
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed C–N bond activation: activated primary amines as alkylating reagents in reductive cross-coupling†
Huifeng
Yue‡
 a,
Chen
Zhu‡
a,
Li
Shen
a,
Qiuyang
Geng
a,
Katharina J.
Hock
a,
Tingting
Yuan
a,
Luigi
Cavallo
*b and
Magnus
Rueping
*ab
a,
Chen
Zhu‡
a,
Li
Shen
a,
Qiuyang
Geng
a,
Katharina J.
Hock
a,
Tingting
Yuan
a,
Luigi
Cavallo
*b and
Magnus
Rueping
*ab
aInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52074 Aachen, Germany. E-mail: magnus.rueping@rwth-aachen.de
bKAUST Catalysis Center, KCC, King Abdullah University of Science and Technology, KAUST, Thuwal 23955-6900, Saudi Arabia
First published on 20th March 2019
Abstract
Nickel-catalyzed reductive cross coupling of activated primary amines with aryl halides under mild reaction conditions has been achieved for the first time. Due to the avoidance of stoichiometric organometallic reagents and external bases, the scope regarding both coupling partners is broad. Thus, a wide range of substrates, natural products and drugs with diverse functional groups are tolerated. Moreover, experimental mechanistic investigations and density functional theory (DFT) calculations in combination with wavefunction analysis have been performed to understand the catalytic cycle in more detail.
1. Introduction
Transition-metal-catalyzed cross-coupling reactions play an important role in modern organic synthesis.1 Among them, reductive cross-coupling which uses two electrophiles as coupling partners represents an attractive catalytic platform for the formation of diverse chemical bonds.2 Due to the avoidance of stoichiometric organometallic reagents this transformation often possesses a broader substrate scope. Traditionally, alkyl halides,3 which may have limited stability and availability, have been used as alkyl electrophiles. Recently, N-hydroxyphthalimide esters, anhydrides, benzyl oxalates, tosylates and mesylates have been reported as alkyl coupling partners.4 However, the use of primary amines as readily available, cheap, green and stable alkylating reagents in reductive cross-coupling has not been described. Recently, bench-stable pyridinium salts have been reported to be attractive substrates in deaminative cross-coupling with boronic acids.5 In this transformation, the radical is generated via a single electron transfer (SET) pathway between the pyridinium salt and the nickel catalyst. In addition, an alkyl radical can be formed by a photoinduced SET process6 or by an elegant metal-free formation of a boron-based electron-donor–acceptor (EDA) complex7 under blue light irradiation. Also, a Lewis base promoted C–N borylation has been reported.8 As part of our continuing efforts in metal catalyzed functional group interconversion and the activation of inert bonds,9 we herein describe the first Ni-catalyzed deaminative reductive cross-coupling of activated primary amines with aryl halides, providing a versatile method for the transformation of amino groups to aryl motifs under mild conditions (Scheme 1).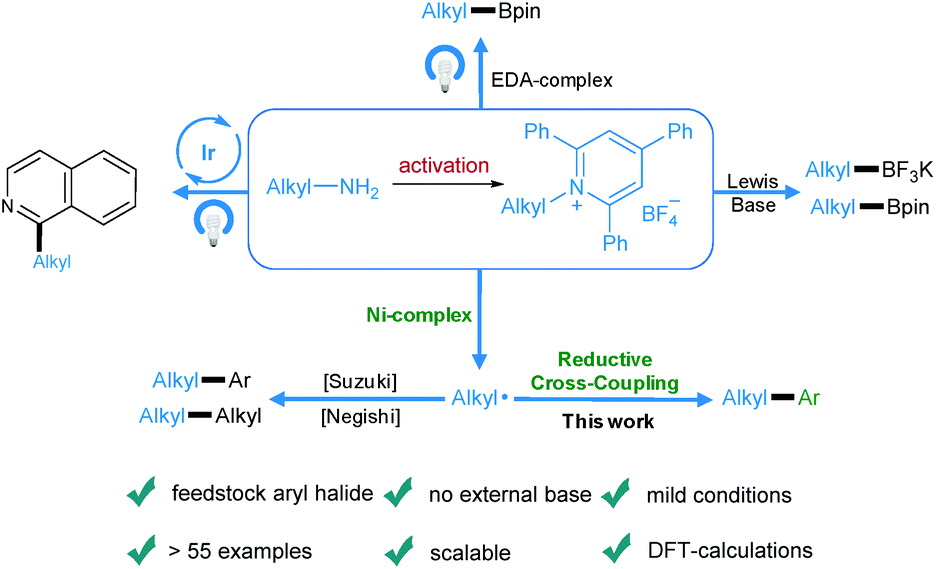 | ||
