
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBorohydride-containing coordination polymers: synthesis, air stability and dehydrogenation†
Kentaro
Kadota
a,
Nghia Tuan
Duong
 b,
Yusuke
Nishiyama
bc,
Easan
Sivaniah
ad,
Susumu
Kitagawa
d and
Satoshi
Horike
*defg
b,
Yusuke
Nishiyama
bc,
Easan
Sivaniah
ad,
Susumu
Kitagawa
d and
Satoshi
Horike
*defg
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
bRIKEN-JEOL Collaboration Center, Tsurumi, Yokohama, Kanagawa 230-0045, Japan
cJEOL RESONANCE Inc., Musashino, Akishima, Tokyo 196-8558, Japan
dInstitute for Integrated Cell-Material Sciences, Institute for Advanced Study, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: horike@icems.kyoto-u.ac.jp
eAIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Yoshida-Honmachi, Sakyo-ku, Kyoto 606-8501, Japan
fDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
gDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
First published on 13th May 2019
Abstract
Control of the reactivity of hydride (H−) in crystal structures has been a challenge because of its strong electron-donating ability and reactivity with protic species. For metal borohydrides, the dehydrogenation activity and air stability are in a trade-off, and control of the reactivity of BH4− has been demanded. For this purpose, we synthesize a series of BH4−-based coordination polymers/metal–organic frameworks. The reactivity of BH4− in the structures is regulated by coordination geometry and neighboring ligands, and one of the compounds [Zn(BH4)2(dipyridylpropane)] exhibits both high dehydrogenation reactivity (1.4 wt% at 179 °C) and high air stability (50 RH% at 25 °C, 7 days). Single crystal X-ray diffraction analysis reveals that Hδ+⋯Hδ− dihydrogen interactions and close packing of hydrophobic ligands are the key for the reactivity and stability. The dehydrogenation mechanism is investigated by temperature-programmed desorption, in situ synchrotron PXRD and solid-state NMR.
Controlling the reactivity of hydride-based ions in the solid state is important for hydrogen-related functions such as hydrogen (H2) storage, heterogeneous hydrogenation catalysis and ion conduction.1–3 Hydride-based ions include hydride (H−) and molecular ions such as borohydride (BH4−) and alanate (AlH4−), and they exhibit a strong electron-donating ability.4 The reactive character causes high sensitivity to moisture in the air, and it has been a challenge to design the dual properties of high reactivity of hydrides and air stability in the solid state. For example, the dehydrogenation reactivity and air stability are in a trade-off for BH4− in metal borohydrides (MBHs). Air-stable NaBH4 releases H2 above 500 °C, whereas Al(BH4)3 is pyrophoric in contact with moisture in the air, and it releases H2 at 60 °C.5,6 The properties of MBHs are affected by some parameters of metal ions – electronegativity and ionic radius.7 A number of studies to tune the properties of MBHs were made (e.g., mixed metal ion MBHs,8–10 hybridization with organic polymers,11 and nano-confinement in porous scaffolds12,13). The reactivity of BH4− in crystal structures depends on hydrogen–hydrogen interactions (e.g., hydrogen bonding and dihydrogen bonding).14,15 For the precise tuning of these interactions, we applied coordination polymers (CPs) and metal–organic frameworks (MOFs).16–19 Tuning the coordination geometry and reactivity of metal ions or bridging ligands can afford the precise configuration of ions/molecules in CP architectures. CP structures are thought to be constructed with BH4−; however it has been unsuccessful because of the high reactivity of BH4−.
