
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriperyleno[3,3,3]propellane triimides: achieving a new generation of quasi-D3h symmetric nanostructures in organic electronics†
Lingling
Lv
a,
Josiah
Roberts
b,
Chengyi
Xiao
a,
Zhenmei
Jia
a,
Wei
Jiang
a,
Guowei
Zhang
a,
Chad
Risko
 *b and
Lei
Zhang
*a
*b and
Lei
Zhang
*a
aBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: zhl@mail.buct.edu.cn
bDepartment of Chemistry & Center for Applied Energy Research, University of Kentucky, Lexington, Kentucky 40506-0055, USA. E-mail: chad.risko@uky.edu
First published on 12th April 2019
Abstract
Rigid three-dimensional (3D) polycyclic aromatic hydrocarbons (PAHs), in particular 3D nanographenes, have garnered interest due to their potential use in semiconductor applications and as models to study through-bond and through-space electronic interactions. Herein we report the development of a novel 3D-symmetric rylene imide building block, triperyleno[3,3,3]propellane triimides (6), that possesses three perylene monoimide subunits fused on a propellane. This building block shows several promising characteristics, including high solubility, large π-surfaces, electron-accepting capabilities, and a variety of reactive sites. Further, the building block is compatible with different reactions to readily yield quasi-D3h symmetric nanostructures (9, 11, and 13) of varied chemistries. For the 3D nanostructures we observed red-shift absorption maxima and amplification of the absorption coefficients when compared to the individual subunits, indicating intramolecular electronic coupling among the subunits. In addition, the microplates of 9 exhibit comparable mobilities in different directions in the range of 10−3 cm2 V−1 s−1, despite the rather limited intermolecular overlap of the π-conjugated moieties. These findings demonstrate that these quasi-D3h symmetric rylene imides have potential as 3D nanostructures for a range of materials applications, including in organic electronic devices.
Introduction
Due to the prospects of distinctive electronic and optical properties, considerable attention has been given to the synthesis of polycyclic aromatic hydrocarbons (PAHs).1–4 Most PAHs can be classified into one- (1D) and two-dimensional (2D) PAHs, such as 1D linear polyacenes and 2D nanographenes, with both of them showing robust and large charge-carrier mobilities in transistors.5–7 In contrast, three-dimensional (3D) PAHs remain an underexplored molecular construct due to numerous synthetic challenges, in particular for rigid 3D-symmetric PAHs,8–11 even though they have been suggested as materials systems for organic electronics, molecular rotors, host–guest chemistries, liquid crystals, and supramolecular assemblies.12–15The classic 3D organic building block triptycene was first synthesized in 1942 by Bartlett et al. (Fig. 1).16 Triptycene and its derivatives, for instance, have been widely incorporated into polymer backbones to disrupt chain packing and increase the free volume, qualities that are beneficial for improving gas diffusion, sensory response, thermal stability, Young's modulus, and luminescence quantum efficiency.17–20 On the other hand, there has been interest in the synthesis of triptycene-extended PAHs, wherein additional five- and six-membered aromatic rings are fused to the triptycene core.21–28 This interest is partly due to the π-extended triptycenes possessing large internal molecular free volume (IMFV), which can provide an approach to prepare nanoporous materials or two-dimensional (2D) polymers.29–32 Importantly, such molecules can serve as key components in molecular electronic materials,33,34 as they can possess extended π-conjugation in three directions in a rigid structure, which will influence the condensed-phase aggregation and alter the stability, solubility, electronic properties, and device performance. For example, Wang et al. recently demonstrated a π-extended triptycene fused with perylene diimides (PDI) as a non-fullerene acceptor for organic solar cells.35 Further, Nuckolls et al. reported a promising class of 3D nanographenes based on triptycene, which were used as electron-extracting layers, which helped to decrease contact resistances in perovskite solar cells and led to power conversion efficiencies greater than 18%.36 Notably, the synthesis of these 3D porous materials was completed by the fusion of triptycenes to the bay positions of PDI, leading to materials that exhibit remarkably stable electron-accepting pseudocapacitive behavior, demonstrating an exceptional benefit derived from the large internal free volume.37
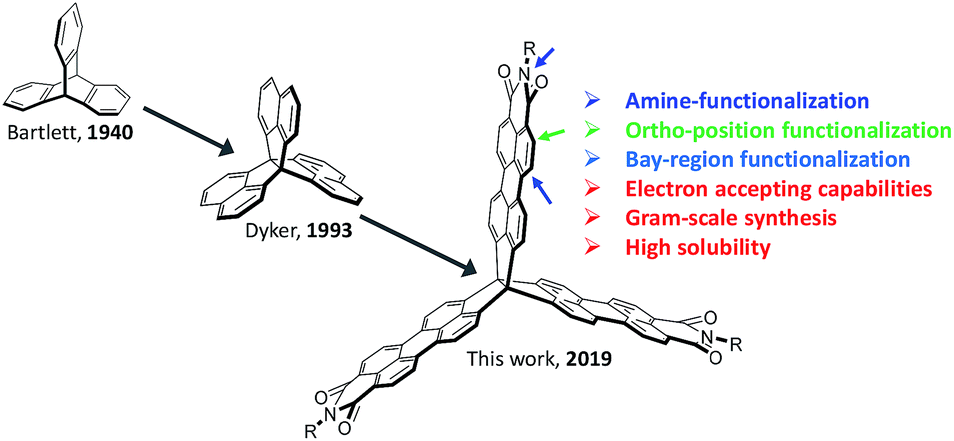 | ||
| Fig. 1 Representation of the structural evolution of 3D-symmetric building blocks for rigid 3D PAHs, from triptycene (left) to trinaphtho[3,3,3]propellane (middle) to triperyleno[3,3,3]propellane triimides (right). | ||
These successful materials applications have encouraged us to develop an extended π-conjugated 3D building block to widen the scope of available building blocks and introduce new properties to the resulting 3D materials. In particular, 3D-symmetric molecules provide an ideal model to investigate through-bond and through-space electronic communication among the different aromatic units, which can play a crucial role in charge-carrier transport in organic semiconductors.38–41 To date, PDI and its derivatives have been extensively used to construct 2D rylene imides and n-type nanographenes with promising applications in organic electronic devices and batteries.42–44 Due to significant synthetic challenges, however, there are only a few literature reports on 3D rylene imides that result in 3D solid-state networks for high performance organic devices.45–47
Herein we report the synthesis and characterization of a novel rigid 3D-symmetric building block, triperyleno[3,3,3]propellane triimides (6), which comprises three perylene monoimide subunits fused on a [3,3,3]propellane, as shown in Fig. 1. Importantly, this system can be synthesized on a gram scale. A noteworthy feature of this molecule is that it can be considered to be composed of three perylene monoimide (PMI) subunits, which provide, in contrast with triptycene, reactive sites at the imide nitrogen atoms, the “bay” positions, and the “ortho” positions for further functionalization, which in cooperation with electron-deficient imide groups can facilitate the creation of n-type PAHs with high electron affinity and solubility. Moreover, the chemically robust nature of PMI allows the use of well-established PDI/PMI synthetic chemistry, which generally produces rigid 3D PAHs with tunable electronic properties with good to excellent yields under very mild reaction conditions. Here, we extend 6 to three additional large rigid quasi-D3h symmetric nanostructures (9, 11 and 13) that contain dibenzocoronene monoimide, dithienocoronene monoimide, and coronene monoimide as subunits, respectively. We also undertook proof-of-principle studies of the charge-carrier transport properties of the rigid 3D PAHs in the solid state.