| Scheme 1 Diverse activation modes of pyridinium salts and the advantages of this novel deaminative reductive cross-coupling protocol. | ||
2. Results and discussion
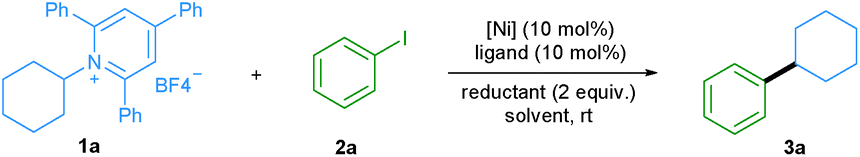
We started our investigation by evaluating the reductive cross-coupling of cyclohexylamine derived pyridinium salt 1a and iodobenzene 2a using NiCl2·dme as the catalyst, bipyridine ligand L1 as the ligand and a reducing agent (Table 1). Since solvents typically play an important role in cross-coupling reactions, we first tested several solvents (entries 1–5) and found DMA to be the optimal solvent. Subsequently the ratio of reactants 1a and 2a was evaluated (entries 6 and 7) and the yield was improved to 61%. Bidentate as well as tridentate N-containing ligands L1–L4 can be applied (entries 8–10) and L1, L2, and L4 showed a similar efficiency, providing the desired product in about 60% yield. With the readily available and cheap bipyridine L2, we examined a series of nickel catalysts, including NiBr2·dme, Ni(OAc)2·4H2O, Ni(acac)2, and NiCl2·6H2O. However lower yields were observed (entries 11–14). The yield was significantly improved to 99% when Mn powder was used as a reductant (entry 16), whereas the utilization of Mg powder gave the desired product in only 8% yield (entry 15). Besides aryl iodides, the protocol was also applied to aryl bromides. When bromobenzene was used, the desired product was also obtained in a good yield (entry 17). Control experiments demonstrated that the nickel catalyst, ligand, and reductant are all essential for the success of this transformation (entries 18–20).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Ni] | Ligand | Reductant | Solvent | Yieldb (%) |
a Reaction conditions: Katritzky pyridinium salt 1a (0.1 mmol), iodobenzene 2a (0.1 mmol), [Ni] (0.01 mmol), ligand (0.01 mmol), reductant (2 equiv.) in 1.0 ml solvent at rt.
b GC yield using decane as the internal standard.
c The ratio of 1a to 2a was set at 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
d The ratio of 1a to 2a was set at 1:2.
e Bromobenzene was used instead of iodobenzene. :1.
d The ratio of 1a to 2a was set at 1:2.
e Bromobenzene was used instead of iodobenzene.
|
|||||
| 1 | NiCl2·dme | L1 | Zn | DMA | 29 |
| 2 | NiCl2·dme | L1 | Zn | DMF | 11 |
