DOI:
10.1039/C9SC00910H
(Edge Article)
Chem. Sci., 2019,
10, 7156-7162
Introduction of the α-ketoamide structure: en route to develop hydrogen peroxide responsive prodrugs†
Received
21st February 2019
, Accepted 28th June 2019
First published on 28th June 2019
Abstract
Leveraging the elevated levels of hydrogen peroxide (H2O2) in cancer, inflammatory diseases and cardiovascular disorders, H2O2-activated promoieties have been widely used in drugs and biomaterials design. However, the overwhelming majority of the promoieties only share the common structure of a H2O2-responsive arylboronic acid/ester moiety with low diversity. We report here an unprecedented strategy to construct novel H2O2-responsive prodrugs based on an α-ketoamide structure. As a proof of concept, we designed and synthesized a panel of α-ketoamide based nitrogen mustard prodrugs, among which KAM-2 showed potent growth inhibitory activity and high selectivity toward cancer cells. The H2O2-trigged decomposition of KAM-2 was validated, and the DNA damaging and apoptosis promoting activity attributed to the released nitrogen mustard were demonstrated. Our work unveils α-ketoamide as a new scaffold for prodrug design and may quickly inspire future developments.
Introduction
Prodrugs and probes, two important and vibrant research subjects in medicinal chemistry and analytical chemistry, are indeed closely related. Probes are usually trigged by specific analytes to release fluorescent signals, and prodrugs are activated by pathological stimuli to liberate drugs.1,2 Importantly, chemists are always encouraged by the analyte-responsive moieties of probes and expand their applications to prodrug design to achieve spatiotemporal control of drug release. Recent examples include the intriguing discoveries of prodrugs activated by β-secretase,3 histone deacetylase (HDAC)/cathepsin L (CTSL),4 matrix metalloproteinase-7 (MMP-7),5 caspase-3,6 γ-glutamyl transferase,7 thioredoxin reductase,8 lysyloxidase,9 photocleavage,10 hypochlorous acid,11etc.
H2O2 has been taken as an important molecule due to its elevated levels in many pathological conditions.12,13 The pioneer work from Chang et al.,14,15 who initiated fluorescent probes bearing a H2O2-responsive boronate trigger in 2004–2005, led to the rapid expansion of this strategy to the design of H2O2-activated prochelators and MMP proinhibitors.16,17 Given the significantly high H2O2 concentrations (5–1000 μM) in cancer cells relative to normal cells (0.001–0.7 μM), Peng et al.,18 for the first time, reported boron-based nitrogen mustard precursors as selective anticancer agents in 2011, followed by the blooming of various prodrugs (over 50 published papers) bearing an arylboronic acid/ester as the H2O2-cleavable trigger between 2012–2019 (Table S1†). Other H2O2-responsive moieties, such as arylsulfonyl ester,19 thiazolidinone,20 and benzothiazole21 have also been reported for constructing small molecule prodrugs, but with very limited examples of each. In addition, sulfide, thioether, thioketal, selenium, and peroxalate ester are also known as H2O2-sensitive scaffolds, but in most cases, their applications are confined to polymers or nanoparticles.22,23 As yet, currently over 90% H2O2-activated small molecule prodrugs still rely on the long-known boron-based triggers. Moreover, several disadvantages have been reported for the boron moiety, such as the high susceptibility of boronic esters to hydrolysis,24 the off-target reactions between boronic acids and biological diols,25 the moderate selectivity toward H2O2 and interference from ONOO−, as well as the potential unknown bioeffects from the by-produced boric acid.21 Considering the extremely low structural diversity of the current H2O2-responsive prodrugs, we have been highly interested in searching for novel chemical structures with H2O2-cleavable property.
We were recently enlightened by the elegant work from Tang et al., who used α-ketoamide-based fluorescent probes for intracellular H2O2 visualization.26–28 The α-ketoamide moiety was demonstrated as a highly specific reaction switch for H2O2. Herein, as a proof-of-concept, we introduce the α-ketoamide group into prodrug design by hybridizing it with nitrogen mustards, affording a panel of α-ketoamide-based nitrogen mustard (KAM) prodrugs. Multiple assays confirmed that one of the prodrugs, KAM-2, was efficiently and preferentially activated by H2O2, and showed potent and selective cytotoxicity to several cancer cell lines, particularly for HL-60 human leukemia cells. The ability of KAM-2 to induce DNA damage and apoptosis was thoroughly studied. To the best of our knowledge, this is the first example to utilize the α-ketoamide moiety as an alternative non-boron H2O2-responsive element for prodrug development.
