (a) 1 (4.4 × 10−5 M) and (b) 6 (4.8 × 10−5 M) in the presence of (R,R)-14 (blue lines: 1, 2 and 4 equiv. for 1, 1, 2 and 3 equiv. for 6) or (S,S)-14 (red lines), measured in dichloromethane at 293 K [inset: plots of induced molar CD versus equivalents of the guest added]. | ||
Alternatively, in the case of 6, the addition of a guest led to a continuous change in the UV spectrum of 6 (Fig. S7b†), and Cotton effects were induced in the absorption region of 6 while the appearance of the CD spectrum was maintained (Fig. 8b). Based on these results, along with those in the 1H NMR spectrum, we considered that there was a change in conformation of 6 from an achiral conformation in the absence of a guest, and thus it could adopt chiral conformations in a complexed state. CD titration curves were obviously sigmoidal (Fig. 8b, inset). The intensity of induced Cotton effects was noteworthy and reminiscent of helical columnar assemblies,9c,d even though there existed only two rings stacked helically. VT-CD measurements showed that induced Cotton effects were attenuated or enhanced with an increase or decrease in temperature, while the appearance of the spectrum was maintained (Fig. S7d†). This was the result of dynamic interconversion between two diastereomeric complexes with (M)- or (P)-helicity. For both 1 and 6, an antipodal guest led to the induction of mirror-imaged Cotton effects. Thus, the dynamic chirality of 1 and 6 was elucidated.
 | ||
| Fig. 9 CD spectra of (a) 2 (4.8 × 10−5 M) and (b) 7 (4.7 × 10−5 M) in the presence of (R,R)-14 (blue lines: 0.5, 1 and 2 equiv. for 2, 1, 2 and 2.5 equiv. for 7) or (S,S)-14 (red lines), measured in dichloromethane at 293 K [inset: plots of induced molar CD versus equivalents of the guest added]. | ||
Alternatively, we found no change in absorption upon the addition of a guest to a solution of 7 (Fig. S8b†). Induced Cotton effects resembled those for a complex of 2 (Fig. 9). Based on these results, we considered that there was no change in the conformation of 7 upon complexation, and that a helical conformation was adopted predominantly regardless of whether the guest was present or not. The guest simply led to a helical-sense preference in a complexed state. Mirrored images were given by the addition of an antipodal guest. Thus, we concluded that helically-chiral conformations existed in a larger ring of [12]PAM, and they were dynamically interconvertible.
 | ||
| Fig. 10 CD spectra of (a) (+)-3 (solid line, first fraction) and (−)-3 (dashed line, second fraction), and (b) (+)-8 (solid line, first fraction) and (−)-8 (dashed line, second fraction), measured in dichloromethane at 293 K. | ||
Conclusions
Based on the assembly of achiral cyclic components, multiple forms were generated: two rings of [6]PAM or a single larger ring of [12]PAM. In a molecule, these achiral pieces were connected by a two-fold covalent bridge to exert chirality. Based on a difference in the bridging positions (1- and 3-, or 1- and 4-positions) on PAMs, two series of chiral molecules were demonstrated (1–5 and 6–8). The bridging position itself contributed to the diversification of chiral molecules, as shown by 3, 4 and 5. As always, it is important to seek to obtain the target molecule selectively. Here, we have considered competitive reactions in the final step of synthesis for the generation of multiple products. Thus, we obtained seven chiral isomers with the same molecular weight. A methodology for obtaining such a series of chiral molecules would be useful for the consideration of chiroptical properties based on definite molecular structures.Dynamic chirality was demonstrated by two helically-stacked rings of [6]PAM (1 and 6). Especially in 6, the intensity of induced Cotton effects was strikingly great, even with only two-molecule stacking, which was comparable to that of helical columnar assemblies of [6]PAMs. Through a successful enantiomeric separation for two interlocked rings of [6]PAM (3 and 8), we observed remarkable Cotton effects and specific optical rotations. Different values of [α]D were obtained for 3 and 8, which had an identical composition. Although there has been no previous report of a chiral structure based on [12]PAM, we observed unique Cotton effects for helically-folded forms in [12]PAM (2 and 7). The enantiomeric conformations of [12]PAM were dynamically interconvertible to each other.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Testa, Helv. Chim. Acta, 2013, 96, 351 CrossRef CAS.
- (a) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418 RSC; (b) J. Yea and S. Ma, Org. Chem. Front., 2014, 1, 1210 RSC.
- (a) T. Kakuta, T. Yamagishi and T. Ogoshi, Chem. Commun., 2017, 53, 5250 RSC; (b) M. Nakazaki, K. Yamamoto, S. Tanaka and H. Kametani, J. Org. Chem., 1977, 42, 287 CrossRef CAS; (c) M. Shibahara, M. Watanabe, T. Iwanaga, T. Matsumoto, K. Ideta and T. Shinmyozu, J. Org. Chem., 2008, 73, 4433 CrossRef CAS PubMed.