| 3 | NiCl2·dme | L1 | Zn | CH3CN | 10 |
| 4 | NiCl2·dme | L1 | Zn | THF | 16 |
| 5 | NiCl2·dme | L1 | Zn | Toluene | 0 |
| 6c | NiCl2·dme | L1 | Zn | DMA | 61 |
| 7d | NiCl2·dme | L1 | Zn | DMA | 36 |
| 8c | NiCl2·dme | L2 | Zn | DMA | 61 |
| 9c | NiCl2·dme | L3 | Zn | DMA | 49 |
| 10c | NiCl2·dme | L4 | Zn | DMA | 60 |
| 11c | NiBr2·dme | L2 | Zn | DMA | 51 |
| 12c | Ni(OAc)2·4H2O | L2 | Zn | DMA | 15 |
| 13c | Ni(acac)2 | L2 | Zn | DMA | 35 |
| 14c | NiCl2·6H2O | L2 | Zn | DMA | 35 |
| 15c | NiCl2·dme | L2 | Mg | DMA | 8 |
| 16c | NiCl2·dme | L2 | Mn | DMA | 99 |
| 17c,e | NiCl2·dme | L2 | Mn | DMA | 81 |
| 18c | — | L2 | Mn | DMA | 0 |
| 19c | NiCl2·dme | — | Mn | DMA | 6 |
| 20c | NiCl2·dme | L2 | — | DMA | 0 |
|
|||||
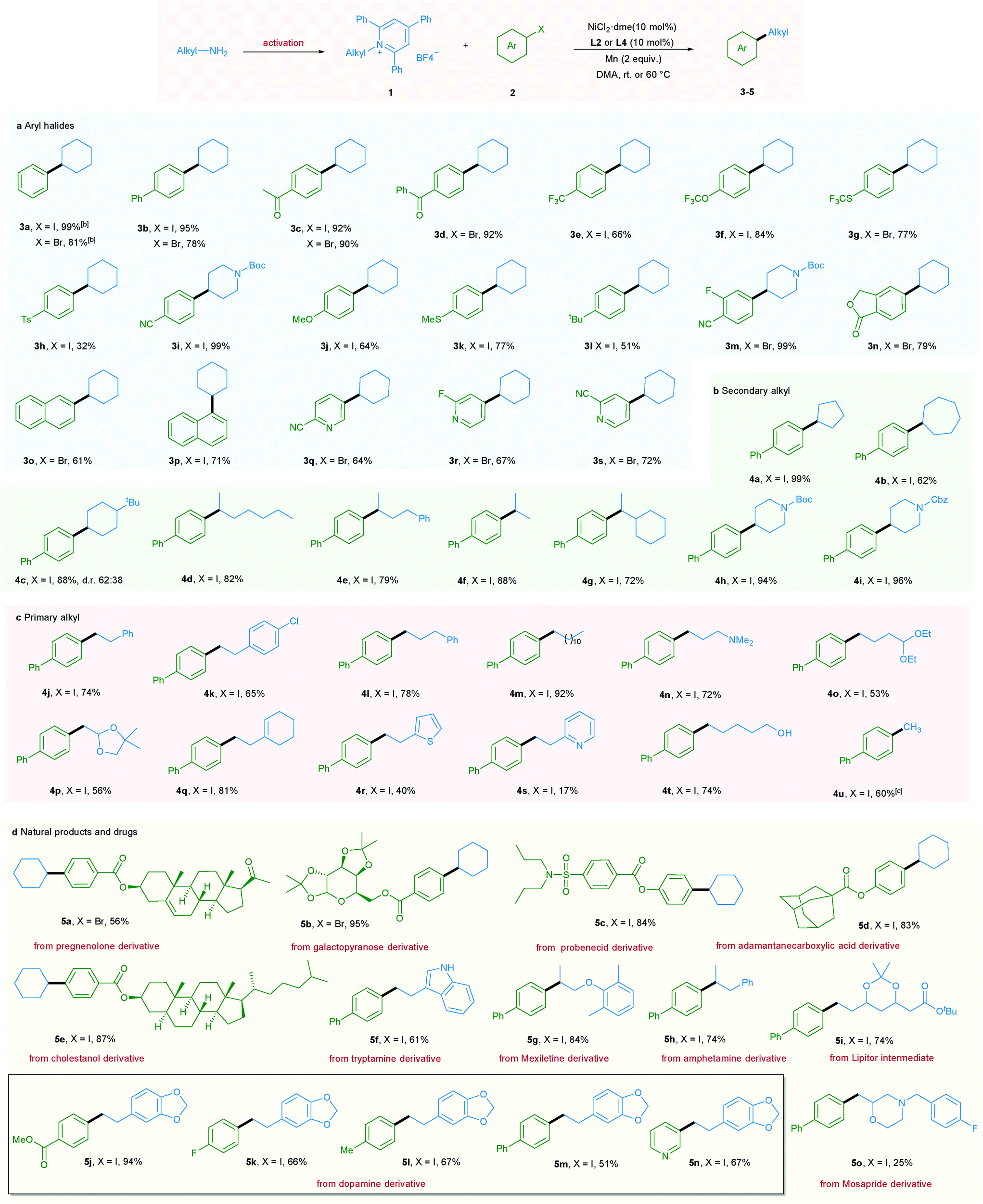
With the optimized reaction conditions in hand, the scope of aryl halides was first evaluated (Table 2a). A wide variety of aryl halides bearing electron-donating, electron-neutral, and electron-withdrawing functional groups could be successfully converted into the corresponding products in good to excellent yields. For example, phenyl and biphenyl iodides and bromides underwent this reaction smoothly, giving the corresponding products in excellent yields (3a and 3b).
|
a Reaction conditions: pyridinium salt 1 (0.30 mmol), aryl halide 2 (0.20 mmol), NiCl2·dme (0.02 mmol), L2 (0.02 mmol, for secondary alkyl) or L4 (0.02 mmol, for primary alkyl), Mn powder (0.40 mmol) in 1.0 ml DMA at rt (for secondary alkyl) or 60 °C (for primary alkyl); yields after purification.
b GC yield.
c The ratio of pyridinium salt to aryl halide 1:3, 100 °C, NMR yield.
|
|---|
|
In addition, a wide range of functional groups including ketone (3c and 3d), trifluoromethyl (3e), trifluoromethoxy (3f), trifluoromethylthio (3g), tosyl (3h), cyano (3i and 3m), methoxy (3j), methylthio (3k), t-butyl (3l), fluoro (3m, 3r and 5k), and ester (5j) were well tolerated under the mild reaction conditions, highlighting the high chemoselectivity of this newly developed deaminative reductive cross-coupling reaction. Use of disubstituted aryl bromide and bicyclic substrates including phthalides and naphthyl halides also gave the products 3m–3p in good yields. It is noteworthy that pharmaceutically relevant 3- and 4- bromopyridines could be applied to this protocol with good to high efficiency (3q–3s, 5n). Next, the scope of pyridinium salts was explored. A wide range of structurally diverse pyridinium salts were suitable substrates for this transformation. Cyclic and acyclic secondary amine substrates could undergo this deaminative arylation reaction in good to excellent yield (4a–4g) and the same applies for N-heterocyclic pyridinium salts (4h and 4i) (Table 2b).