Results and discussion
Design and synthesis of KAM prodrugs
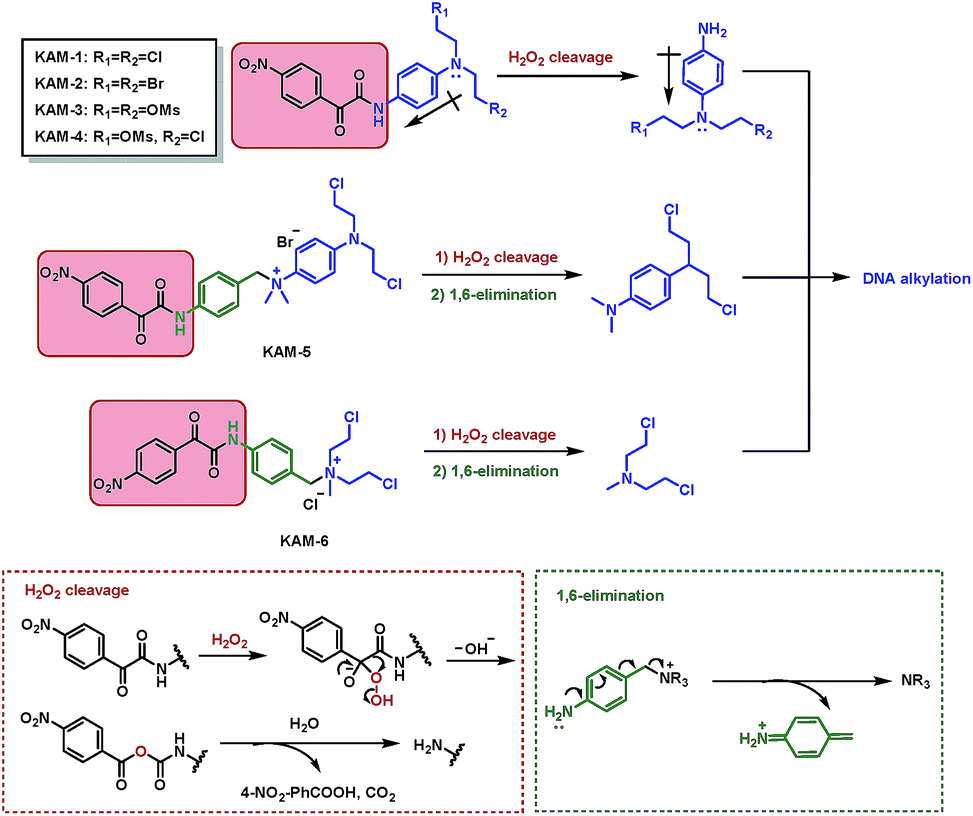
The structures of KAM prodrugs are illustrated in Scheme 1. It is speculated that the α-ketoamide moiety in KAM-1–KAM-4 acts as an electron-withdrawing group and shields the activity of nitrogen mustards.18,29–31 Upon the nucleophilic attack by H2O2 anion and the following Baeyer–Villiger rearrangement,27 the promoiety finally hydrolyses into aniline nitrogen mustards with increased nitrogen electron density and restored DNA alkylating activity. To enrich the structural diversity, quaternary ammonia salts KAM-5 and KAM-6 were also designed as masked mustard prodrugs. The activation process of KAM-5 and KAM-6 are similar to the mechanism described above but require an additional spontaneous 1,6-elimination.32
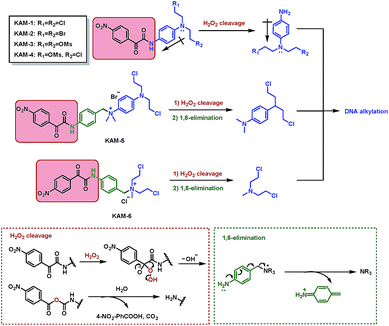 |
| | Scheme 1 Structures of KAM prodrugs and the mechanisms of H2O2-responsive activation to generate DNA alkylating agents. | |
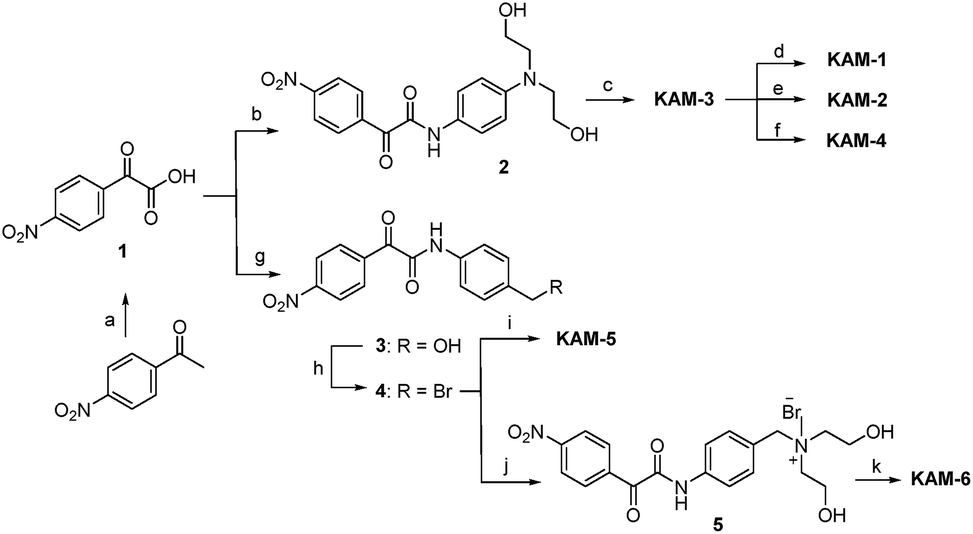
The synthetic route (Scheme 2) of the target compounds commenced with a SeO2-mediated oxidation of acetophenone in pyridine to afford phenylglyoxylic acid 1, which was coupled with N-4-aminophenyl-diethanolamine or (4-aminophenyl)methanol in the presence of EDCI and DIEA to give α-ketoamides 2 or 3, respectively. The two hydroxy groups of 2 were methylated to generate KAM-3. Further dichlorination, dibromination, and monochlorination of KAM-3 furnished KAM-1, KAM-2, and KAM-4, respectively. Bromination of the hydroxy group of 3 using CBr4 and PPh3 yielded 4, which was then directly reacted with aniline nitrogen mustard to give KAM-5. Alternatively, nucleophilic substitution of 4 with N-methyl-diethanolamine, followed by chlorination with SOCl2 generated KAM-6. Detailed synthetic procedures and spectroscopic data are provided in the ESI.†
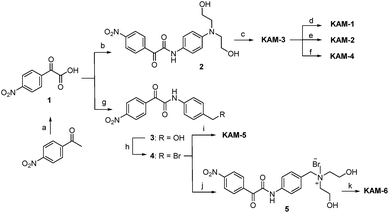 |
| | Scheme 2 Synthesis of KAM prodrugs. Reactants and conditions: (a) SeO2, pyridine, 90 °C, N2, 35%; (b) N-4-aminophenyl diethanolamine, EDCI, DIEA, DMF, 42%; (c) MsCl, DMAP, TEA, DCM, reflux, 70%; (d) LiCl (5 eq.), DMF, 60 °C, 65%; (e) NaBr (5 eq.), DMF, 60 °C, 67%; (f) LiCl (1 eq.), MeCN, 60 °C, 23%; (g) (4-aminophenyl)methanol, EDCI, DIEA, DMF, 33%; (h) CBr4, PPh3, DCM, 0 °C–r.t., 50%; (i) N-(4-N′,N′-dimethylaminophenyl)-bis(2-chloroethyl)amine, MeCN, 43%; (j) N-methyl-diethanolamine, CHCl3, reflux, 76%; (k) SOCl2, r.t., 62%. | |
Anticancer activity screening
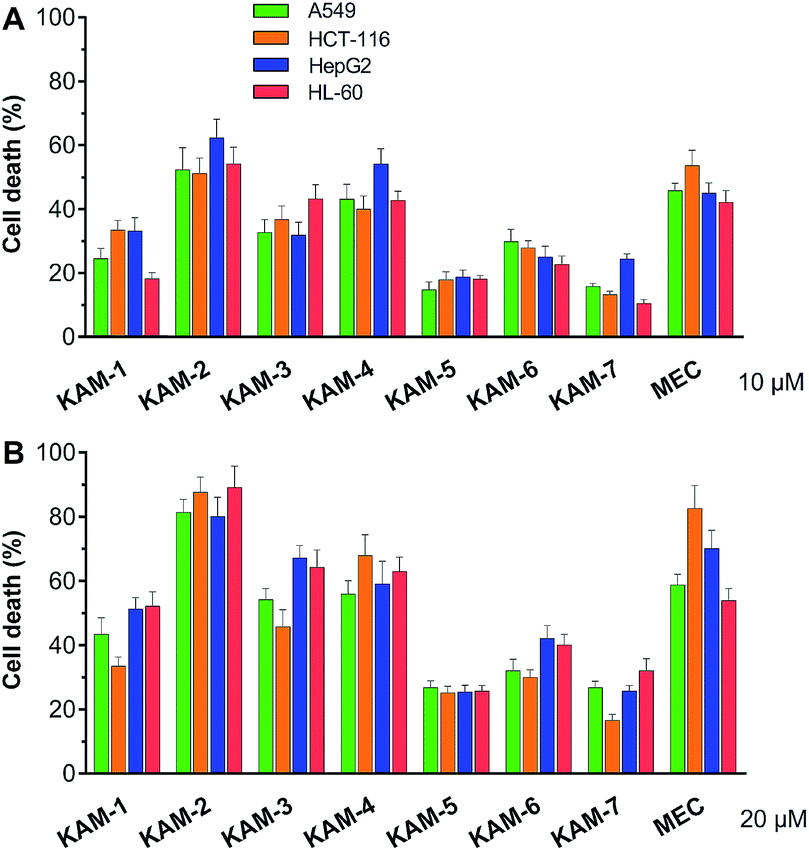
The antiproliferative activity of KAM prodrugs was screened against four human cancer cell lines (A549, HepG2, HCT-116, and HL-60), which have all been well-employed in H2O2 responsive prodrug studies.33–35 The classical nitrogen mustard mechlorethamine (MEC) was selected as the positive control. Evidently, KAM-2 exhibited the most potent activity among all the candidates (Fig. 1). At a dose of 10 μM, the inhibition rates of KAM-2 in all cancer cells were >50%, which further exceeded 80% at 20 μM. Structurally similar prodrugs KAM-3 and KAM-4 showed decreased cytotoxicity compared with KAM-2. The activity of chlorine counterpart, KAM-1, was further compromised. These results suggest that the leaving groups of mustard significantly affect the anticancer activity of KAM prodrugs. Ammonia salts KAM-5 and KAM-6 showed limited inhibition effects, particularly for KAM-5, possibly due to their lower membrane permeability and absorption.30 This assumption was preliminarily tested by calculating the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and membrane permeability values of KAM prodrugs, and the results are summarized in Table S2.† Considering the essential role of the nitro group of KAM in H2O2-initiated nucleophilic attack,26,27 a non-nitro analogue KAM-7 was synthesized (ESI†) as a negative control, which was supposed to be resistant to H2O2. As expected, KAM-7 displayed diminished cytotoxicity against cancer cells (Fig. 1), demonstrating the role of H2O2 in prodrug activation.