Results and discussion
Synthesis and molecular characterization
Scheme 1 shows the synthesis of the rigid 3D-symmetric rylene imide. The key chemistry is the synthesis of the 3D perylenetetracarboxylic trianhydrides (5), which is unlike the synthesis of perylene dicarboxylic monoanhydride via decarboxylation of 3,4,9,10-perylenetetracarboxylic dianhydrides at high temperature.48,49 We designed an efficient synthetic protocol based on acid-promoted cyclodehydrogenation of binaphthyl derivatives bearing dicarboxylic anhydride at the peri-position to prepare the key intermediate compound 5. We chose trinaphtho[3.3.3]propellane (TNP, 1) as the starting material.50–52 Bromination of 1 provided compound 2 as a mixture of regioisomers in a high yield (∼98%). Without isolation, 2 could be readily converted to triborate 3 in a 75% yield. A Suzuki coupling of 4-bromo-1,8-naphthalic anhydride with 3 was followed by ring closure with AlCl3 as oxidant to afford the dicarboxylic anhydride 5 as a red solid (∼30% yield in two steps from 4). Finally, the desired building block 6 was achieved through direct imidization of 5 with an amine in N,N-dimethylformamide (DMF).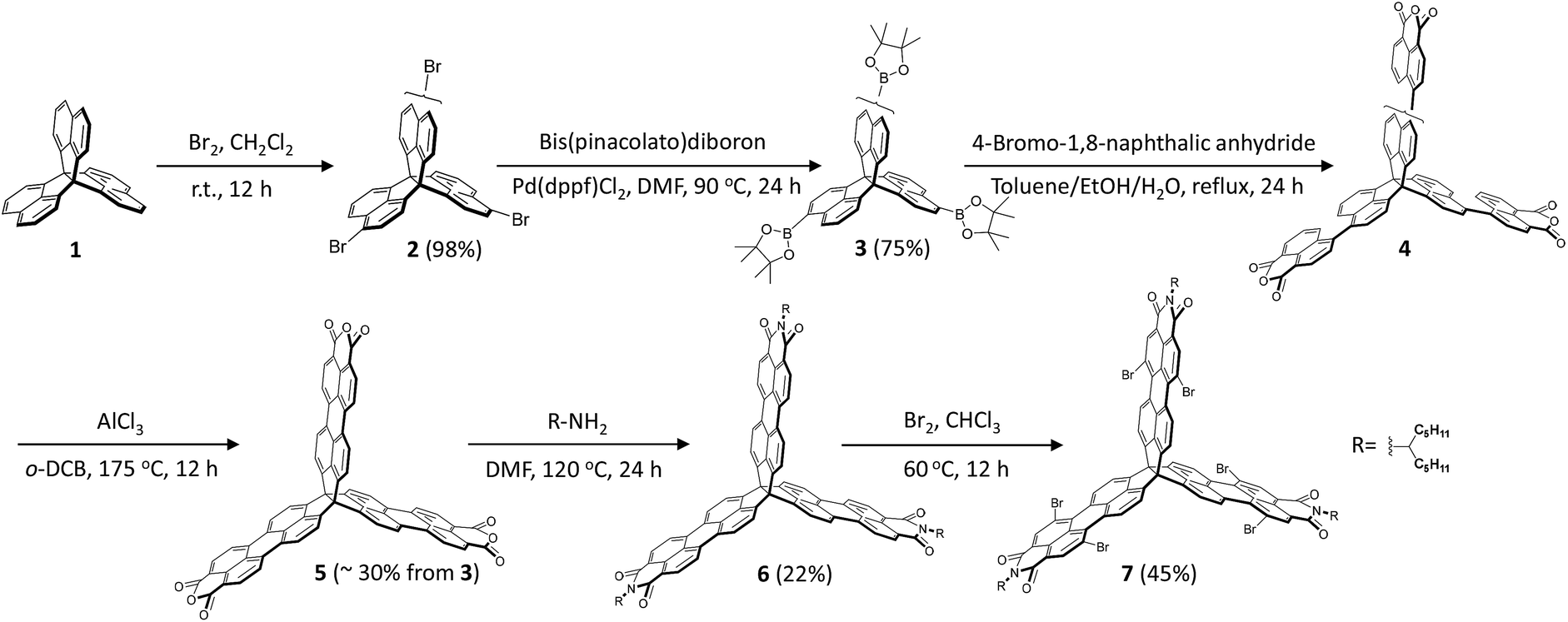 | ||
| Scheme 1 Synthetic steps to 3D-symmetric building block 6 and its brominated 7. | ||
Synthetically, the three PMI subunits offer the opportunity for further synthetic modification. Thus, we subjected 6 to typical PMI reactions to functionalize the bay positions. In analogy with the standard PMI chemistry, bromination of 6 in chloroform gave hexabrominated 7 in 45% yield. To explore the scope of the Pd-catalyzed cross-coupling strategy, reactions of 7 with different substrates (4-tert-butylphenylboronic acid, 2-trimethyltin-5-triisopropylsilylthiophene, and n-butyl-acetylene) were studied (Scheme 2). We found that the desired 8, 10, and 12 could be obtained after 10 hours in excellent to moderate yields (40–60%). We next examined the cyclization reaction of 8, 10, and 12. Substrates 8 and 10 could undergo 6-fold cyclization smoothly to produce the desired product 9 in 75% yield and 11 in 75% yield under standard photo-induced cyclization conditions. During our initial synthetic effort for compound 13, we observed no cyclization products under the typical alkyne cyclization conditions with either PtCl2 or some Brønsted acids.53–55 Finally, the cyclization product 13 was obtained by treatment of 12 with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in 25% yield. It should be emphasized that the t-butyl, n-butyl, and triisopropylsilyl (TIPS) groups are necessary to ensure adequate solubility for efficient cyclization. The feasibility of the synthetic strategy suggests that the bromine-containing building block 7 is amenable to a wide range of large and rigid 3D nanostructures with tunable geometries through PMI/PDI chemistry. These rigid 3D-symmetric compounds are soluble in common solvents and could be characterized by spectroscopic analysis, including HMBC and 1D NOE.