It should be mentioned that when we applied primary alkyl pyridinium salts to this protocol, the reaction did not occur. However, simply switching the ligand from L2 to L4 and slightly raising the reaction temperature to 60 °C allowed this transformation to occur smoothly. A series of primary alkyl pyridinium salts bearing diverse functional groups such as amine, acetal, dioxole, cyclohexenyl, thiophene, and pyridine were suitable coupling partners for this deaminative reductive cross-coupling, leading to products 4n–4s. Notably, chloro (4k), unprotected OH and indole NH groups were also tolerated (4t and 5f), providing the option for further functionalization. Moreover, methylation reaction, which is challenging in reductive cross-coupling, was also realized via the utilization of methyl pyridinium salts (Table 2c). Importantly, our newly developed protocol could also be readily extended to a wide range of complex molecules derived from natural products and drugs. As such pregnenolone, galactopyranose, probenecid, adamantane carboxylic acid, and cholestanol derivatives could be transformed to the corresponding products 5a–5e in good to excellent yield. Moreover, a series of pyridinium salts derived from drugs or drug intermediates, including tryptamine, mexiletine, amphetamine, Lipitor intermediate, and dopamine, all underwent the mild coupling protocol with good to excellent efficiency (5f–5n). Use of Mosapride derived pyridinium salts gave product 5o in a lower yield (Table 2d).
Additionally, a gram-scale reaction was successfully conducted using 1a and 4-iodobiphenyl 2b in the presence of only 5 mol% nickel catalyst and the desired product 3b was obtained in 96% yield (Scheme 2a), demonstrating the practicability of our newly developed deaminative reductive cross-coupling methodology. Also, byproduct 6, which is potentially a useful organic base, could be isolated in 85% yield. To shed light on the mechanism of this transformation, an experiment was conducted with the radical trapping reagent TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy, 2 equiv.).5a,c The reaction was suppressed and no product 5m was detected (Scheme 2b).