P and membrane permeability values of KAM prodrugs, and the results are summarized in Table S2.† Considering the essential role of the nitro group of KAM in H2O2-initiated nucleophilic attack,26,27 a non-nitro analogue KAM-7 was synthesized (ESI†) as a negative control, which was supposed to be resistant to H2O2. As expected, KAM-7 displayed diminished cytotoxicity against cancer cells (Fig. 1), demonstrating the role of H2O2 in prodrug activation.
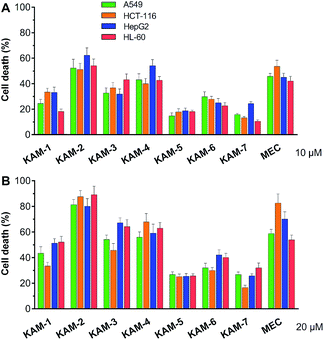 |
| | Fig. 1 Cytotoxicity of KAM prodrugs against four human cancer cell lines. Cells were incubated with indicated compounds at 10 (A) and 20 μM (B) for 72 h, and the percentages of cell death were assessed by MTT and CCK-8 assays. Mean ± SD, n = 3. | |
The anticancer activity of KAM-2 was then more closely studied by measuring its IC50 values, employing three additional H2O2-overproducing cancer cell lines (SW620, MCF-7, PANC-1)36–38 plus one normal line, peripheral blood mononuclear cell (PBMC) (Fig. S1†). As summarized in Table 1, KAM-2 showed lower IC50 values than MEC in cancer cells, except for PANC-1, which was particularly sensitive to MEC. The lowest IC50 (5.942 μM) of KAM-2 was seen in HL-60 cell. Encouragingly, KAM-2 proved its excellent selectivity toward cancer cells since the IC50 value in PBMC was larger than 50 μM. In contrast, MEC remained cytotoxic in PBMC with an IC50 value of 11.50 μM.
Table 1 IC50 values (μM) of KAM-2 and MEC against selected cell linesa
| Cell lines |
IC50 (μM) |
|
KAM-2
|
MEC |
|
Cells were incubated with indicated compounds at different concentrations for 72 h, and IC50 values were measured by MTT and CCK-8 assays. Mean ± SD, n = 3.
|
| A549 |
8.82 ± 0.83 |
10.07 ± 0.71 |
| HCT-116 |
8.01 ± 0.85 |
13.25 ± 1.57 |
| HepG2 |
8.84 ± 1.24 |
10.68 ± 1.52 |
| HL-60 |
5.94 ± 0.94 |
12.60 ± 3.47 |
| MCF-7 |
12.19 ± 1.55 |
24.26 ± 1.23 |
| PANC-1 |
16.25 ± 1.15 |
2.19 ± 0.08 |
| SW620 |
9.10 ± 0.27 |
18.50 ± 0.07 |
| PBMC |
>50 |
11.50 ± 0.94 |
H2O2-responsive behaviours
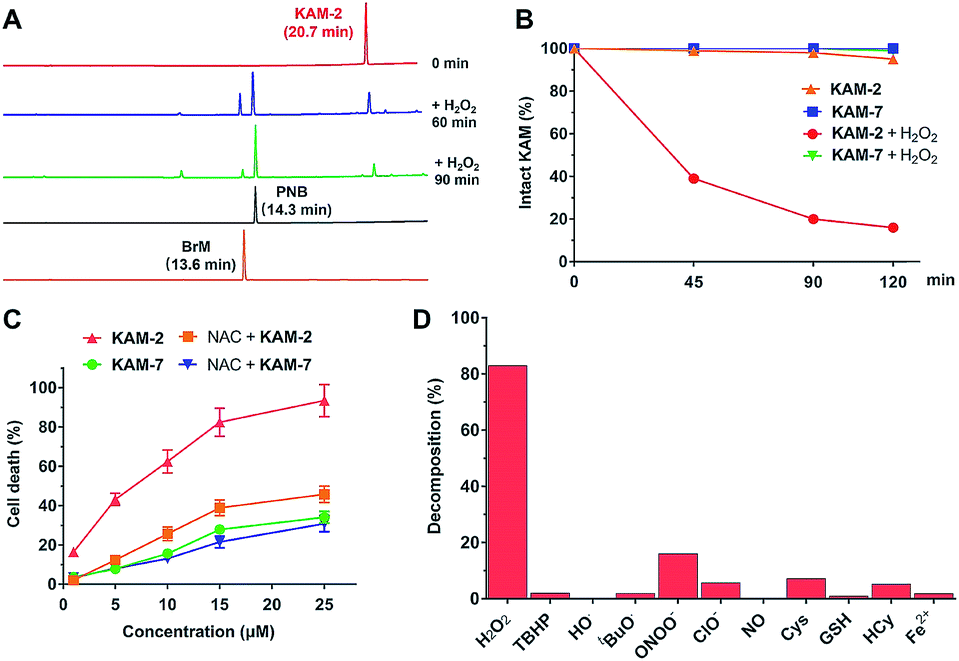
All the KAM prodrugs decomposed quickly upon H2O2 treatment (Fig. S2A†). Considering the good cytotoxicity and selectivity of KAM-2 toward cancer cells, its H2O2-responsive behaviours were in-depth studied. KAM-2 exhibited a decent stability in the absence of H2O2 at pH 4, 7, 10, respectively (Fig. S2B and S2C†). H2O2 rapidly trigged the decomposition of KAM-2 in a H2O2 equivalent-dependent way (Fig. S2D†). As monitored by HPLC, the released nitrogen mustard N,N-bis(2-bromoethyl)benzene-1,4-diamine (BrM), as well as the by-product p-nitrobenzoic acid (PNB), were clearly observed 60 min after H2O2 treatment (Fig. 2A). In sharp contrast, KAM-7 remained intact regardless of H2O2 presence (Fig. 2B and S2E†). The decomposition of KAM-2 in H2O2 was further studied by 1H NMR and mass spectroscopy, both of which unambiguously confirmed the prodrug activation behaviour (Fig. S3 and S4†). It is worth noting that in both HPLC and NMR studies, the signal of BrM shrank with time and became barely visible after long time treatment with H2O2. This is highly possible due to the instability of the phenylenediamine mustard toward H2O2 oxidation39,40 under the in vitro study conditions, as shown in Fig. S5 and S6.† To provide a visual demonstration of the mustard release from KAM-2, BrM was prepared as a yellow solution, which turned dark upon H2O2 addition. The same colour change was observed with KAM-2/H2O2, but not with KAM-7/H2O2 (Fig. S7†). Moreover, in HL-60 cells, H2O2 scavenger N-acetyl-cysteine (NAC) pretreatment remarkably decreased the cytotoxicity of KAM-2, but not KAM-7 (Fig. 2C). All the above results clearly demonstrated the H2O2-responsive activation of KAM-2. In regards of selectivity, KAM-2 was resistant to HO˙, tBuO˙, TBHB, NO, GSH, and Fe2+, very stable in ClO−, Cys and Hcy. Slight decomposition was observed in ONOO−, but to a much lesser extent than in H2O2 (Fig. 2D).