The attempts to incorporate reactive BH4− into CP structures readily lead to the reduction of metal ions or decomposition of organic ligands. There are many reports on polymeric crystal structures consisting of metal ions and bridging BH4−.20 On the other hand, the extended BH4−-based crystal structures constructed from metal ions and organic bridging ligands are limited: ([Mg(BH4)2(pyrazine)2] and [Th(OTerMes)2(BH4)2(4,4′-bipyridyl)]) were the only examples reported in the Cambridge Crystallographic Data Centre (CCDC) database.21,22 As shown in previous studies, neutral N-donor ligands are suitable to incorporate BH4− as a counter anion in CP structures. We then used commercially available Mg(BH4)2 and Ca(BH4)2 for CP synthesis using neutral N-donor ligands under Ar. The solution reaction using Mg(BH4)2 and 4,4′-bipyridyl in acetonitrile (MeCN) afforded a polymeric structure. Ca(BH4)2 with poor solubility in organic solvents is not suitable for solution reactions. A solvent-free mechanochemical reaction of Ca(BH4)2 and pyrazine produced crystalline powder of CP. On the other hand, the synthetic attempts using other pyridyl-based ligands afforded amorphous powder or no solid product. This is because of a weak coordination interaction between hard Mg2+ or Ca2+ and soft N-donor ligands according to Hard and Soft Acids and Bases (HSAB) theory. The theory says that soft transition metal ions (e.g., Zn2+ and Mn2+) form stable coordination bonds with N-donor ligands. [PPh4Zn(BH4)3] and [Mn(BH4)2·3THF]·NaBH4 were therefore prepared as starting materials.23 Bulky PPh4+ cations were incorporated to increase the solubility of the salt. Transition metal ion-based MBH precursors successfully form coordination bonds with pyridyl and imidazolate ligands, and 10 CP crystals were isolated as summarized in Fig. 1. In general, the structural analysis from a single crystal for MBH is difficult, and it is usually hard to determine the exact position of hydrogen atoms in BH4−.24,25 In the present cases, high-quality single-crystal diffraction data enable us to locate the hydrogen atoms of BH4− in a Fourier map and refine freely without restraints. This is beneficial to understand the local structure of BH4− and the resulting properties.
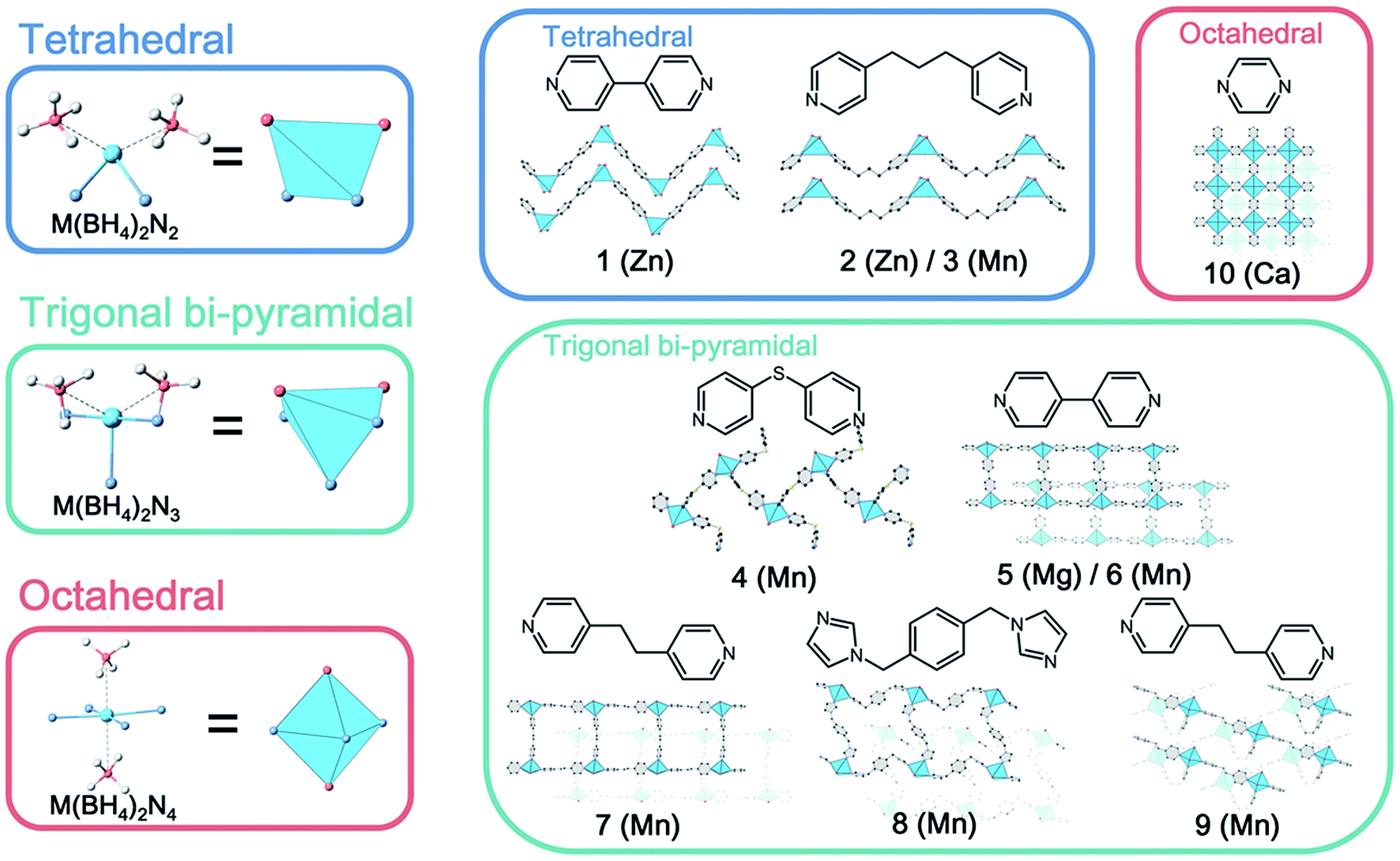 | ||
| Fig. 1 Crystal structures of BH4−-based CPs. Tetrahedral (1–3), trigonal bi-pyramidal (4–9) and octahedral (10) geometries, and constituent metal ions and ligands are represented, respectively. | ||