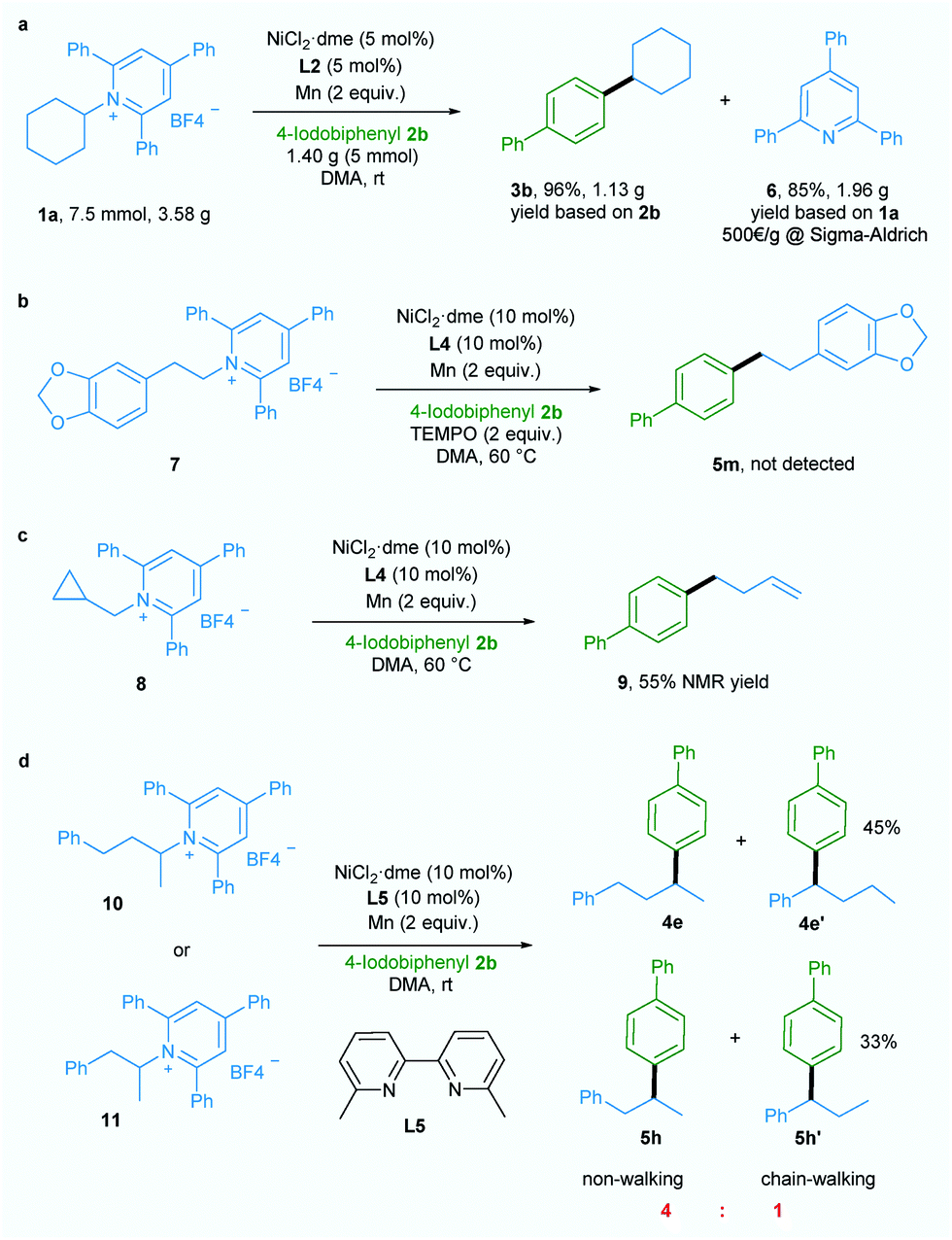 | ||
| Scheme 2 Gram-scale reaction and mechanism investigation. | ||
Also, ring-opened product 9 was generated in 55% yield when a substrate bearing a cyclopropane motif 8 was used (Scheme 2c).5a,c Both these results suggest the involvement of an alkyl radical in this transformation. When ligand L5 which is effective for chain-walking reductive cross-coupling10 was used in our catalytic system, non-walking and chain-walking products (4e and 4e′, 5h and 5h′) were obtained with a ratio of 4:1, suggesting an oxidative addition of aryl halide to Ni0 to give a NiII intermediate prior to alkyl radical generation. Since the NiIII intermediate generated from addition of the alkyl radical to the NiII intermediate is less likely to undergo the chain-walking step due to steric hindrance the non-chain-walking product is the major product.
Furthermore, detailed DFT calculations were performed to rationalize our newly designed catalytic reaction (Scheme 3; computational methods, see ESI†). As a model system, we investigated the reaction of phenyl bromide with A1 in the presence of NiCl2·dme, bpy as the ligand and Mn as the reducing agent. The reaction starts with the complexation of the ligand bpy to the NiII precatalyst, followed by reduction to form the active Ni0 catalyst B (Scheme S1 in ESI†). The catalytic process is initiated by oxidative addition of phenyl bromide to Ni0via transition state B-TS with an energy barrier of 11.7 kcal mol−1. The formed NiII intermediate C is reduced by Mn, leading to intermediate D with a free energy gain of 6.9 kcal mol−1. In the next step, A1 is coordinated to D, followed by SET reduction of A1 to generate radical A2 and NiII intermediate F. The radical A2 is prone to undergo C–N bond cleavage with an energy barrier of 19.9 kcal mol−1, liberating the alkyl radical A3 and the aromatic pyridine A4. At this point, alkyl radical A3 adds to NiII intermediate F to form NiIII intermediate G. Subsequently, the C–C bond cross-coupling product A5 is formed via reductive elimination from NiIII with an energy barrier of 4.9 kcal mol−1.