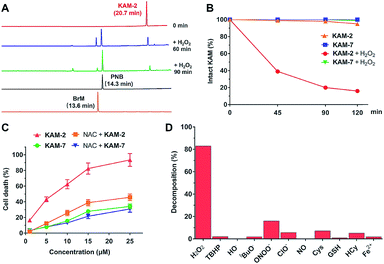 |
| | Fig. 2 H2O2-responsive behaviours of KAM-2. (A) Decomposition of 100 μM KAM-2 in presence of H2O2 (2 eq.) as monitored by RP-HPLC (254 nm). (B) Decomposition of KAM-2 and KAM-7 (100 μM) with or without H2O2 (10 eq.). (C) The effects of NAC on the cytotoxicity of KAM-2 and KAM-7 in HL-60 cells after 72 h incubation. (D) Selectivity of KAM-2 toward H2O2 as determined by measuring its decomposition ratios under various stimuli. | |
Proapoptotic mechanism of KAM-2
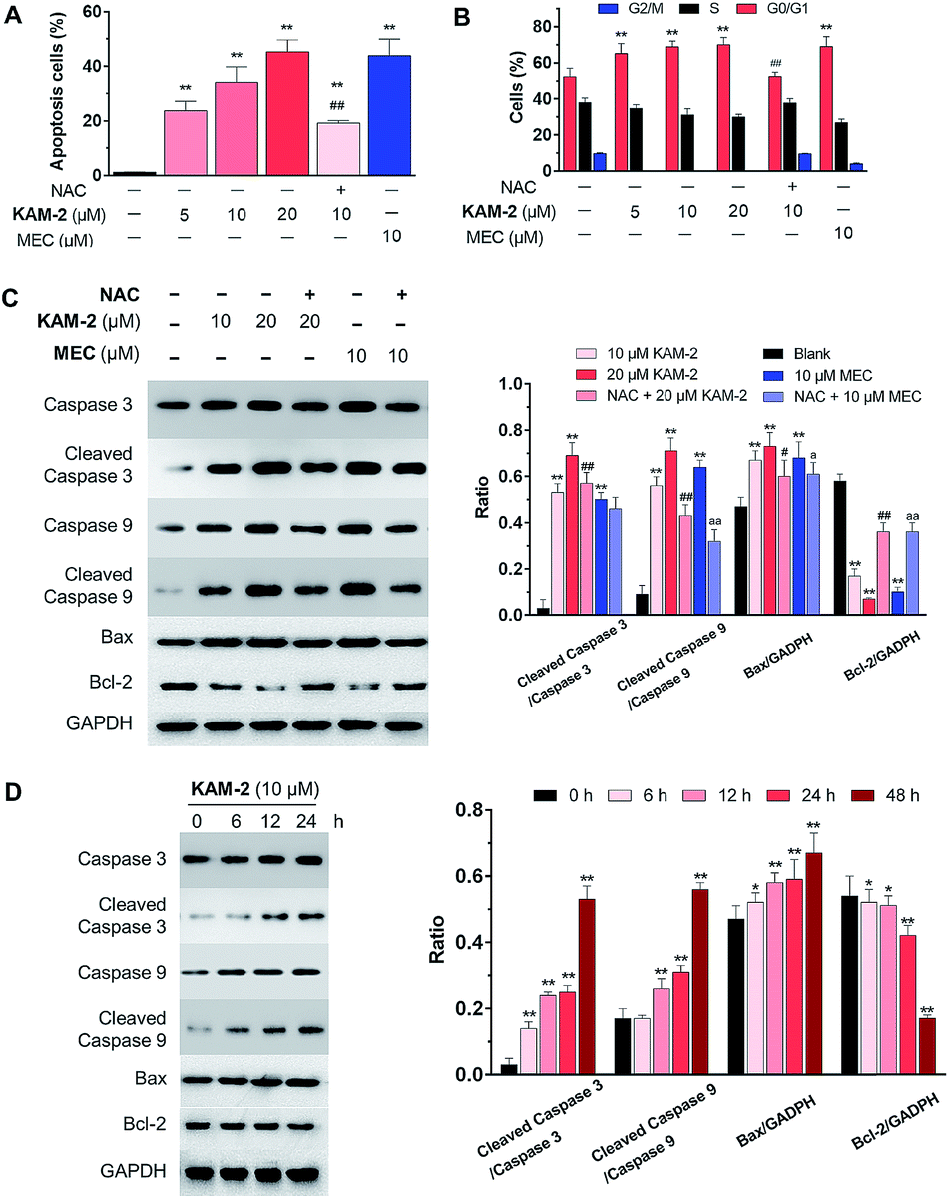
The anticancer activity of KAM-2 was further evaluated in HL-60 cells using MEC as positive control. It was found that KAM-2 treatment for 48 h induced apoptosis (Fig. 3A and S8A†) of HL-60 cells in a dose-dependent manner. Notably, NAC pretreatment significantly decreased the activity of KAM-2. Neither KAM-7 nor NAC showed apoptosis-inducing activity under the same condition (Fig. S8B†). Moreover, KAM-2 arrested cell cycle in G0/G1 phase and NAC reversed the cell cycle arrest (Fig. 3B and S9†). Modest effects of KAM-2 on the loss of mitochondrial membrane potential were also observed (Fig. S10†). Western blot studies revealed that KAM-2 induced the cleavage of caspase 3 and caspase 9, elevated the expression of Bax, and suppressed the expression of Bcl-2 in a dose (Fig. 3C) and time (Fig. 3D) dependent way, similar to the effects of MEC. The above results suggest that KAM-2 acts at least partially through the mitochondria-mediated apoptosis pathway.
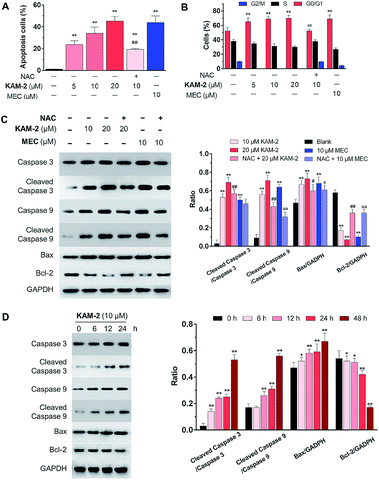 |
| | Fig. 3 (A) Apoptotic effects of KAM-2 measured by flow cytometry using annexin V-FITC/PI staining. HL-60 cells were treated with compounds at indicated concentrations for 48 h, or pre-incubated with ROS scavenger NAC (20 mM) for 1 h, followed by treatment with 10 μM KAM-2 for 48 h. Mean ± SD, n = 3. (B) Effects of KAM-2 on cell cycle arrest. HL-60 cells were treated with compounds at indicated concentrations for 48 h, or pre-incubated with NAC (20 mM) for 1 h, followed by treatment with 10 μM KAM-2 for 48 h. The percentage of cells in each cell cycle phase was analyzed by flow cytometry, mean ± SD, n = 3. (C) Dose-dependent effects of KAM-2 on the expression of apoptosis-related proteins by Western blot. HL-60 cells were treated with compounds at indicated concentrations for 48 h or pre-incubated with NAC (20 mM) for 1 h, followed by treatment with compounds for 48 h. Mean ± SD, n = 3. **p < 0.01 vs. blank; #p < 0.05 vs. 20 μM KAM-2 treatment; ##p < 0.01 vs. 20 μM KAM-2 treatment; ap < 0.05 vs. 10 μM MEC treatment; aap < 0.01 vs. 10 μM MEC treatment. (D) Time-dependent effects of KAM-2 on the expression of apoptosis-related proteins by Western blot. HL-60 cells were treated with 10 μM KAM-2 for 0, 6, 12, 24, and 48 h. Mean ± SD, n = 3. *p < 0.05 vs. 0 h; **p < 0.01 vs. 0 h. | |
DNA cross-linking and damaging effects of KAM-2
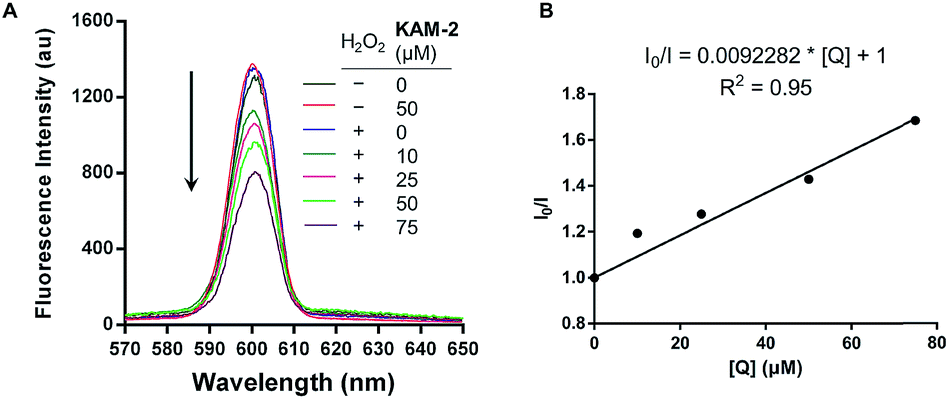
In addition to the above disclosed mechanism, the nitrogen mustard released from KAM-2 upon H2O2 activation is supposed to exert its cytotoxicity by DNA interstrand cross-linking (ICL).41 This was first examined by a fluorescence quenching experiment. It is known that ethidium bromide (EB) interacts with DNA to yield maximum fluorescence emission at 600 nm (Ex = 300 nm), which is supposed to be quenched due to competitive binding of nitrogen mustard to DNA. As expected, KAM-2 decreased the fluorescence intensity at 600 nm in the presence of H2O2 (Fig. 4).
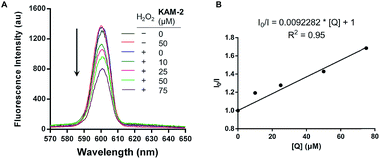 |
| | Fig. 4 (A) Fluorescence quenching of EB/DNA by KAM-2. Various concentrations of KAM-2 was added into EB-DNA solution (2.5 μM EB and 50 μM linearized pBR322 plasmid DNA) with or without H2O2 (10 mM) in Tris–HCl/NaCl buffer. Ex = 300 nm (B) Stern–Volmer plots of fluorescence quenching of EB-DNA by KAM-2. I0 and I are the emission intensity in the absence and presence of the KAM-2, respectively. Slope (Ksv) is the quenching rate constant, and [Q] is the concentration of the quencher KAM-2. | |
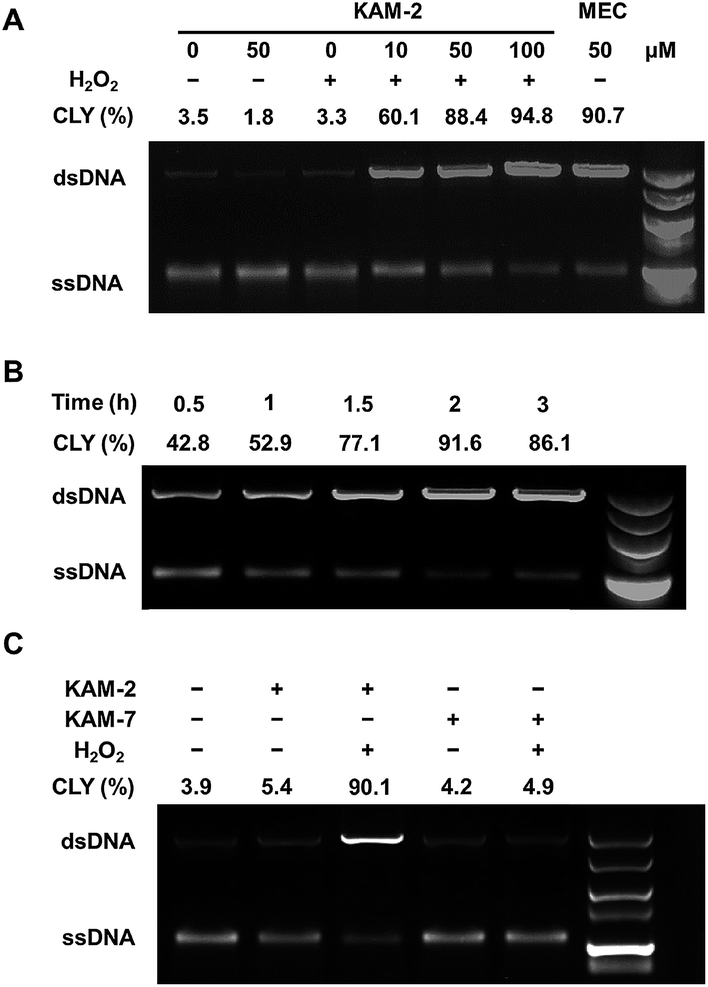
The DCL ability of activated KAM-2 was further confirmed by denaturing alkaline agarose gel electrophoresis.42 As shown in Fig. 5A and B, KAM-2 or H2O2 alone had no effects on linearized pBR322 plasmid DNA. However, in the presence of 10 mM H2O2, KAM-2 treatment induced DNA ICL (from 3.3% to 94.8%) as indicated by the intensified bands of double strand DNA (dsDNA) in both a dose and time dependent way. In contrast, KAM-7 combined with H2O2 was unable to yield dsDNA (Fig. 5C). These results again, strongly support the H2O2-triggered activation of KAM-2 and consequent generation of nitrogen mustard.
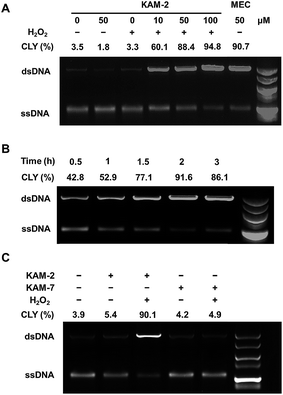 |
| | Fig. 5 DNA interstrand cross-linking (ICL) ability of KAM-2. (A) Denaturing alkaline agarose gel electrophoresis performed with 1 μg linearized pBR322 plasmid DNA and indicated concentrations of KAM-2 or MEC with or without H2O2 (10 mM) for 2 h at 37 °C. (B) Denaturing alkaline agarose gel electrophoresis performed with 1 μg linearized pBR322 plasmid DNA, 50 μM KAM-2, and 10 mM H2O2 at 37 °C for indicated time. (C) Denaturing alkaline agarose gel electrophoresis performed with 1 μg pBR322, 50 μM KAM-2 or KAM-7 with or without H2O2 (10 mM) for 2 h at 37 °C. CLY: cross-linking yield. | |
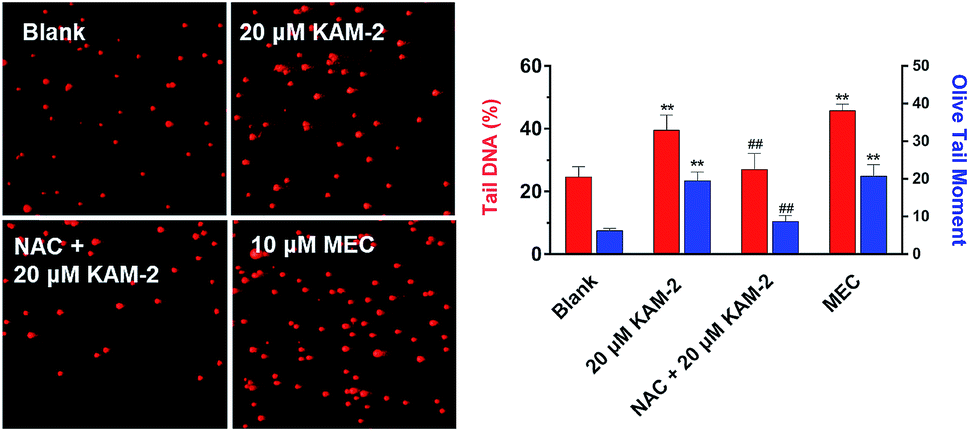
Next, the DNA damaging activity of KAM-2 was assessed in HL-60 cells using comet assay. KAM-2 treatment resulted in obvious cellular DNA damage as visually shown in Fig. 6. NAC preincubation protected HL-60 cells from tail DNA formation due to diminished activation of KAM-2. KAM-7 or NAC alone had no effects on DNA damage (Fig. S11†).
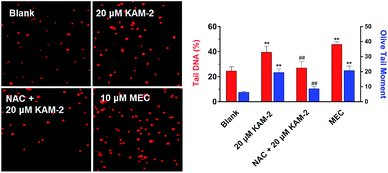 |
| | Fig. 6 Effects of KAM-2 on cell damage assessed by comet assay. HL-60 cells were treated with 20 μM KAM-2 or 10 μM MEC for 48 h, or pre-incubated with NAC (20 mM) for 1 h, followed by treatment with 10 μM KAM-2 for 48 h. Alkaline comet electrophoresis was performed. Quantitative analysis was conducted with the comet analysis software CASP. The tail DNA% and olive tail moment were employed to evaluate DNA damage. Mean ± SD, n = 3. **p < 0.01 vs. blank; ##p < 0.01 vs. 20 μM KAM-2 treatment. | |
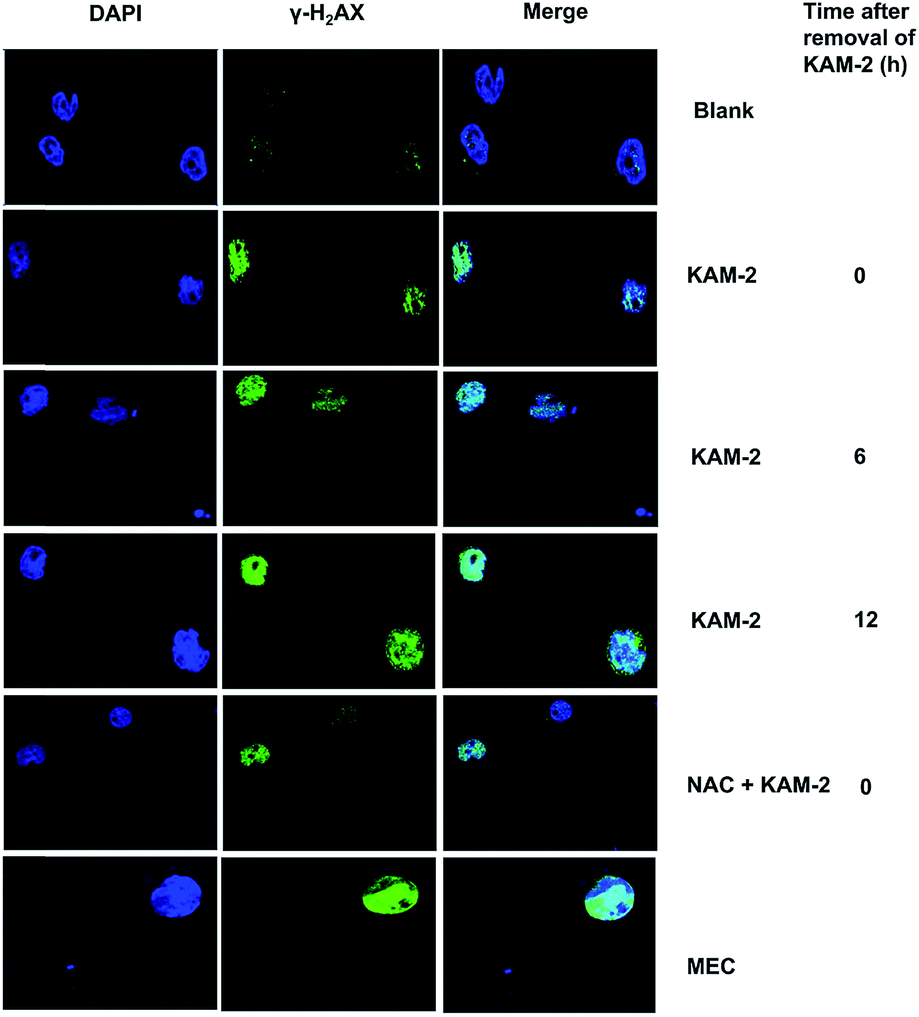
Following DNA double-strand break (DSB), histone H2AX is rapidly phosphorylated at Ser139 to yield γ-H2AX as a golden biomarker of DNA damage response (DDR).43 The expression of γ-H2AX upon KAM-2 treatment was therefore examined using immunofluorescence microscopy. Incubation of HL-60 cells with KAM-2 for 12 h remarkably increased the level of γ-H2AX (Fig. 7). Notably, when KAM-2 was removed and cells were allowed to grow without KAM-2 for an additional 6 or 12 h, elevated expression of γ-H2AX was still observed. However, NAC pretreatment mitigated the effects of KAM-2 on γ-H2AX formation. Compared with KAM-2, KAM-7 and NAC had negliable effects on γ-H2AX formation as shown in Fig. S12.†
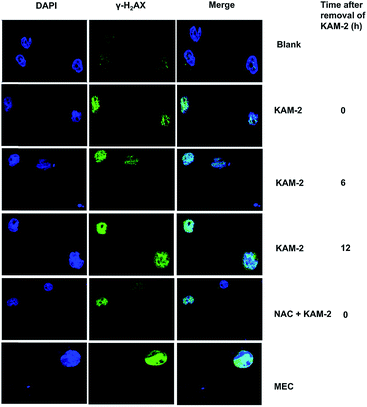 |
| | Fig. 7 Immunofluorescence of DNA cleavage protein γH2AX. HL-60 cells were treated with KAM-2 or MEC (10 μM) for 12 h, or pre-incubated with ROS scavenger NAC (20 mM) for 1 h, followed by treatment with KAM-2 for 12 h. For some groups, cells were incubated for an additional 6 or 12 h after removal of KAM-2. | |
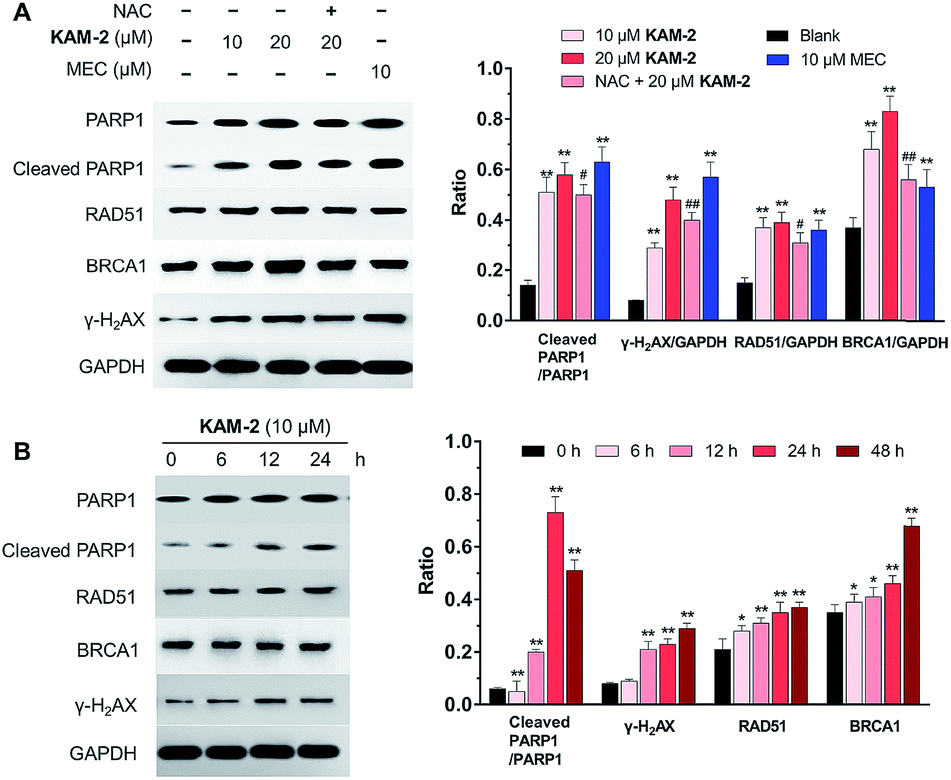
In addition to γ-H2AX, poly(ADP-ribose) polymerase-1 (PARP1), RAD51 and BRCA1 are activated by DNA DSB and involved in DNA repair.44 The changes of PARP1, RAD51, BRCA1, and γ-H2AX were quantified by Western blot. Clearly, KAM-2 induced the cleavage of PARP1 and increased the expression of γ-H2AX, RAD51 and BRCA1 in a dose (Fig. 8A) and time (Fig. 8B) dependent way. In contrast, KAM-7 or NAC treatment led to no alterations of the expressions of cleaved PARP1 or γ-H2AX (Fig. S13†).
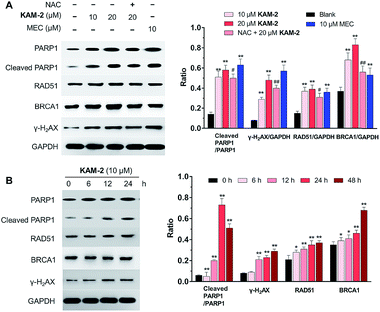 |
| | Fig. 8 (A) Concentration-dependent effects of KAM-2 on the expression of DDR-related proteins by Western blot. HL-60 cells were treated with compounds at indicated concentrations for 48 h or pre-incubated with NAC (20 mM) for 1 h, followed by treatment with compounds for 48 h. Mean ± SD, n = 3. **p < 0.01 vs. blank; #p < 0.05 vs. 20 μM KAM-2 treatment; ##p < 0.01 vs. 20 μM KAM-2 treatment. (B) Time-dependent effects of KAM-2 on the expression of DDR-related proteins by Western blot. HL-60 cells were treated with 10 μM KAM-2 for 0, 6, 12, 24, and 48 h. Mean ± SD, n = 3. *p < 0.05 vs. 0 h; **p < 0.01 vs. 0 h. | |
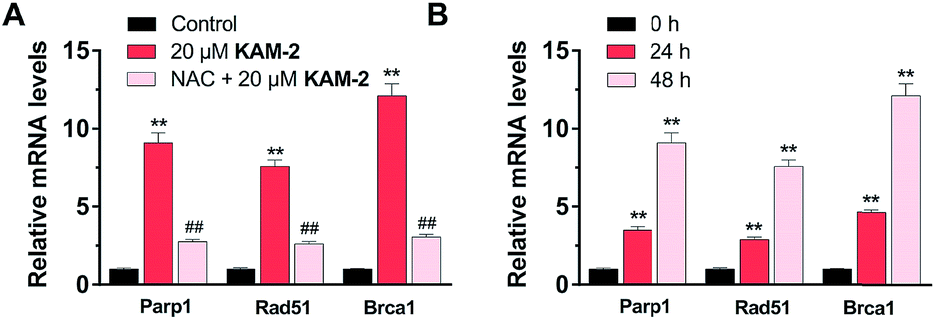
Finally, the effects of KAM-2 on the expression of Parp1, Rad51 and Brca1 mRNAs were investigated in HL-60 cells. Real-time PCR assay confirmed that KAM-2 concentration (Fig. 9A) and time (Fig. 9B) dependently elevated the mRNA expression levels of Parp-1, Rad51 and Brca1. Importantly, NAC pretreatment mitigated the effects of KAM-2 on gene expression.
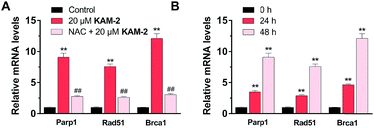 |
| | Fig. 9 The concentration- (A) and time-dependent (B) effects of 20 μM KAM-2 on the expression DDR-related mRNA levels tested by real-time PCR. Mean ± SD, n = 3. **p < 0.01 vs. control or 0 h treatment; ##p < 0.01 vs. 20 μM KAM-2 treatment. | |
Conclusions
In summary, we for the first time present an α-ketoamide scaffold to build up H2O2-activated prodrugs, using nitrogen mustard as the model drug. Our studies identified KAM-2 as a novel anticancer prodrug deserving further investigations. KAM-2 obviously inhibited the growth of different cancer cell lines with diminished cytotoxicity in normal PBMC cells. The H2O2-induced decomposition of KAM-2 was confirmed using various analytical methods. The nitrogen mustard released from KAM-2 exhibited DNA ICL ability, led to DNA damage in HL-60 cells, and increased the expression of DDR related proteins and mRNAs. Additional mechanism studies suggested that KAM-2 induced HL-60 apoptosis in a mitochondrial-dependent manner. In addition to the emerged boron-based H2O2-responsive prodrugs, our work showcases the establishment of a new platform for prodrug design inspired from analytical probes. Considering the close involvement of H2O2 in many pathological diseases, it is anticipated that our results will actively promote the development of both small molecule and nanoparticle-based agents in related fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by National Natural Science Foundation of China (21877052, 81602960), Natural Science Foundation of Jiangsu Province (BK20180030, BK20161028), and the Fundamental Research Funds for the Central Universities (JUSRP51712B).
Notes and references
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559 CrossRef CAS PubMed.
- S. H. Alamudi and Y. T. Chang, Chem. Commun., 2018, 54, 13641 RSC.
- D. S. Folk and K. J. Franz, J. Am. Chem. Soc., 2010, 132, 4994 CrossRef CAS PubMed.
- N. Ueki, S. Lee, N. S. Sampson and M. J. Hayman, Nat. Commun., 2013, 4, 2735 CrossRef PubMed.
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770 CrossRef CAS PubMed.
- S. W. Chung, J. U. Choi, Y. S. Cho, H. R. Kim, T. H. Won, P. Dimitrion, O. C. Jeon, S. W. Kim, I. S. Kim, S. Y. Kim and Y. Byun, Adv. Sci., 2018, 5, 1800368 CrossRef PubMed.
- S. Bakthavatsalam, M. L. Sleeper, A. Dharani, D. J. George, T. Zhang and K. J. Franz, Angew. Chem., Int. Ed., 2018, 57, 12780 CrossRef CAS PubMed.
- X. Li, Y. Hou, X. Meng, C. Ge, H. Ma, J. Li and J. Fang, Angew. Chem., Int. Ed., 2018, 57, 6141 CrossRef CAS PubMed.
- F. Kang, J. Zhu, J. Wu, T. Lv, H. Xiang, J. Tian, Y. Zhang and Z. Huang, Chem. Sci., 2018, 9, 6893 RSC.
- S. B. Salunke, J. A. Malla and P. Talukdar, Angew. Chem., Int. Ed. Engl., 2019, 58, 5354 CrossRef CAS PubMed.
- P. Wei, L. Liu, Y. Wen, G. Zhao, F. Xue, W. Yuan, R. Li, Y. Zhong, M. Zhang and T. Yi, Angew. Chem., Int. Ed., 2019, 58, 4547 CrossRef CAS PubMed.
- R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5839 CrossRef CAS PubMed.
- B. D'Autreaux and M. B. Toledano, Nat. Rev. Mol. Cell Biol., 2007, 8, 813 CrossRef PubMed.
- M. C. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392 CrossRef CAS PubMed.
- E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652 CrossRef CAS PubMed.
- L. K. Charkoudian, D. M. Pham and K. J. Franz, J. Am. Chem. Soc., 2006, 128, 12424 CrossRef CAS PubMed.
- J. L. M. Jourden and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 6795 CrossRef PubMed.
- Y. Kuang, K. Baakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278 CrossRef CAS PubMed.
- K. B. Daniel, J. L. M. Jourden, K. E. Negoescu and S. M. Cohen, J. Biol. Inorg Chem., 2011, 16, 313 CrossRef CAS PubMed.
- C. Perez, J.-P. Monserrat, Y. Chen and S. M. Cohen, Chem. Commun., 2015, 51, 7116 RSC.
- C. K. Wang, J. Cheng, X. G. Liang, C. Tan, Q. Jiang, Y. Z. Hu, Y. M. Lu, K. Fukunaga, F. Han and X. Li, Theranostics, 2017, 7, 3803 CrossRef CAS PubMed.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553 CrossRef CAS PubMed.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed.
- C. Achilli, A. Ciana, M. Fagnoni, C. Balduini and G. Minetti, Cent. Eur. J. Chem., 2013, 11, 137 CAS.
- Y. Furikado, T. Nagahata, T. Okamoto, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, Chem.–Eur. J., 2014, 20, 13194 CrossRef CAS PubMed.
- C. Gao, Y. Tian, R. Zhang, J. Jing and X. Zhang, Anal. Chem., 2017, 89, 12945 CrossRef CAS PubMed.
- X. Xie, X. Yang, T. Wu, Y. Li, M. Li, Q. Tan, X. Wang and B. Tang, Anal. Chem., 2016, 88, 8019 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035 RSC.
- S. Cao, Y. Wang and X. Peng, Chem.–Eur. J., 2012, 18, 3850 CrossRef CAS PubMed.
- W. Chen, K. Balakrishnan, Y. Kuang, Y. Han, M. Fu, V. Gandhi and X. Peng, J. Med. Chem., 2014, 57, 4498 CrossRef CAS PubMed.
- W. Chen, H. Fan, K. Balakrishnan, Y. Wang, H. Sun, Y. Fan, V. Gandhi, L. A. Arnold and X. Peng, J. Med. Chem., 2018, 61, 9132 CrossRef CAS PubMed.
- A. Alouane, R. Labruere, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492 CrossRef CAS PubMed.
- R. Kumar, J. Han, H. J. Lim, W. X. Ren, J. Y. Lim, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836 CrossRef CAS PubMed.
- H. W. Liu, X. X. Hu, K. Li, Y. Liu, Q. Rong, L. Zhu, L. Yuan, F. L. Qu, X. B. Zhang and W. Tan, Chem. Sci., 2017, 8, 7689 RSC.
- H. Hagen, P. Marzenell, E. Jentzsch, F. Wenz, M. R. Veldwijk and A. Mokhir, J. Med. Chem., 2012, 55, 924 CrossRef CAS PubMed.
- J. Noh, B. Kwon, E. Han, M. Park, W. Yang, W. Cho, W. Yoo, G. Khang and D. Lee, Nat. Commun., 2015, 6, 6907 CrossRef CAS PubMed.
- C. Q. Luo, Y. X. Zhou, T. J. Zhou, L. Xing, P. F. Cui, M. Sun, L. Jin, N. Lu and H. L. Jiang, J. Controlled Release, 2018, 274, 56 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS.
- Z. Can, A. Uzer, K. Turkekul, E. Ercag and R. Apak, Anal. Chem., 2015, 87, 9589 CrossRef CAS PubMed.
- T. B. Zanoni, F. Hudari, A. Munnia, M. Peluso, R. W. Godschalk, M. V. Zanoni, G. J. den Hartog, A. Bast, S. B. Barros, S. S. Maria-Engler, G. J. Hageman and D. P. de Oliveira, Toxicol. Lett., 2015, 239, 194 CrossRef CAS PubMed.
- R. K. Singh, S. Kumar, D. N. Prasad and T. R. Bhardwaj, Eur. J. Med. Chem., 2018, 151, 401 CrossRef CAS PubMed.
- J. J. Tepe and R. M. Williams, J. Am. Chem. Soc., 1999, 121, 2951 CrossRef CAS.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, J. Biol. Chem., 1998, 273, 5858 CrossRef CAS PubMed.
- S. Ganesan and A. F. Keating, Toxicol. Appl. Pharmacol., 2015, 282, 252 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00910h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Pengfei
Zhang
a,
Jing
Hu
a,
Junjie
Fu
c,
Pengfei
Zhang
a,
Jing
Hu
a,
Junjie
Fu