As shown in Fig. 1, each product is classified according to the local coordination environments at the M2+ center (tetrahedral, trigonal bi-pyramidal and octahedral, where BH4− is assumed to occupy one site). As the coordination number of ligands increases, the M–B distance increases (tetrahedral: 2.339 Å, trigonal bi-pyramidal: 2.499 Å, and octahedral: 2.696 Å, Table S3†). Electron-donation from N-donor ligands to metal ions decreases the electronic interaction between M and BH4−.21 The coordination mode of BH4− to metal ions was clearly determined, and most of them were bidentate. In addition, BH4− in the structures shows dihydrogen bonding with the hydrogen atoms of bridging ligands. A dihydrogen bond is an interaction between negatively charged hydrogen and positively charged hydrogen (e.g., B–Hδ−···Hδ+–C, normally <2.4 Å), and affects the crystal packing and physical properties.26 The number of dihydrogen bonds varies in the range of 0 to 3 as summarized in Table S3.† Four packing structures were observed, one-dimensional (1D) chain (1, 2, 3, 4), 1D ladder (5, 6, 7, 8), two-dimensional (2D) bilayer (9) and 2D sheet (10). The CPs with a higher coordination number of the ligands show higher structural dimensionality. 5, 6, 7 and 9 contain MeCN as guest molecules, and the permanent porosity of 5 and 6 was determined by N2 and CO2 adsorption (Fig. S27 and S28†).
Bulk powder samples were prepared under the same reaction conditions, and phase purity was confirmed by PXRD and infrared (IR) spectroscopy under Ar (Fig. S24 and S25†). The thermal properties were characterized by thermogravimetric analysis (TGA) under Ar (Fig. S26†). As various types of chemical environments of BH4− were observed in 1–10, the dependence of the air stability on the local structure was examined. Each powder sample (30 mg) was exposed to humidified air (relative humidity (RH) 50% at 25 °C, Fig. S29†), and time-course IR spectra and PXRD patterns were recorded to monitor BH4−. Reference MBHs, NaBH4 and Mg(BH4)2 were also evaluated for comparison. After air exposure of Mg(BH4)2 for 3 hours, no peak is observed in the B–H region in the IR spectrum (Fig. 2A). The broad peak at 3500 cm−1 indicates the hydrolysis of BH4−. Although NaBH4 is considered as an air-stable MBH, it showed deliquescence in 2 hours under the conditions and hydrolyzed to form Na2[B4O5(OH)4]·3H2O after air exposure for 7 days (Fig. S30†). Humidified conditions are harsh enough to hydrolyze BH4− in most of the MBHs.
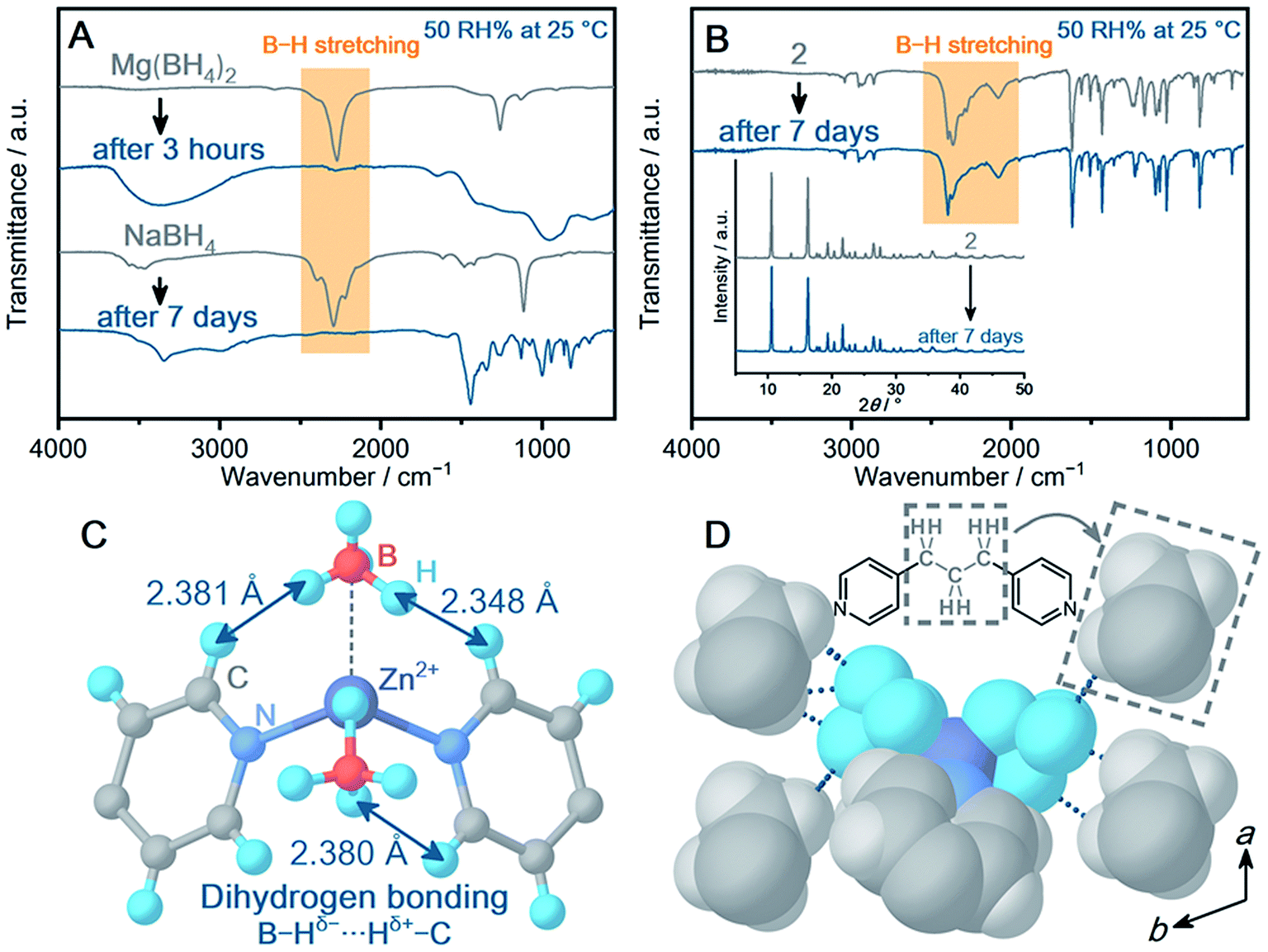 | ||
| Fig. 2 (A) IR spectra of Mg(BH4)2 and NaBH4 before and after the air exposure (50 RH% at 25 °C). (B) IR spectra and PXRD patterns (inset) of 2 before and after the air exposure (50 RH% at 25 °C) for 7 days. (C) Local coordination geometry of BH4− in 2 from single-crystal X-ray diffraction analysis. The arrows represent the dihydrogen bonds between B–Hδ− and Hδ+–C. (D) Packing structure of 2. The short contacts between the hydrogen atoms of BH4− and the aliphatic group of the surrounding dpp (–CH2CH2CH2–) are displayed as a blue dotted line. The pyridyl rings of the neighbouring dpp are omitted for clarity. | ||
Some crystalline powder samples changed to amorphous in a few minutes (5, Mg2+ and 10, Ca2+) or 1 day (3–4, 6–9, Mn2+). Notably, among the BH4−-CPs, Zn2+-based CPs (1 and 2) exhibited high air stability. In particular, 2 exhibits IR spectra identical before and after air exposure for 7 days in Fig. 2B. In addition, 2 retains highly crystalline peaks in PXRD (Fig. 2B, inset). The results demonstrate improved air stability of 2, and the mechanisms are discussed based on the crystal structure of 2. Although many structural parameters affect the air stability, we classify the main contributions to the high air stability of 2 as the (i) metal–ligand coordination bond (ii) dihydrogen bonding and (iii) packing structure. 2 shows higher air stability than the isostructural Mn2+-based 3. In general, a metal–ligand bond is an essential parameter to determine the air stability of CPs.27 The Zn–N bond in tetrahedral geometry is known to construct highly stable CPs.28 Meanwhile, limited examples of CPs with Mn–N in tetrahedral geometry have been reported, and most of them are sensitive to air.29,30 The tendency of air stability depending on metal ions is also suggested by a survey of the CCDC database.31Fig. 3C displays the dihydrogen bond between BH4− and the neighboring dipyridylpropane (dpp) in the structure of 2. In addition to the X-ray diffraction analysis, the geometry optimization on 2 utilizing DFT calculation suggests that dihydrogen bonds form as well (Fig. S46†). In general, dihydrogen bonding stabilizes a crystal structure by the electrostatic interaction between oppositely charged hydrogen atoms,32,33 and the interaction lowers the electron donating ability of BH4−. The intermolecular interaction and packing structure are also essential for air stability, because they affect the diffusion of H2O molecules in the structure. BH4− is surrounded by the hydrophobic propyl group (2.545–3.010 Å) as shown in Fig. 2D, which avoids the attack of H2O. In addition, the concentration of hydrophobic species around BH4− of 2 was compared with that of other CPs showing different air stability. The number of carbon atoms away from the boron atoms of BH4− within 5 Å was counted for 2, 9 and 10 (Fig. S47†). 2 shows a higher concentration of carbon atoms than 9 and 10 (the total number of carbon atoms within 5 Å, 2: 47, 9: 39, 10: 16), which also contributes to the high air stability of 2.34
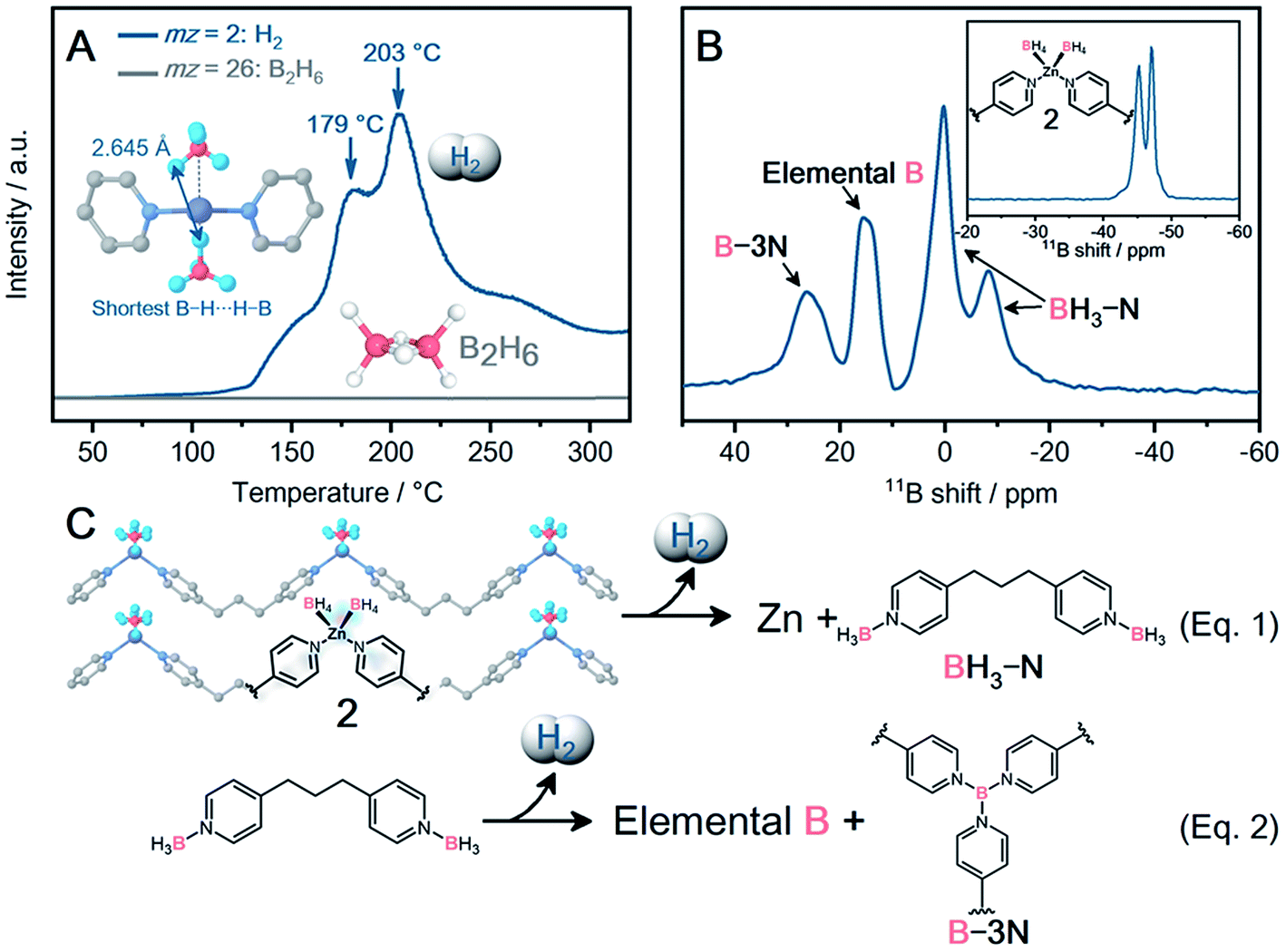 | ||
| Fig. 3 (A) TPD profile of 2 under Ar. The release of H2 (blue line) and B2H6 (gray line) was monitored. Inset: the shortest B–H⋯H–B distance is displayed. (B) Solid-state 11B MAS NMR spectra of 2 before (inset) and after dehydrogenation. (C) Schematic illustration of the proposed dehydrogenation mechanism of 2. | ||
As 2 exhibited exceptionally high air stability, the dehydrogenation properties of 2 were characterized. The TGA profile under Ar exhibits a characteristic weight loss of 1.4 wt% at 170 °C with a second weight loss at 260 °C (Fig. S26†). The released chemical species at 170 °C were analyzed using temperature-programmed desorption (TPD) as shown in Fig. 3A. The release of H2 (mz = 2) starts from 108 °C and peaks are observed at 179 and 203 °C in the TPD profile, corresponding to the weight loss (1.4 wt%) at 170 °C in TGA. The observed dehydrogenation temperature for 2 is higher than that of [NaZn(BH4)3] (103 °C),8 which is due to the stabilization effect of BH4− in 2. In spite of the higher air stability of 2 than NaBH4, 2 exhibits a lower dehydrogenation temperature than NaBH4 (170 vs. 505 °C). Note that dpp ligands decrease the amount of H2 release per weight, and the 1.4 wt% of H2 is smaller than that of the MBHs. The close contact of each BH4− enables dehydrogenation at a lower temperature. The inset of Fig. 3A displays the closest distance between B–H⋯H–B (2.645 Å), which is comparable to that of Zn2+-based MBHs ([LiZn2(BH4)5]: 2.482 Å; [NaZn(BH4)3]: 2.291 Å).8 Tetrahedral geometry in 2 is suitable to arrange BH4− in close proximity. The variable temperature synchrotron PXRD experiments indicate the structural expansion of 2 upon heating (25–226 °C, Fig. S38 and S39†). 2 does not exhibit an amorphous phase before dehydrogenation at 179 °C, and metallic Zn peaks are observed above 180 °C. The dehydrogenation of two BH4− produces one molecule of H2 and subsequently reduces Zn2+ to metallic Zn.
The environment of BH4− after the dehydrogenation was characterized by solid-state 11B magic angle spinning (MAS) nuclear magnetic resonance (NMR). The 11B NMR spectrum of pristine 2 shows two peaks at −45 and −47 ppm, and both peaks correspond to crystallographically independent BH4− (Fig. 3B, inset).8 The dehydrogenized 2 displays 11B peaks at −8, 0, 15, and 30 ppm in Fig. 3B, and was further characterized by using the 2D multi-quantum (MQ) MAS NMR spectrum (Fig. S42†). To identify the resultant boron species, a 2D 1H-{11B} through-bond heteronuclear multiple quantum coherence (HMQC) experiment was performed (Fig. S43†). The 11B peaks observed at −8 and 0 ppm correlate with the proton at 2 ppm, indicative of a B–H bond. These boron species are assigned to the [(BH3)2dpp] complexes.35 Meanwhile, the remaining two 11B peaks (15, 30 ppm) do not exhibit a clear correlation, indicating no direct bond between these boron species and hydrogen atoms. The 11B peak at 15 ppm corresponds to elemental boron and matches well with the simulated one (Fig. S44†), while the 11B peak at 30 ppm corresponds to B-3N species (e.g., [B(dpp)x]).36–38 The 2D 1H/13C heteronuclear correlation spectrum clearly exhibits the 1H and 13C peaks corresponding to the aromatic ring and aliphatic chain of dpp, indicating that dpp does not decompose during dehydrogenation (Fig. S45†).
The first peak of H2 release at 179 °C in TPD corresponds to the dehydrogenation of BH4− (eqn (1)). The release of toxic B2H6 is suppressed by the complexation with the N-donor dpp ligand to form [(BH3)2dpp] as revealed by solid-state NMR and IR spectra (Fig. S40†). The second peak of H2 release at 203 °C in TPD corresponds to the further dehydrogenation of [(BH3)2dpp]. Solid-state NMR suggests that [(BH3)2dpp] partly forms elemental boron and B-3N species such as [B(dpp)x] through the dehydrogenation (eqn (2)). 2 arranged BH4− and nitrogen atoms of dpp close to each other in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (B⋯N distance: 3.450 and 3.462 Å, Fig. S35†). 2 maintains the crystal structure before dehydrogenation at 179 °C as observed by in situ PXRD. The results indicate that the pre-organized environment of BH4− in 2 is preserved before the dehydrogenation, leading to the release of pure H2 without B2H6.
:1 ratio (B⋯N distance: 3.450 and 3.462 Å, Fig. S35†). 2 maintains the crystal structure before dehydrogenation at 179 °C as observed by in situ PXRD. The results indicate that the pre-organized environment of BH4− in 2 is preserved before the dehydrogenation, leading to the release of pure H2 without B2H6.
Conclusions
By considering suitable combinations of metal ions and N-donor ligands, we have constructed 10 coordination polymer crystals involving reactive BH4− with various coordination geometries. In particular, [Zn(BH4)2(dipyridylpropane)] (2) demonstrated both high dehydrogenation reactivity and high air stability. The crystal structure of 2 was intact under humidified conditions (50 RH% at 25 °C, 7 days) which indicates exceptionally high air stability as compared with conventional MBHs. This is due to the strong metal–ligand bond, the electrostatic stabilization by dihydrogen bonding, and close packing of the hydrophobic group with BH4−. In spite of the stabilization of BH4−, the dehydrogenation reactivity was maintained (pure H2, 1.4 wt%, 179 °C). Tetrahedral geometry in 2 arranged two BH4− closely, which accelerated the dehydrogenation. We demonstrated that crystal engineering of coordination polymers and metal–organic frameworks expands the library of hydride-based crystal structures and their properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Hiroyasu Sato from RIGAKU Corporation and Ms Nanae Shimanaka for their assistance in solving the crystal structure. The authors thank Dr Shogo Kawaguchi at the Super Photon Ring (SPring-8) for the synchrotron PXRD measurements. The work was supported by the Japan Society of the Promotion of Science (JSPS) for a Grant-in-Aid for Scientific Research (B) (JP18H02032) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and Strategic International Collaborative Research Program (SICORP) and Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from the Japan Science and Technology, Japan.References
- T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Nat. Rev. Mater., 2016, 1, 16059 CrossRef CAS.
- G. Kobayashi, Y. Hinuma, S. Matsuoka, A. Watanabe, M. Iqbal, M. Hirayama, M. Yonemura, T. Kamiyama, I. Tanaka and R. Kanno, Science, 2016, 351, 1314–1317 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS.
- J. F. Mao and D. H. Gregory, Energies, 2015, 8, 430–453 CrossRef CAS.
- Y. Nakamori, H. W. Li, K. Kikuchi, M. Aoki, K. Miwa, S. Towata and S. Orimo, J. Alloys Compd., 2007, 446–447, 296–300 CrossRef CAS.
- Y. Nakamori, K. Miwa, A. Ninomiya, H. Li, N. Ohba, S. Towata, A. Züttel and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045126 CrossRef.
- D. Ravnsbæk, Y. Filinchuk, Y. Cerenius, H. J. Jakobsen, F. Besenbacher, J. Skibsted and T. R. Jensen, Angew. Chem., Int. Ed., 2009, 121, 6787–6791 CrossRef.
- T. Jaroń, P. A. Orłowski, W. Wegner, K. J. Fijałkowski, P. J. Leszczyński and W. Grochala, Angew. Chem., Int. Ed., 2015, 54, 1236–1239 CrossRef PubMed.
- R. Černý, P. Schouwink, Y. Sadikin, K. Stare, L. u. Smrčok, B. Richter and T. R. Jensen, Inorg. Chem., 2013, 52, 9941–9947 CrossRef PubMed.
- J. Huang, Y. Yan, L. Ouyang, H. Wang, J. Liu and M. Zhu, Dalton Trans., 2014, 43, 410–413 RSC.
- E. Callini, P. Á. Szilágyi, M. Paskevicius, N. P. Stadie, J. Réhault, C. E. Buckley, A. Borgschulte and A. Züttel, Chem. Sci., 2016, 7, 666–672 RSC.
- P. N. Suwarno, A. Nale, T. M. Eggenhuisen, M. Oschatz, J. P. Embs, A. Remhof and P. E. de Jongh, J. Phys. Chem. C, 2017, 121, 4197–4205 CrossRef CAS PubMed.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS.
- D. J. Wolstenholme, J. L. Dobson and G. S. McGrady, Dalton Trans., 2015, 44, 9718–9731 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CAS.
- V. D. Makhaev, Russ. Chem. Rev., 2000, 69, 727–746 CrossRef CAS.
- M. J. Ingleson, J. P. Barrio, J. Bacsa, A. Steiner, G. R. Darling, J. T. A. Jones, Y. Z. Khimyak and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2009, 48, 2012–2016 CrossRef CAS PubMed.
- J. McKinven, G. S. Nichol and P. L. Arnold, Dalton Trans., 2014, 43, 17416–17421 RSC.
- V. D. Makhaev, A. P. Borisov, T. P. Gnilomedova, É. B. Lobkovskii and A. N. Chekhlov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1987, 36, 1582–1586 CrossRef.
- Y. Filinchuk, R. Černý and H. Hagemann, Chem. Mater., 2009, 21, 925–933 CrossRef CAS.
- Y. Filinchuk, D. Chernyshov and R. Cerny, J. Phys. Chem. C, 2008, 112, 10579–10584 CrossRef CAS.
- N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- K. Kadota, E. Sivaniah, S. Bureekaew, S. Kitagawa and S. Horike, Inorg. Chem., 2017, 56, 8744–8747 CrossRef CAS PubMed.
- K. Müller-Buschbaum and F. Schonfeld, CSD Communication, 2014 Search PubMed.
- In CCDC database, the reported examples of [Zn(X)2(pyridyl)2] are greater than twice of the hydrated geometry, [Zn(X)2(pyridyl)2(H2O)2] (1306 vs. 814 hits). By contrast, Mn2+ prefers the hydrated structure (unhydrated: 28 hits, hydrated: 375 hits).
- J. Fanfrlik, M. Lepsik, D. Horinek, Z. Havlas and P. Hobza, ChemPhysChem, 2006, 7, 1100–1105 CrossRef CAS PubMed.
- I. V. Glukhov, K. A. Lyssenko, A. A. Korlyukov and M. Y. Antipin, Russ. Chem. Bull., 2005, 54, 547–559 CrossRef CAS.
- X. Zhao, H. Yang, E. T. Nguyen, J. Padilla, X. Chen, P. Feng and X. Bu, J. Am. Chem. Soc., 2018, 140, 13566–13569 CrossRef CAS PubMed.
- P. Veeraraghavan Ramachandran, A. S. Kulkarni, Y. Zhao and J. Mei, Chem. Commun., 2016, 52, 11885–11888 RSC.
- G. Xia, Q. Gu, Y. Guo and X. Yu, J. Mater. Chem., 2012, 22, 7300–7307 RSC.
- C. L. Turner, R. E. Taylor and R. B. Kaner, J. Phys. Chem. C, 2015, 119, 13807–13813 CrossRef CAS.
- P. M. I. Irene, A. C. Armando and C. Rosalinda, Magn. Reson. Chem., 1993, 31, 189–193 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1855660–1855667 and 1855670–1855672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00731h |
| This journal is © The Royal Society of Chemistry 2019 |