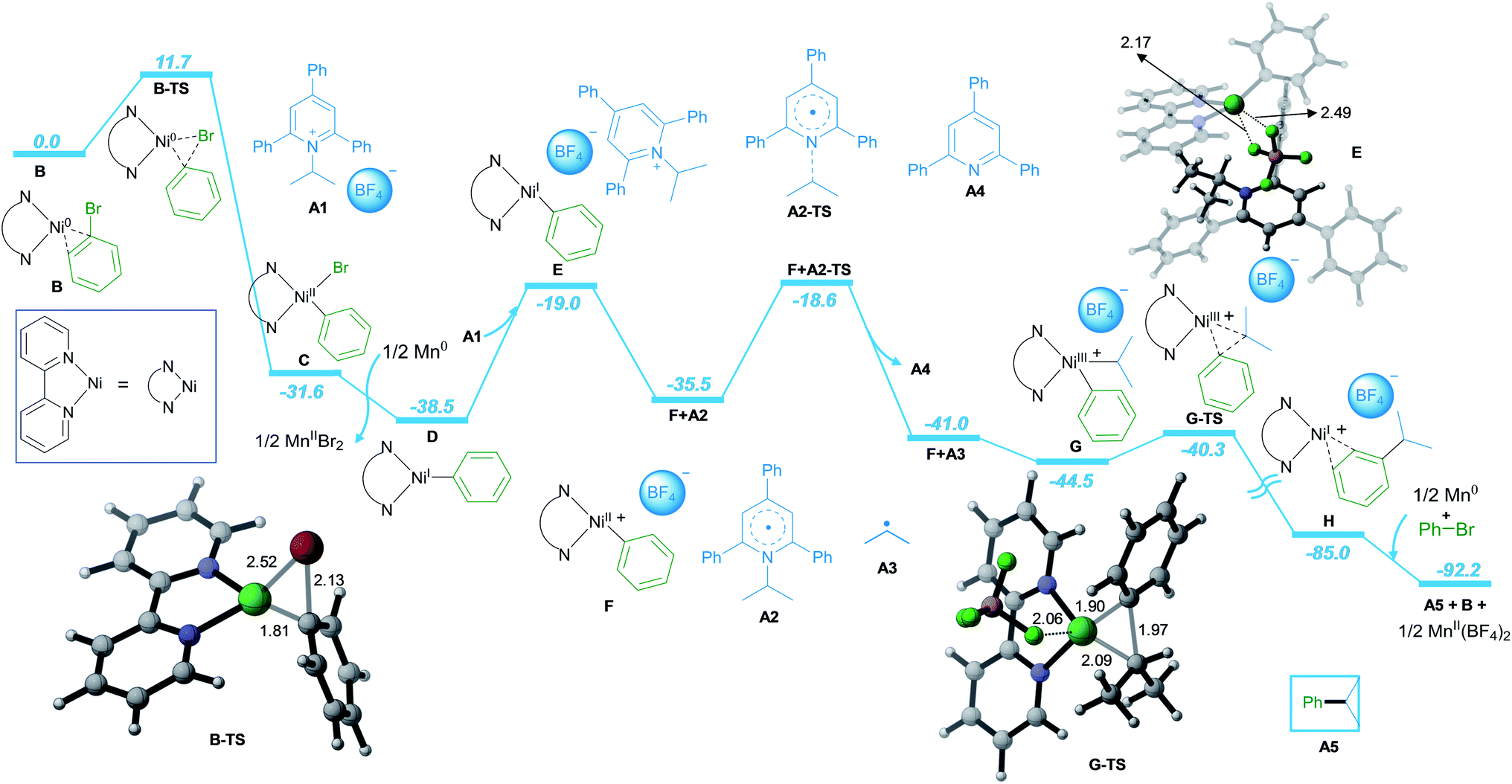 | ||
| Scheme 3 DFT-Computed energy profile for the nickel-catalyzed reductive cross-coupling reaction of aryl halides and pyridinium salts. Free energies in solution (in kcal mol−1) at the SMD(DMA)-M06/Def2-QZVPP//ωB97xD/Def2-TZVP(Ni,Mn)/Def2-SVP (non-metal) level are displayed. Selected DFT optimized geometries are listed. Bond lengths are in Å. | ||
Finally, the NiI intermediate H is further reduced by Mn to regenerate the Ni0 active catalyst B and initiate the next catalytic cycle.
Subsequently, we focused on the origin of the SET reduction of the pyridinium salt and the generation of the alkyl radical.11 The molecular orbital plots (Fig. 1a) show that the SOMO of E corresponds to the singly occupied MO predominantly localized on the dz2-orbital of Ni. At the same time, the LUMO of E corresponds to a π-orbital delocalized around the central nitrogen-containing aromatic ring of the pyridinium salt. However, after A2 is displaced away from the Ni species, the SOMO becomes localized on the central aromatic ring of the pyridinium salt. This indicates that upon separation of the Ni and pyridyl fragments the unpaired electron transfers from Ni to the pyridinium salt and delocalizes around the central aromatic ring. Spin density analysis further supported this process. The unpaired electron density is localized on the Ni-center when the pyridinium salt is coordinated to the NiI-complex, as in E (Fig. 1b, left), while it is transferred to the central pyridine ring as the pyridine moiety A2 dissociates from the Ni complex (Fig. 1b, middle). Subsequently, the C–N bond dissociates and the spin density is further transferred to the sp3-carbon atom, indicating generation of the alkyl radical (Fig. 1b, right). Additionally, cyclic voltammetry (CV) measurements of the pyridinium salt were conducted (see ESI†) and reversible peaks at −0.94 V vs. SCE were observed, suggesting the existence of stable radical intermediate A2. In order to understand the C–N bond dissociation, we performed localized orbital locator (LOL) – π analysis12 and multi-center bond order calculations (Iring)13 for the whole process (Fig. 1c). Before the SET reduction, the central pyridine ring has full aromaticity (Iring = 0.05 in A1, for comparison, Iring = 0.05 in benzene and pyridine12c). After the reduction the aromaticity is lost and the free energy is 3.0 kcal mol−1 higher (Iring = 0 in A2). With the C–N bond dissociating, the aromaticity is partially regained (c.f. Iring = 0.01 in transition state A2TS). After the generation of alkyl radicals, the aromaticity of A3 is fully restored (Iring = 0.05 in A3) with a free energy gain of 5.5 kcal mol−1.