- (a) J. OuYang and J. Crassous, Coord. Chem. Rev., 2018, 376, 533 CrossRef CAS; (b) K. Kato, Y. Segawa and K. Itami, Synlett, 2019, 30, 370 CrossRef CAS; (c) M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2016, 45, 1542 RSC; (d) J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824 RSC; (e) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS PubMed; (f) S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3223 CrossRef CAS.
- (a) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266 RSC; (b) N. H. Evans, Chem.–Eur. J., 2018, 24, 3101 CrossRef CAS PubMed.
- (a) R. Schmieder, G. Hübner, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 3528 CrossRef CAS; (b) C. Reuter, W. Wienand, C. Schmuck and F. Vögtle, Chem.–Eur. J., 2001, 7, 1728 CrossRef CAS PubMed; (c) N. Kameta, K. Hiratani and Y. Nagawa, Chem. Commun., 2004, 466 RSC; (d) L. D. Movsisyan, M. Franz, F. Hampel, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, J. Am. Chem. Soc., 2016, 138, 1366 CrossRef CAS PubMed.
- (a) Y. Kaida, Y. Okamoto, J.-C. Chambron, D. K. Mitchell and J. P. Sauvage, Tetrahedron Lett., 1993, 34, 1019 CrossRef CAS; (b) C. Yamamoto, Y. Okamoto, T. Schmidt, R. Jäger and F. Vögtle, J. Am. Chem. Soc., 1997, 119, 10547 CrossRef CAS; (c) E. Alcalde, L. Pérez-García, S. Ramos, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 2007, 13, 3964 CrossRef CAS PubMed.
- (a) F. B. L. Cougnon, K. Caprice, M. Pupier, A. Bauzá and A. Frontera, J. Am. Chem. Soc., 2018, 140, 12442 CrossRef CAS PubMed; (b) S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Angew. Chem., Int. Ed., 2017, 56, 11166 CrossRef CAS PubMed; (c) J. Guo, P. C. Mayers, G. A. Breault and C. A. Hunter, Nat. Chem., 2010, 2, 218 CrossRef CAS PubMed; (d) M. Feigel, R. Ladberg, S. Engels, R. Herbst-Irmer and R. Fröhlich, Angew. Chem., Int. Ed., 2006, 45, 5698 CrossRef CAS PubMed; (e) O. Lukin and F. Vögtle, Angew. Chem., Int. Ed., 2005, 44, 1456 CrossRef CAS PubMed; (f) G. Rapenne, C. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1996, 118, 10932 CrossRef CAS.
- (a) H. Yamagishi, T. Fukino, D. Hashizume, T. Mori, Y. Inoue, T. Hikima, M. Takata and T. Aida, J. Am. Chem. Soc., 2015, 137, 7628 CrossRef CAS PubMed; (b) A. Pop, M. O. Vysotsky, M. Saadioui and V. Böhmer, Chem. Commun., 2003, 1124 RSC; (c) X. Zhou, G. Liu, K. Yamato, Y. Shen, R. Cheng, X. Wei, W. Bai, Y. Gao, H. Li, Y. Liu, F. Liu, D. M. Czajkowsky, J. Wang, M. J. Dabney, Z. Cai, J. Hu, F. V. Bright, L. He, X. C. Zeng, Z. Shao and B. Gong, Nat. Commun., 2012, 3, 949 CrossRef PubMed; (d) Y. Zhong, Y. Yang, Y. Shen, W. Xu, Q. Wang, A. L. Connor, X. Zhou, L. He, X. C. Zeng, Z. Shao, Z. Lu and B. Gong, J. Am. Chem. Soc., 2017, 139, 15950 CrossRef CAS PubMed.
- T. Prakasam, M. Lusi, E. Nauha, J.-C. Olsen, M. Sy, C. Platas-Iglesias, L. J. Charbonnière and A. Trabolsi, Chem. Commun., 2015, 51, 5840 RSC.
- (a) P. R. Ashton, A. S. Reder, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1993, 115, 5286 CrossRef CAS; (b) A. Theil, C. Mauve, M.-T. Adeline, A. Marinetti and J.-P. Sauvage, Angew. Chem., Int. Ed., 2006, 45, 2104 CrossRef CAS; (c) Y. Okada, Z. Miao, M. Akiba and J. Nishimura, Tetrahedron Lett., 2006, 47, 2699 CrossRef CAS; (d) Y. Nakatani, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2010, 49, 5463 CrossRef CAS PubMed; (e) R. Mitra, M. Thiele, F. Octa-Smolin, M. C. Letzel and J. Niemeyer, Chem. Commun., 2016, 52, 5977 RSC.