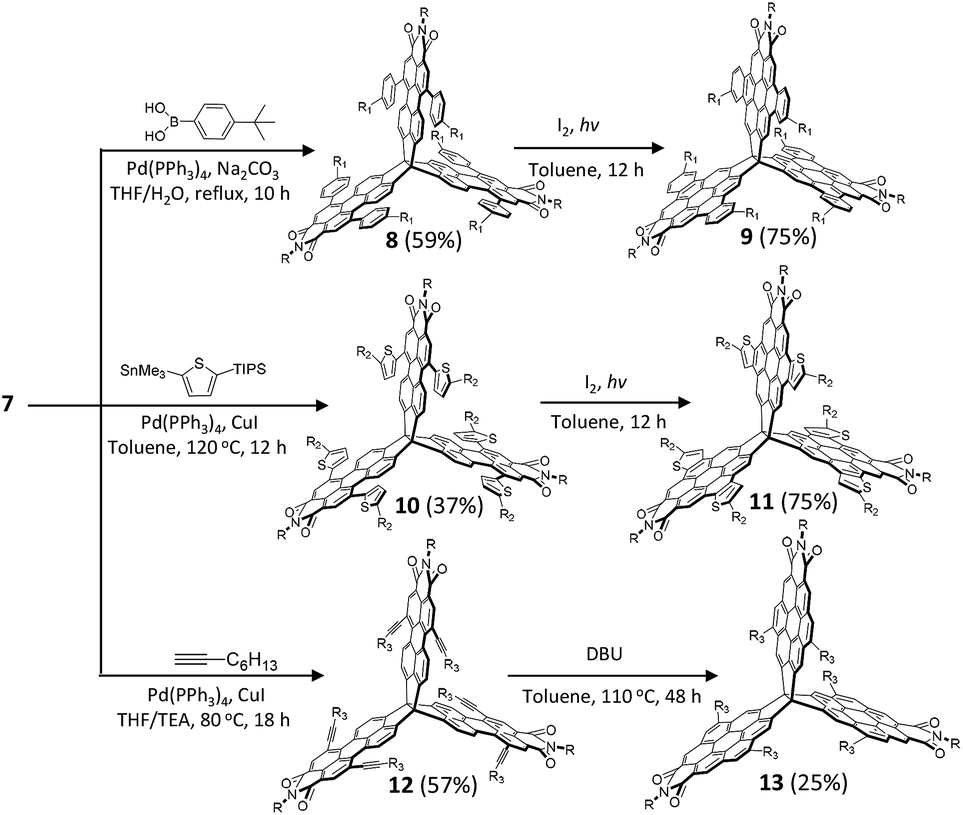 | ||
| Scheme 2 Synthesis of quasi-D3h symmetric nanostructures based on 6. | ||
X-ray crystallography
Single crystals of 9 suitable for X-ray diffraction analysis were grown by slow diffusion of methanol into toluene solution at room temperature. 9 crystallizes in the P21/c space group with four molecules per unit cell. As shown in Fig. 2A and B, the molecule adopts a quasi-D3h symmetric conformation with a dihedral angle of 116° between the individual subunits. The central carbon–carbon bond (denoted as C1–C2) length of the bridging ethylane unit in 9 is quite long, 1.62 Å, a feature due to the large PDI sizes and bulky alkyl chains, which combine to provide considerable strain to the central carbon–carbon bond. The surrounding carbon–carbon bond lengths are 1.51–1.53 Å, which are close to that of typical sp3–sp3 carbon–carbon bonds. 9 has a maximal distance of 2.1 nm between the ends of two subunits and a width of 1.2 nm and a length of 1.6 nm across one subunit.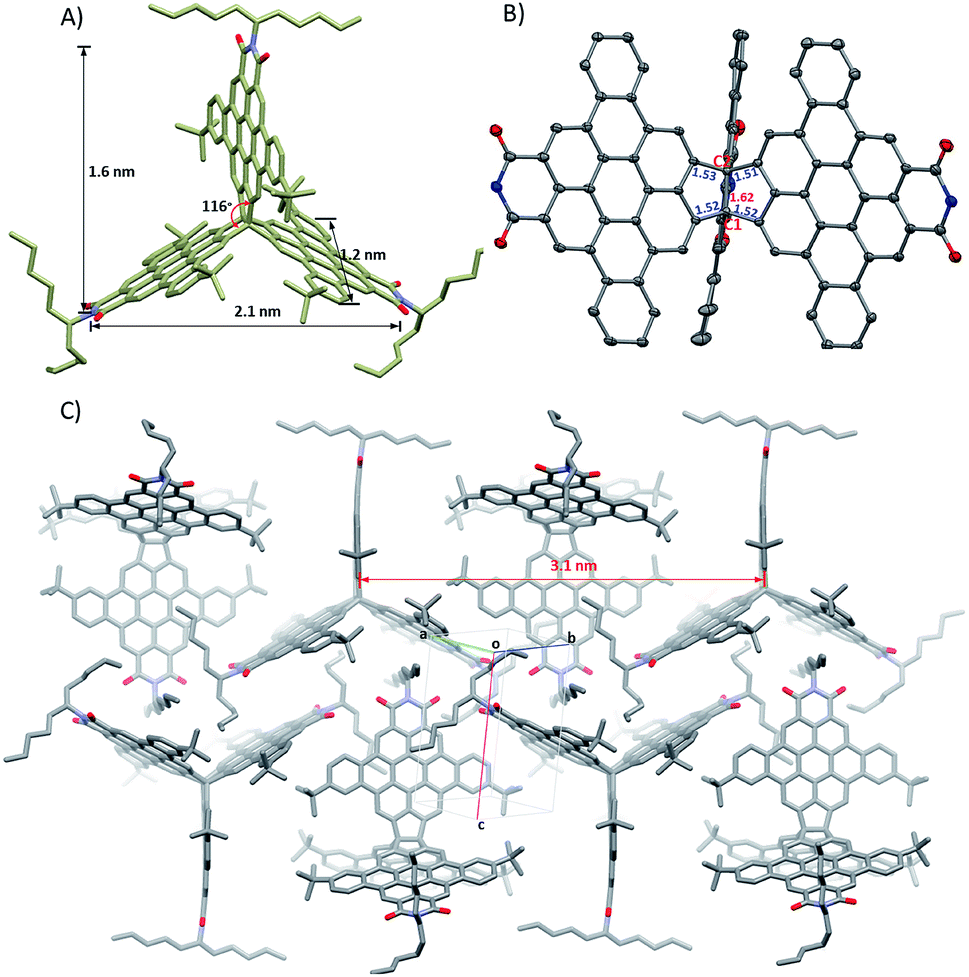 | ||
| Fig. 2 (A) Crystal structure of 9, (B) ORTEP structure with 30% probability of 9 with selected bond lengths (Å) of the central C1–C2 bond and the surrounding C–C bonds (butyl substituents and alkyl chains are omitted for clarity), and (C) crystal packing of 9. | ||
The molecules in the crystal pack in 2D layers. As shown in Fig. 2C, in an individual layer, two molecules are parallel to each other, with a distance of 3.1 nm between their symmetry axes, with the third molecule situated in the centre of this cavity and its symmetry axis perpendicular to that of neighbor molecule. Hence, there are no large voids in the layers. This crystal packing is stabilized via multiple short C–H⋯π contacts (in the range of 2.72–2.87 Å) among the alkyl chains on one molecule and the PDI monomers on adjacent molecules; no intermolecular π⋯π interactions are observed due to the alkyl chains, which block the overlap between the π-faces of the subunits. This packing contrasts with triptycene derivatives, which usually crystallize into layer structures that are driven by intermolecular π⋯π interactions between the individual acene subunits and produce large voids between the individual subunits.24,29
Electronic structure and optical and redox properties