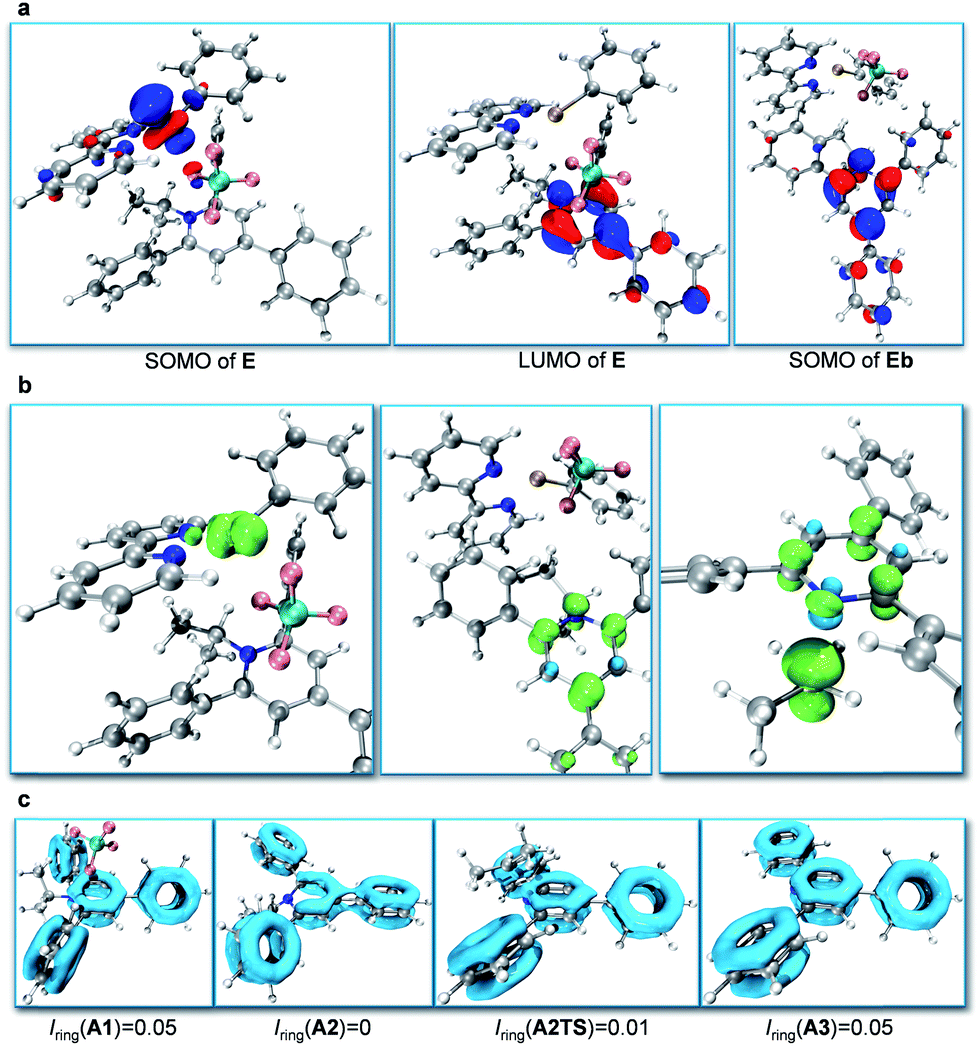 | ||
| Fig. 1 (a) Molecular orbital plots: SOMO of E (left), LUMO of E (middle) and SOMO of Eb (right). (b) Spin density plots of E (left), Eb (middle) and A2TS (right). (c) Localized orbital locator (LOL) – π plots of A1, A2, A2TS, and A3 with multi-center bond order of the central nitrogen-containing aromatic ring Iring listed below. | ||
3. Conclusions
In summary, we have developed a new deaminative reductive cross-coupling of aryl halides with pyridinium salts derived from readily available alkyl amines. Due to the avoidance of stoichiometric organometallic reagents and external bases, the scope is rather broad as demonstrated using the many different substrates employed (>55 examples). In addition, the chemoselectivity of this protocol is good and functional groups, including cyano, methoxy, methylthio, fluoro as well as chloro, unprotected OH and indole NH groups, are well tolerated. Importantly, the cross-coupling reaction can be scaled-up using a lower amount of nickel catalyst without diminishing the yield, demonstrating the practicability of this protocol. Furthermore, experimental mechanistic investigations and DFT calculations combined with wavefunction analysis have been conducted to gain insight into the catalytic process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. Y. acknowledges the China Scholarship Council. C. Z. acknowledges the King Abdullah University of Science and Technology (KAUST) for support and the KAUST Supercomputing Laboratory for providing computational resources of the supercomputer Shaheen II.Notes and references
- (a) T. J. Colacot, New Trends in Cross-Coupling: Theory and Applications, RSC Publishing, Cambridge, 2014 RSC; (b) A. d. Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, 2014 CrossRef.