- (a) H.-R. Tseng, S. A. Vignon, P. C. Celestre, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 2003, 9, 543 CrossRef CAS PubMed; (b) S. A. Vignon, J. Wong, H.-R. Tseng and J. F. Stoddart, Org. Lett., 2004, 6, 1095 CrossRef CAS PubMed.
- (a) Y. Liu, S. A. Vignon, X. Zhang, K. N. Houk and J. F. Stoddart, Chem. Commun., 2005, 3927 RSC; (b) C. Reuter, A. Mohry, A. Sobanski and F. Vögtle, Chem.–Eur. J., 2000, 6, 1674 CrossRef CAS PubMed.
- (a) W. Zhang, A. Abdulkarim, F. E. Golling, H. J. Räder and K. Müllen, Angew. Chem., Int. Ed., 2017, 56, 2645 CrossRef CAS PubMed; (b) T. Prakasam, M. Lusi, M. Elhabiri, C. Platas-Iglesias, J.-C. Olsen, Z. Asfari, S. Cianférani-Sanglier, F. Debaene, L. J. Charbonnière and A. Trabolsi, Angew. Chem., Int. Ed., 2013, 52, 9956 CrossRef CAS PubMed; (c) N. C. Habermehl, D. J. Eisler, C. W. Kirby, N. L.-S. Yue and R. J. Puddephatt, Organometallics, 2006, 25, 2921 CrossRef CAS.
- (a) A. Bogdan, M. O. Vysotsky, T. Ikai, Y. Okamoto and V. Böhmer, Chem.–Eur. J., 2004, 10, 3324 CrossRef CAS PubMed; (b) O. Molokanova, A. Bogdan, M. O. Vysotsky, M. Bolte, T. Ikai, Y. Okamoto and V. Böhmer, Chem.–Eur. J., 2007, 13, 6157 CrossRef CAS PubMed.
- (a) R. Katoono, Y. Obara, K. Kusaka and T. Suzuki, Chem. Commun., 2018, 54, 735 RSC; (b) R. Katoono, S. Kawai and T. Suzuki, Chem. Sci., 2016, 7, 3240 RSC.
- (a) A. L. Sadowy and R. R. Tykwinski, in Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, ch. 6, pp. 185–231 Search PubMed; (b) C. S. Jones, M. J. O'Connor and M. M. Haley, in Acetylene Chemistry. Chemistry, Biology and Material Science, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, ch. 8, pp. 303–385 Search PubMed.
- (a) J. Zhang, D. J. Pesak, J. L. Ludwick and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 4227 CrossRef CAS; (b) H. A. Staab and K. Neunhoeffer, Synthesis, 1974,(6), 424 CrossRef CAS.
- Z. Wu, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1992, 114, 8732 CrossRef.
- (a) C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1984, 106, 3043 CrossRef CAS; (b) D. G. Hamilton, J. E. Davies, L. Prodi and J. K. M. Sanders, Chem.–Eur. J., 1998, 4, 608 CrossRef CAS; (c) M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897 RSC; (d) G. Götz, X. Zhu, A. Mishra, J.-L. Segura, E. Mena-Osteritz and P. Bäuerle, Chem.–Eur. J., 2015, 21, 7193 CrossRef PubMed.
- (a) C. Schweez and S. Höger, Chem.–Eur. J., 2018, 24, 12006 CrossRef CAS PubMed; (b) Ö. Ünsal and A. Godt, Chem.–Eur. J., 1999, 5, 1728 CrossRef; (c) S.-H. Li, H.-Y. Zhang, X. Xu and Y. Liu, Nat. Commun., 2015, 6, 7590 CrossRef PubMed.
- In a preliminary synthetic trial, we confirmed that a set of four products 1–4 were obtained from an approx. 1:1 mixture of 9a and 9b.
- It was impossible to separate the racemates 4 even though we tried many conditions with several columns and eluents: CHIRALPAK IA, IB, IC, ID, IE, IF, IG and IH, commercially available from DAICEL Co., Japan.
- S. Akine, TitrationFit, program for analyses of host–guest complexation, Kanazawa University, Kanazawa, Japan, 2013 Search PubMed.
- Below room temperature, Cotton effects were slightly attenuated (Fig. S6c†). This was a specific case for highly-flexible model 13. This result suggested that higher-ordered complexation through hydrogen bonding was preferred at lower temperatures to reach a situation similar to that in the presence of abundant equivalents of guest.
- (a) J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793 CrossRef CAS PubMed; (b) R. B. Prince, S. A. Barnes and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 2758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI_1: details of energy-minimized structures, NMR and CD spectroscopic data (Fig. S1–S9), and experimental details of new compound synthesis (Scheme S1–S4), ESI_2: copies of 1H and 13C NMR, IR and mass spectra of new compounds. See DOI: 10.1039/c9sc00972h |
| This journal is © The Royal Society of Chemistry 2019 |