To gain insight into the electronic and optical properties of these molecules, the electronic structure was evaluated via density functional theory (DFT) calculations at the OT-ωB97X-D/6-31g(d,p) level of theory (further details are given in the ESI†).56–58 Calculations were carried out for the four full molecules (6, 9, 11, and 13) and for each of the four monomeric units. The frontier HOMO and LUMO of 6, 9, 11 and 13 are assorted linear combinations of the HOMO and LUMO (respectively) of the individual monomers. As shown in Fig. 3, the HOMO is delocalized across all three units in 6, 9, 11, and 13. For 6 and 9, the HOMO-1 and HOMO-2 are energetically degenerate, are approximately 0.3 eV energetically stabilized when compared to the HOMO, and are delocalized on only two monomer units (Table S2†). The pattern differs in 11 and 13, where the degeneracy of the HOMO-1 and HOMO-2 is broken, with the HOMO-1 also delocalized over all three units (as with the HOMO), while the HOMO-2 spans only two monomers; here, the HOMO is approximately 0.2 eV higher in energy than the HOMO-1. All four molecules share the same pattern for the frontier LUMO, LUMO+1, and LUMO+2, where the LUMO and LUMO+1 are degenerate and delocalized over two of the three monomers while the LUMO+2 is delocalized over all three monomers.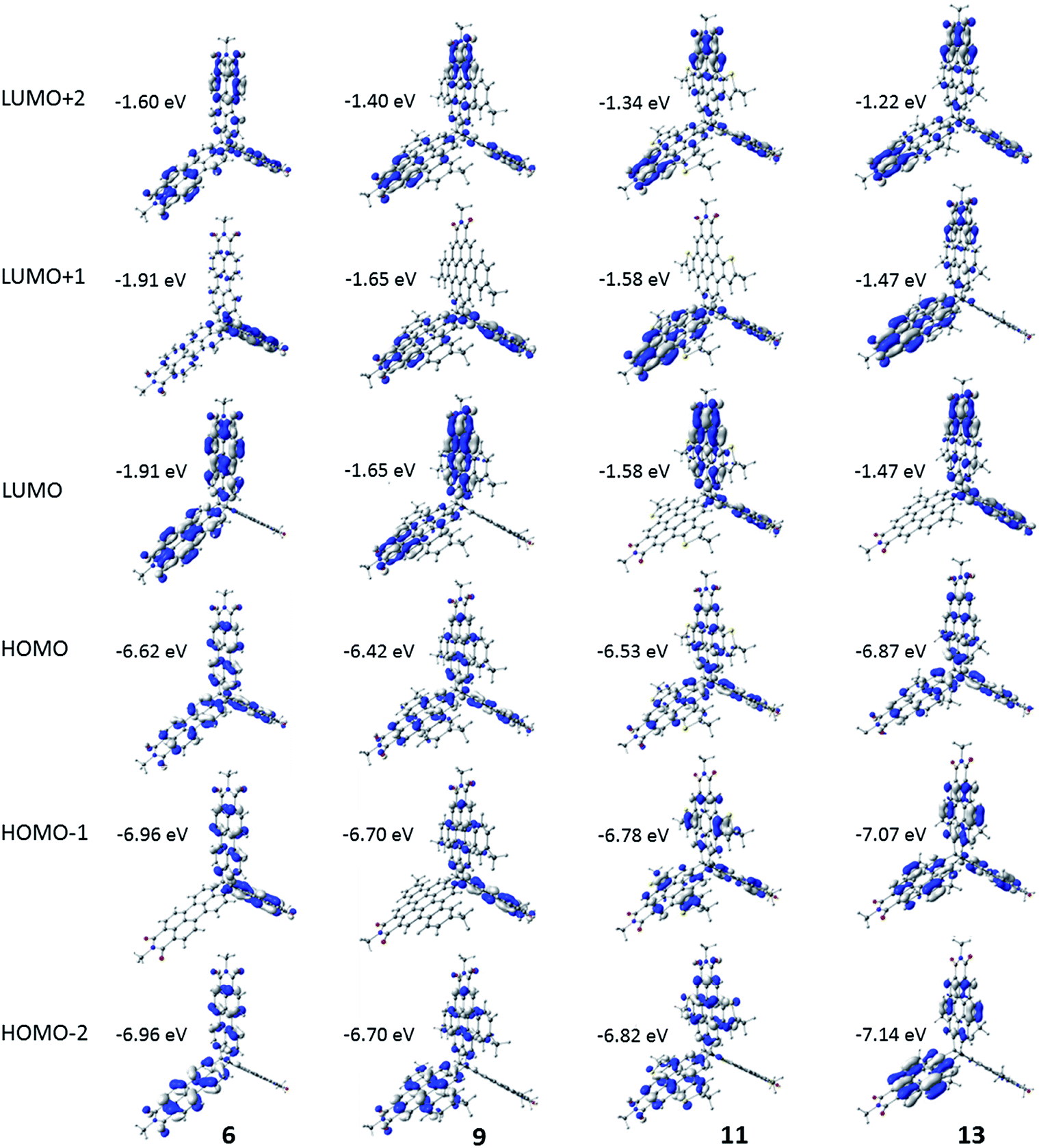 | ||
| Fig. 3 Pictorial representations of selected frontier molecular orbitals of 6, 9, 11, and 13 as determined at the OT-ωB97X-D/6-31g(d,p) level of theory. Pictorial representations of the monomeric units are shown in the ESI.† | ||
UV-vis absorption spectra of 6, 9, 11, and 13 in chloroform are shown in Fig. 4. In the high-energy (short wavelength) region, the absorption spectra of these compounds, except 6, show well-defined vibronic fine structures. In the low-energy (longer wavelength) region, the absorption spectra of the bay-extended compounds (9, 11 and 13) show a clear blue-shift relative to the parent 6, and there is a red-shift in the absorption in the order of 13 > 9–11. This trend is consistent with those previously observed in bay-extended PDI55 and PMI.44,59 It should be noted that compound 6 exhibits the characteristic absorption of typical perylene imide with maxima at 554 nm (2.24 eV), which is slightly red-shifted by 46 nm (0.20 eV) compared to that of PMI (508 nm; 2.44 eV), indicating that there is some electronic communication among the PMI subunits. Remarkably, at the longest wavelength absorption (554 nm), the maximum absorption coefficient is 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 844 M−1 cm−1, which is significantly larger than the sum of the three PMI units (3 × 33305 M−1 cm−1 = 99915 M−1 cm−1) (Fig. S1 in the ESI†), a phenomenon that has been observed in triptycene-based 3D graphene nanostructures.36 A similar red-shift and amplification of the absorption coefficient (122700 M−1 cm−1) at the maximum absorption wavelength (433 nm) of 13 are also found in contrast to the coronene monoimide subunit (418 nm, 33020 M−1 cm−1).59 Compounds 6 and 9 show strong emission bands at 588 nm and 514 nm, with fluorescence quantum yields of 85% and 89%, which are higher than those of 11 (505 nm, 22%) and 13 (418 nm, 32%).
844 M−1 cm−1, which is significantly larger than the sum of the three PMI units (3 × 33305 M−1 cm−1 = 99915 M−1 cm−1) (Fig. S1 in the ESI†), a phenomenon that has been observed in triptycene-based 3D graphene nanostructures.36 A similar red-shift and amplification of the absorption coefficient (122700 M−1 cm−1) at the maximum absorption wavelength (433 nm) of 13 are also found in contrast to the coronene monoimide subunit (418 nm, 33020 M−1 cm−1).59 Compounds 6 and 9 show strong emission bands at 588 nm and 514 nm, with fluorescence quantum yields of 85% and 89%, which are higher than those of 11 (505 nm, 22%) and 13 (418 nm, 32%).
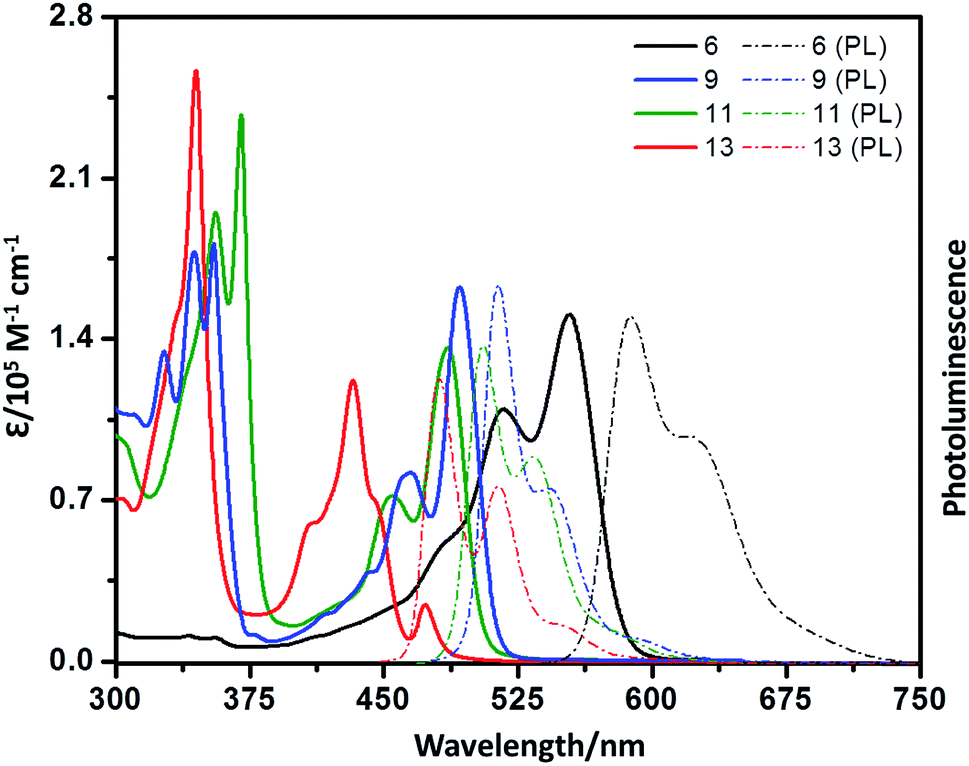 | ||
| Fig. 4 UV/vis absorption (solid line) and photoluminescence spectra (dotted line) of 6, 9, 11, and 13 in chloroform (∼10–5 M). | ||
Time-dependent DFT (TDDFT) calculations at the OT-ωB97X-D/6-31g(d,p) level of theory were performed to dig deeper into the nature of the molecular excited-state characteristics. For the full molecules, the excited-states contain significant contributions from several one-electron transitions (Table S2†), and state degeneracy makes the description of the excitations somewhat complex. Further, for 6, 11 and 13, the low-lying excitations with appreciable oscillator strengths are composed of two degenerate excitations, while for 9 this degeneracy is broken. As reported by Müllen and co-workers, the experimental spectrum of 13 does show a low-energy, low-oscillator strength transition that can be described as having PMI → coronene character.43,44,59 For 6 and 13, natural transition orbital (NTO) analyses show that the first excitations with large intensity are combinations of two transitions where the holes and electrons are delocalized on two monomers, while the second excitation is composed of a combination of two monomer-localized transitions. In 9, the pattern is reversed, though the lowest-energy transition has the holes and electrons mainly on one monomer. For 11, the two degenerate transitions are composed of two mostly monomer-localized hole–electron pairs, though in each there is some extension onto the second monomer. Overall, there is generally a good agreement among the UV-vis absorption spectra and computed TDDFT excitations.
Adiabatic ionization potentials and electron affinities (AIP and AEA, respectively) and spin densities were determined by DFT. Both the AIP and AEA decrease when moving from the monomer to the full 3D molecule, with the AEA decreasing more; we note that the decrease of each value is very similar across all four molecules (Table S2†). The spin densities, which reveal the distribution of the excess charge in the oxidized or reduced states, of the monomers display some density on the terminal sp3 carbons in both the anion and cation (Fig. S9†). In the full molecules, the radical-cation is fully delocalized – a feature consistent with the fact that the HOMO for each molecule was delocalized over each monomer unit. The radical-anion, on the other hand, is only partially delocalized. We recall that the LUMO and LUMO+1 are degenerate orbitals that are both delocalized across only two subunits; we deduce that the nature of the radical-anion spin density arises from a linear combination of this orbital character.
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) studies were performed in dichloromethane with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte, and the onset oxidation-reduction potentials were determined relative to Fc/Fc+ (4.8 eV) (Fig. 5). In contrast to the CV of PMI exhibiting two reduction waves and one oxidation wave, the CV of 6 reveals a splitting into four reduction waves and three oxidation waves. The observed splitting is due to the coulombic interactions from the electrons in different PMI subunits, which indicates electronic communication between different PMI subunits – a result consistent with evaluation of the DFT-derived MO for the neutral molecules and spin densities of the radical-cation and radical-anion states. The first half-wave reduction potential of 6 is −1.42 V, which is slightly more positive than that (−1.46 V) of PMI, suggesting weak conjugation between the individual PMI subunits,60 again consistent with the radical-anion spin densities and AEA. The CV of 9 also shows four reduction waves and three oxidation waves, and 11 exhibits four reduction waves and two oxidation waves. Interestingly, 13 shows only two reduction waves and two oxidation waves. Moreover, we observed that the oxidation waves of these compounds nearly overlap, which is further evidence that electron communication between the individual subunits is relatively weak. According to their onset potentials, electron affinities/ionization potentials (EA/IP) of 6, 9, 11, and 13 are estimated to be −3.46/−5.56, −3.29/−5.70, −3.31/−5.74, and −3.13/−5.89 eV, respectively (relative to Fc/Fc+, 4.8 eV).
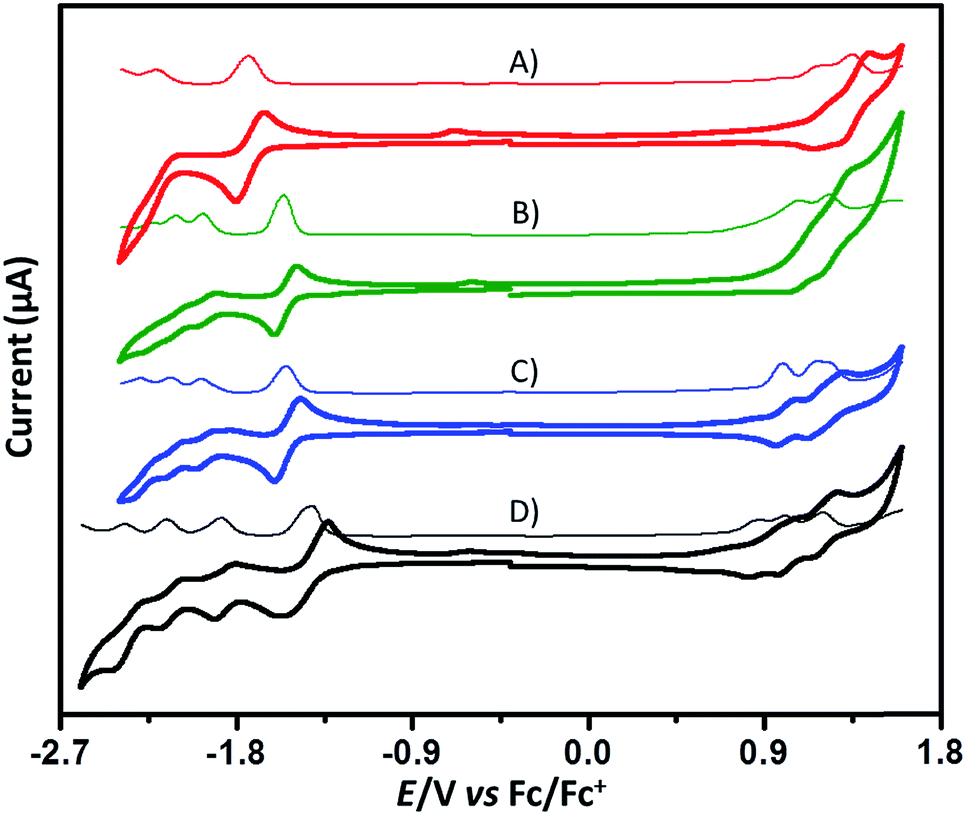 | ||
| Fig. 5 Cyclic voltammograms (bold solid line) and differential pulse voltammograms (thin solid line) of 13 (A), 11 (B), 9 (C), and 6 (D), performed in nitrogen-purged dichloromethane with TBAPF6 (0.1 M). | ||
Semiconductor properties
As a proof-of-concept, we evaluated the intrinsic charge-carrier mobility of 9 in the form of single-crystalline plates, which were prepared by slow diffusion of isopropyl alcohol into a concentrated toluene solution. Fig. 6A shows the SEM images of 9 that form rhombic plates. The SAED pattern of 9 could be indexed based on the single crystal data, which indicate that the (100) plane of the crystal is parallel to the surface of the substrate (Fig. 6B). The single crystalline field-effect transistors were fabricated by an “organic nanowire mask” technique with four electrodes to explore the transport properties along the different directions of the plates (Fig. 6C).61Fig. 6D shows the transfer characteristics of microplates of 9 tested under ambient conditions, which exhibit hole charge-carrier transport behavior. The conducting channels of 1 ↔ 2, 2 ↔ 3, 3 ↔ 4, and 4 ↔ 1 exhibit very similar mobilities of 4.92 × 10−3, 4.55 × 10−3, 4.95 × 10−3, and 4.51 × 10−3 cm2 V−1 s−1, respectively (Table S5†). Although these mobilities are by no means large, 10−3 cm2 V−1 s−1 is in fact quite good considering the limited intermolecular overlap of the π-conjugated units.33 Importantly, the distinctive structure with a quasi-D3h symmetric geometry could serve as a model to investigate the anisotropic charge-carrier transport in different lattice planes.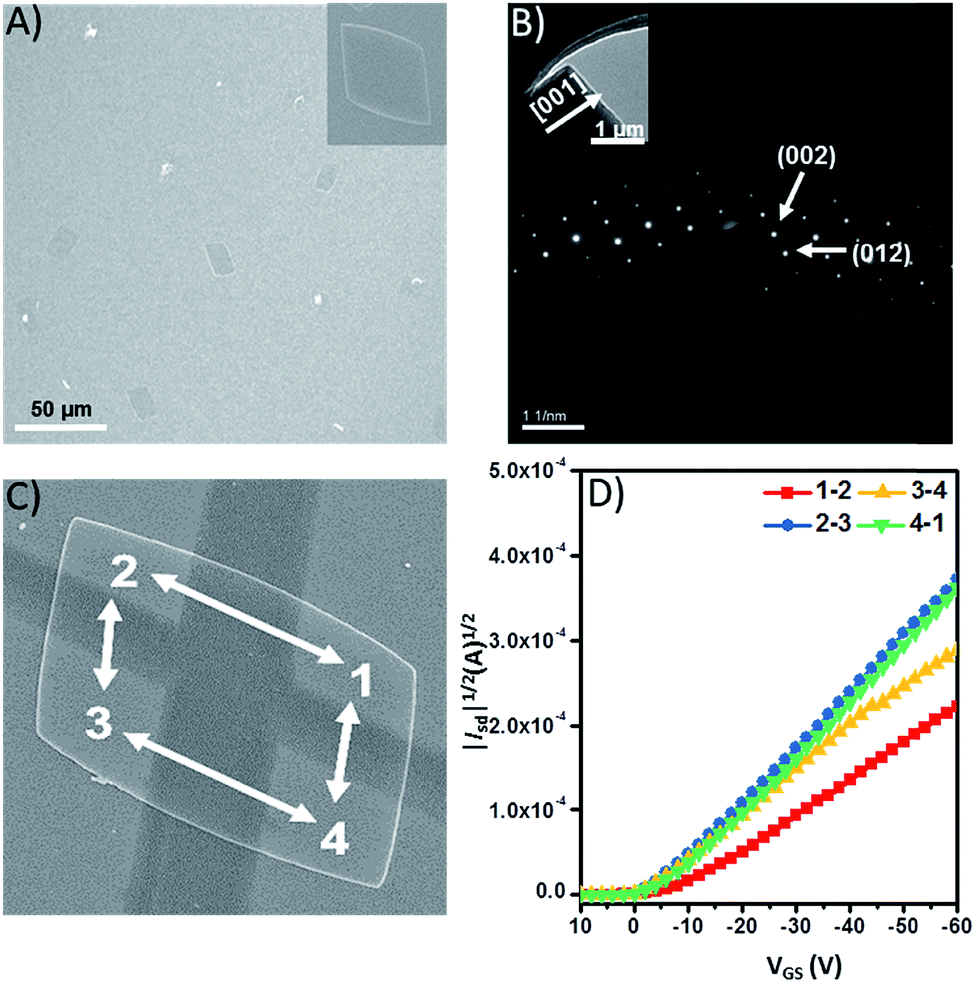 | ||
| Fig. 6 (A) SEM images of rhombic plates of 9 (inset: an individual single-crystalline plate), (B) the selected-area electron diffraction (SAED) patterns of plates of 9 (inset: TEM image of a single-crystalline plate), (C) SEM image of the corresponding transistor with four electrodes to probe the charge transport along (001) and (010) directions, and (D) the transfer curves of the device based on 9. | ||
Conclusions
In summary, we described the straightforward synthesis of 3D-symmetric rylene imide, triperyleno[3,3,3]propellane triimides, that shows attractive properties, such as high solubility, large π-surfaces, electron accepting ability, and many reactive sites. Through reaction with different substrates, we demonstrated that this rylene imide is compatible with different reactions, such as Pd-catalyzed cross-coupling reactions and cyclodehydrogenation reactions, which can be applied to synthesize a variety of complex 3D nanostructures with different molecular geometries. Here, a series of large, rigid 3D-symmetric nanostructures were successfully synthesized by functionalization of the bay positions of the building block. Compared to their subunits, these molecules show red-shifted and amplified absorptions. There is notable electronic communication among the monomer units, which influences the nature of the excited states and redox characteristics. The combination of facile synthetic accessibility, high chemical stability, molecular diversification, and tunable electronic properties makes the 3D rylene imide and its derivatives promising materials for use in organic electronic devices. Further studies to explore the relationship between the molecular geometry and the intramolecular electronic communication as well as for synthesis of even larger, rigid 3D PAHs for organic devices are now in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. Z. thanks the National Science Foundation of China (NSFC) (21672020 and 21871022) and the Beijing Natural Science Foundation (2182049). L. L. thanks the Distinguished Scientist Program at BUCT (buctylkxj02). C. X. thanks the China Postdoctoral Science Foundation (2017M610744). C. R. acknowledges funding from the National Science Foundation Designing Materials to Revolutionize and Engineer our Future (NSF DMREF) program under Award DMR-1627428. Supercomputing resources on the Lipscomb High Performance Computing Cluster were provided by the University of Kentucky Information Technology Department and Center for Computational Sciences (CCS). We thank Dr Sean Parkin (University of Kentucky) for crystal structure refinement.Notes and references
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582–2591 CrossRef CAS PubMed.
- S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed.
- A. Narita, X. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7645 CrossRef CAS PubMed.
- D. Kuck, Chem. Rev., 2006, 106, 4885–4925 CrossRef CAS PubMed.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS PubMed.
- B. VanVeller, D. J. Schipper and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 7282–7285 CrossRef CAS PubMed.
- A. M. Dilmaç, E. Spuling, A. d. Meijere and S. Bräse, Angew. Chem., Int. Ed., 2017, 56, 5684–5718 CrossRef PubMed.
- Y. Han, Z. Meng, Y.-X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026–2040 CrossRef CAS PubMed.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. V. Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- J. Hoogboom and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 15058–15059 CrossRef CAS PubMed.
- Z. Zhu and T. M. Swager, J. Am. Chem. Soc., 2002, 124, 9670–9671 CrossRef CAS PubMed.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- Y. Kim, J. Bouffard, S. E. Kooi and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 13726–13731 CrossRef CAS PubMed.
- T. M. Long and T. M. Swager, J. Am. Chem. Soc., 2003, 125, 14113–14119 CrossRef CAS PubMed.
- Z. Chen and T. M. Swager, Macromolecules, 2008, 41, 6880–6885 CrossRef CAS.
- H. Ma, J. Chen, L. Tan, J. Bu, Y. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS.
- B. Pei, W. Chan and A. W. M. Lee, Org. Lett., 2011, 13, 1774–1777 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2015, 21, 17308–17313 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051–6056 CrossRef CAS PubMed.
- B. Kohl, M. V. Bohnwagner, F. Rominger, H. Wadepohl, A. Dreuw and M. Mastalerz, Chem.–Eur. J., 2016, 22, 646–655 CrossRef CAS PubMed.
- K. Baumgärtner, A. L. M. Chincha, A. Dreuw, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15594–15598 CrossRef PubMed.
- C. Dengiz, S. P. Luppino, G. D. Gutierrez and T. M. Swager, J. Org. Chem., 2017, 82, 7470–7480 CrossRef CAS PubMed.
- N. G. White and M. J. MacLachlan, J. Org. Chem., 2015, 80, 8390–8397 CrossRef CAS PubMed.
- D. Reinhard, F. Rominger and M. Mastalerz, J. Org. Chem., 2015, 80, 9342–9348 CrossRef CAS PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- C. L. Hilton, C. R. Jamison, H. K. Zane and B. T. King, J. Org. Chem., 2009, 74, 405–407 CrossRef CAS PubMed.
- R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134–14141 CrossRef CAS PubMed.
- D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450–3453 CrossRef CAS PubMed.
- E. H. Menke, V. Lami, Y. Vaynzof and M. Mastalerz, Chem. Commun., 2016, 52, 1048–1051 RSC.
- P. Biegger, S. Stolz, S. N. Intorp, Y. Zhang, J. U. Engelhart, F. Rominger, K. I. Hardcastle, U. Lemmer, X. Qian, M. Hamburger and U. H. F. Bunz, J. Org. Chem., 2015, 80, 582–589 CrossRef CAS PubMed.
- D. Meng, H. Fu, B. Fan, J. Zhang, Y. Li, Y. Sun and Z. Wang, Chem.–Asian J., 2017, 12, 1286–1290 CrossRef CAS PubMed.
- S. R. Peurifoy, E. Castro, F. Liu, X. Zhu, F. Ng, S. Jockusch, M. L. Steigerwald, L. Echegoyen, C. Nuckolls and T. J. Sisto, J. Am. Chem. Soc., 2018, 140, 9341–9345 CrossRef CAS PubMed.
- S. R. Peurifoy, J. C. Russell, T. J. Sisto, Y. Yang, X. Roy and C. Nuckolls, J. Am. Chem. Soc., 2018, 140, 10960–10964 CrossRef CAS PubMed.
- S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100–13104 CrossRef CAS PubMed.
- S. V. Rosokha, I. S. Neretin, D. Sun and J. K. Kochi, J. Am. Chem. Soc., 2006, 128, 9394–9407 CrossRef CAS PubMed.
- T. Kodama, Y. Hirao, T. Nishiuchi and T. Kubo, ChemPlusChem, 2017, 82, 1006–1009 CrossRef CAS.
- M. R. Talipov, T. S. Navale and R. Rathore, Angew. Chem., Int. Ed., 2015, 54, 14468–14472 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Acc. Chem. Res., 2014, 47, 3135–3147 CrossRef CAS PubMed.
- L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC.
- Y. Avlasevich, C. Li and K. Müllen, J. Mater. Chem., 2010, 20, 3814–3826 RSC.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- G. Gao, N. Liang, H. Geng, W. Jiang, H. Fu, J. Feng, J. Hou, X. Feng and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15914–15920 CrossRef CAS PubMed.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- K. M. Lefler, D. T. Co and M. R. Wasielewski, J. Phys. Chem. Lett., 2012, 3, 3798–3805 CrossRef CAS PubMed.
- M. T. Vagnini, M. W. Mara, M. R. Harpham, J. Huang, M. L. Shelby, L. X. Chen and M. R. Wasielewski, Chem. Sci., 2013, 4, 3863–3873 RSC.
- G. Dyker, J. Körning, P. G. Jones and P. Bubenitschek, Angew. Chem., Int. Ed., 1993, 32, 1733–1735 CrossRef.
- G. Dyker, T. Kerl, J. Körning, P. Bubenitschek and P. G. Jones, Tetrahedron, 2000, 56, 8665–8668 CrossRef CAS.
- T. Kubo, S. Miyazaki, T. Kodama, M. Aoba, Y. Hirao and H. Kurata, Chem. Commun., 2015, 51, 3801–3803 RSC.
- A. Mukherjee, K. Pati and R.-S. Liu, J. Org. Chem., 2009, 74, 6311–6314 CrossRef CAS PubMed.
- M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895–7896 CrossRef CAS.
- C. L. Eversloh, C. Li and K. Müllen, Org. Lett., 2011, 15, 4148–4150 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- U. Rohr, C. Kohl, K. Müllen, A. Craatsb and J. Warman, J. Mater. Chem., 2001, 11, 1789–1799 RSC.
- Y. Wu, J. Han, M. Hong, M. D. Krzyaniak, A. K. Blackburn, I. R. Fernando, D. D. Cao, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 515–523 CrossRef CAS PubMed.
- L. Jiang, J. Gao, E. Wang, H. Li, Z. Wang and W. Hu, Adv. Mater., 2008, 20, 2735–2740 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterization of the compounds; 1H NMR, 13C NMR, HMBC, and 1D NOE spectra of the compounds; crystal data for 9; UV spectra and CV curves; theoretical calculation details; device fabrication and characterization. CCDC 1894754. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00849g |
| This journal is © The Royal Society of Chemistry 2019 |