- (a) C. E. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed; (b) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS PubMed.
- Examples of reductive cross couplings with N-hydroxyphthalimide esters: (a) K. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed; (b) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 CrossRef CAS PubMed; (c) N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150–2153 CrossRef CAS PubMed; (d) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338–1341 CrossRef CAS PubMed; (e) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809–814 RSC; anhydrides: (f) H. Chen, L. Hu, W. Ji, L. Yao and X. Liao, ACS Catal., 2018, 8, 10479–10485 CrossRef CAS; benzyl oxalates: (g) X.-B. Yan, C.-L. Li, W.-J. Jin, P. Guo and X.-Z. Shu, Chem. Sci., 2018, 9, 4529–4534 RSC; tosylates and mesylates: (h) J. H. Liu, C. T. Yang, X. Y. Lu, Z. Q. Zhang, L. Xu, M. Cui, X. Lu, B. Xiao, Y. Fu and L. Liu, Chem.–Eur. J., 2014, 20, 15334–15338 CrossRef CAS PubMed.
- For leading work, see: (a) C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313–5316 CrossRef CAS PubMed; (b) J. Liao, W. Guan, B. P. Boscoe, J. W. Tucker, J. W. Tomlin, M. R. Garnsey and M. P. Watson, Org. Lett., 2018, 20, 3030–3033 CrossRef CAS PubMed; (c) S. Plunkett, C. H. Basch, S. O. Santana and M. P. Watson, J. Am. Chem. Soc., 2019, 141, 2257–2262 CrossRef CAS PubMed.
- (a) F. J. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336–12339 CrossRef CAS PubMed; (b) X. Jiang, M.-M. Zhang, W. Xiong, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 2402–2406 CrossRef CAS PubMed; (c) F. J. Klauck, H. Yoon, M. J. James, M. Lautens and F. Glorius, ACS Catal., 2018, 9, 236–241 CrossRef; (d) M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362–11367 CrossRef CAS.
- (a) J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed; (b) F. Sandfort, F. Strieth-Kalthoff, F. J. Klauck, M. J. James and F. Glorius, Chem.–Eur. J., 2018, 24, 17210–17214 CrossRef CAS PubMed.
- J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227–15231 CrossRef CAS PubMed.
- Examples: (a) M. Leiendecker, C. C. Hsiao, L. Guo, N. Alandini and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 12912–12915 CrossRef CAS PubMed; (b) L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef CAS PubMed; (c) L. Guo, C.-C. Hsiao, H. Yue, X. Liu and M. Rueping, ACS Catal., 2016, 6, 4438–4442 CrossRef CAS; (d) L. Guo, X. Liu, C. Baumann and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 15415–15419 CrossRef CAS PubMed; (e) X. Liu, C. C. Hsiao, I. Kalvet, M. Leiendecker, L. Guo, F. Schoenebeck and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6093–6098 CrossRef CAS PubMed; (f) H. Yue, L. Guo, S. C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef CAS PubMed; (g) H. Yue, L. Guo, H. H. Liao, Y. Cai, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 4282–4285 CrossRef CAS PubMed; (h) H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371–1375 CrossRef CAS PubMed.
- F. Chen, K. Chen, Y. Zhang, Y. He, Y.-M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929–13935 CrossRef CAS PubMed.
- (a) Molecular orbital plot, spin density plot, and LOL-π plot were visualized by VMD, see: W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS; (b) Molecular orbitals, spin density, LOL–π, and multi-center bond order were analyzed by Multiwfn 3.6. see: T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) H. Schmider and A. Becke, J. Mol. Struct.: THEOCHEM, 2000, 527, 51–61 CrossRef CAS; (b) V. Tsirelson and A. Stash, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 780–785 CrossRef; (c) H. Jacobsen, Can. J. Chem., 2008, 86, 695–702 CrossRef CAS.
- (a) M. Giambiagi, M. S. de Giambiagi and K. C. Mundim, Struct. Chem., 1990, 1, 423–427 CrossRef CAS; (b) P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS; (c) E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00783k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |