
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of unsymmetrical B2E2 and B2E3 heterocycles by borylene insertion into boradichalcogeniranes†
Siyuan
Liu
ab,
Marc-André
Légaré
 ab,
Alexander
Hofmann
ab,
Anna
Rempel
ab,
Stephan
Hagspiel
ab and
Holger
Braunschweig
*ab
ab,
Alexander
Hofmann
ab,
Anna
Rempel
ab,
Stephan
Hagspiel
ab and
Holger
Braunschweig
*ab
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 19th March 2019
Abstract
We report the selective insertion of a range of borylene fragments into the E–E bonds (E = S, Se, Te) of cyclic boron dichalcogenides. This method provides facile synthetic access to a variety of symmetrical and unsymmetrical four- and five-membered rings.
Introduction
1,3-Dithietanes1 – four-membered [C2S2] cyclic molecules – comprise a scarce class of compounds. This, however, is at odds with the fact that the cyclic 1,3-dithietyl moiety is found in bioactive compounds2 such as antibacterial2b and hepatoprotective agents2c,d and pesticides.2e Even more rare are the heavier analogues of 1,3-dithietanes, 1,3-diselenetane and 1,3-ditelluretane, which typically arise from the dimerization of a handful of selenones and tellurones.3 In the main group, Group 13 analogues of these heterocycles are known but remain limited in scope due to a dearth of options for their selective synthesis.4Molecular compounds of aluminium and gallium that feature a central planar M2E2 ring surrounded by organic substituents may be accessed by reaction of dihydride or dimethyl precursors with elemental chalcogens4a–f or by double σ-bond metathesis with H2S4g or E(SiMe3)2.4h Recently Moya-Cabrera and coworkers succeeded in synthesizing a unique mixed-chalcogen Al2OTe ring by step-wise reaction of stoichiometric H2O and Te with a β-diketiminato aluminium dihydride.5 In the rather less well-explored area of low-valent Group 13 chemistry Schnöckel et al. synthesized the cyclic compound [(μ-S)AlI(NEt3)]2 by oxidizing an Al(I) iodide precursor with S8,6 while Power et al. activated N2O and S8 with a β-diketiminate-stabilized Ga(I) compound to yield the corresponding gallium oxide and sulfide dimers.7 For boron, strained diamino-B2E2 rings were obtained by Forstner and Muetterties from the reaction of B(NEt2)3 with H2S,8 and later by Nöth through thermolysis or photolysis of the [2 + 2] cycloaddition products of E![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CE (E = O, S, Se) with a bulky iminoborane.9 Interestingly, while a theoretical investigation led by Marder identified crucial factors for the rarity of B2O2 rings,9c such cyclic compounds can be formed by the hydrolysis of haloboron complexes,9c,d the dimerization of diboroxanes9e and, by the controlled dimerization of a platinum oxoboryl complex.9f Indeed, the chemistry of B–O heterocycles is predominantly the field of six-membered boroxine (B3O3) rings.
CE (E = O, S, Se) with a bulky iminoborane.9 Interestingly, while a theoretical investigation led by Marder identified crucial factors for the rarity of B2O2 rings,9c such cyclic compounds can be formed by the hydrolysis of haloboron complexes,9c,d the dimerization of diboroxanes9e and, by the controlled dimerization of a platinum oxoboryl complex.9f Indeed, the chemistry of B–O heterocycles is predominantly the field of six-membered boroxine (B3O3) rings.
More recently, our group reported the convergent reactivity of low-valent boron species ([(CAAC)(NC)BB(CN)(CAAC)] (CAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene) and cyclo-[(CAAC)B(μ-CN)]4) with sulfur and selenium to give, among other products, [cyclo-(CAAC(NC)B(μ-S))2].10 The outcome of the reaction is highly dependent on the stoichiometry of the reagents used, yielding larger and smaller rings when different amounts of chalcogen were employed. We have also observed the slow, yet selective, dimerization of monomeric, N-heterocyclic carbene (NHC)-stabilized borachalcones [IMe(tBu)BE] (IMe = dimethylimidazolylidene; E = S, Se) into four-membered heterocycles.11 To our knowledge, however, unsymmetrically substituted four- and five-membered cyclic chalcogenides of boron are yet to be reported.
We have recently reported the reactivity of the N-heterocyclic-carbene-stabilized manganese borylene complex [Cp(OC)2MnBtBu(IMe)]12b with chalcogens (S, Se, Te),13 selectively affording a range of unusual boron chalcogenides, including a family of novel free and metal-bound boradichalcogenanes ([Cp(OC)2Mn{κ1-cyclo-TeTeB(tBu)(IMe)}] (1) and [cyclo-EEBtBu(IMe)] (E = Se (2a), S (2b))).11 We now report that these complexes selectively react with a number of different transition metal borylene complexes to afford a range of unsymmetrically substituted B2E2 four-membered rings under mild conditions (Fig. 1).
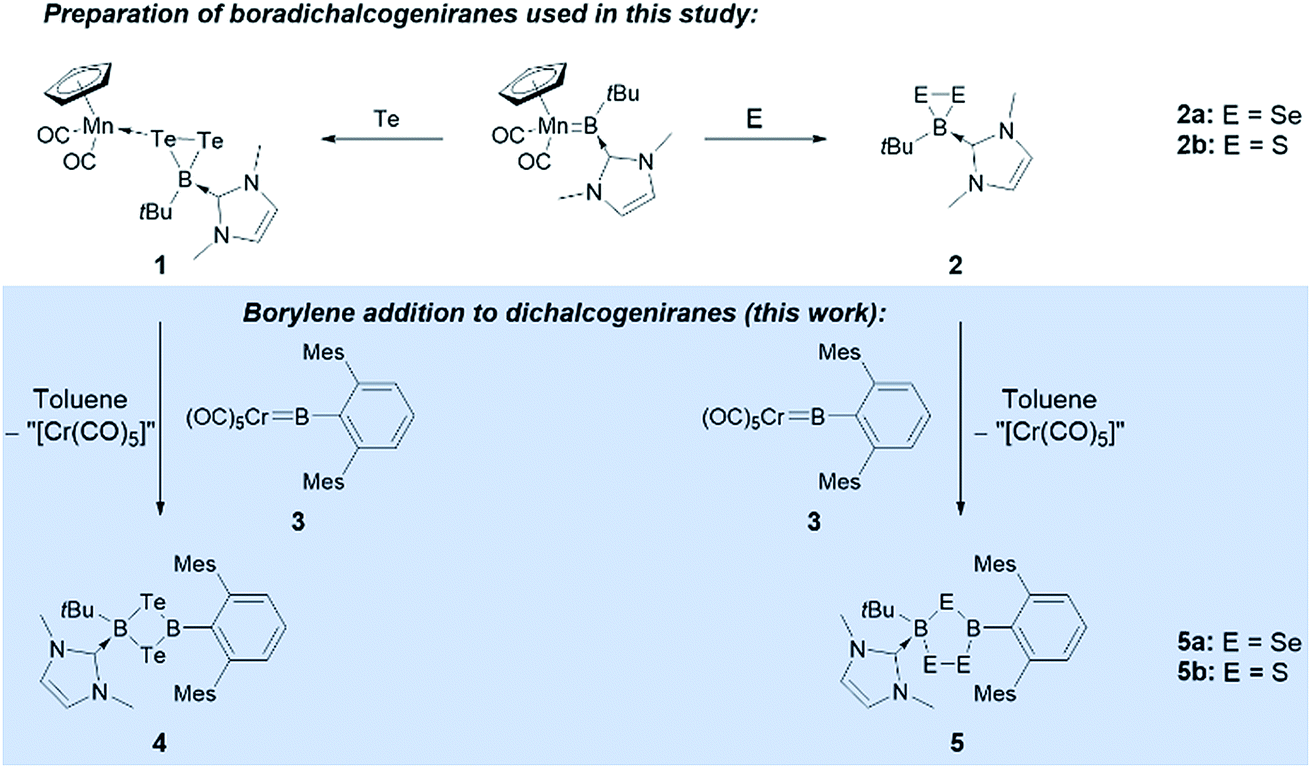 | ||
| Fig. 1 Synthesis of boradichalcogeniranes (and a manganese adduct thereof) from a manganese borylene complex, and reaction of these chalcogenides with chromium borylene complex 3. | ||
Results and discussion
Borylene complexes of the group VI metals figure among the most studied metal-stabilized sources of borylene fragments.14 In recent years, borylene complexes of chromium, molybdenum and tungsten have been shown to engage in metal-to-metal,14h,i,n,o metal-to-carbon14g,j and metal-to-nitrogen14k borylene transfer, in C–H activation14l and in [2 + 1] addition reactions.14m Towards our study of the reactivity of boradichalcogeniranes with low-valent boron fragments, the sterically protected borylene complex [(OC)5CrBTp] (3) (Tp = 2,6-bis(2,4,6-trimethylphenyl)phenyl) was deemed a good starting point. Treatment of boraditellurirane complex 1 with one equivalent of 3 in toluene allowed us to isolate cyclo-[(tBu)(IMe)B–Te–B(Tp)–Te] (4) after recrystallization from a toluene/pentane mixture (yield: 24%). High-resolution mass spectrometry (HRMS) and single-crystal X-ray diffraction analysis allowed us to identify 4 as an NHC-stabilized 1,3-ditellura-2,4-diboretane arising from the insertion of a [BTp] fragment into the Te–Te bond of 1. To the best of our knowledge, 4 represents the first example of such a four-membered B2Te2 ring (Fig. 2).
 | ||
| Fig. 2 POV-Ray depictions of the crystallographically determined structures of compounds 4 (top), 5a (bottom left) and 5b (bottom right). Atomic displacement ellipsoids depicted at the 50% probability level. Hydrogen atoms and ellipsoids of peripheral groups omitted for clarity. Selected bond distances (Å) and angles (°): 4: B1–C(Tp) 1.569(3), B1–Te1 2.153(2), B1–Te2 2.139(2), B2–C(tBu) 1.637(4), B2–C(IMe) 1.624(3), B2–Te1 2.358(2), B2–Te2 2.331(2), Te1–B1–Te2 102.7(1), B1–Te1–B2 82.38(9), Te1–B2–Te2 91.23(9), B1–Te2–B2 83.33(9); 5a: B1–C(Tp) 1.585(5), B1–Se1 1.919(5), B1–Se2 1.921(4), B2–C(tBu) 1.641(7), B2–C(IMe) 1.630(7), B2–Se1 2.115(4), B2–Se3 2.106(4), Se1–B1–Se2 119.4(3), C(Tp)–B1–Se1 120.3(3), C(Tp)–B1–Se2 120.2(3), C(tBu)–B1–C(IMe) 112.7(3), Se1–B1–Se3 105.4(2), B2–Se3–Se2 102.7(1), Se3–Se2–B1 100.7(2), B1–Se1–B2 105.5(2). | ||
This new species features two resonances in its 11B NMR spectrum (δ = −33.0 and 61.1) which differ considerably from those of both starting materials (δ = −2.0 (1) and 147 (3)), as expected for its unsymmetrical substitution pattern. The new signal at −33.0 ppm is attributable to the sp3-hybridized tetrahedral boron atom that is coordinated by an IMe ligand (B2), while the peak at 61.1 ppm can be assigned to the BTp moiety. In the solid state, 4 features a near-planar four-membered cycle which is close to perpendicular with the CBC plane of the (tBu)B2(IMe) group. The Tp substituent is rotated approximatively 15° from perpendicularity with the B2E2 cycle. Interestingly, the B–Te distances are different for both boron centers. Indeed, the TpB1–Te bonds are significantly shorter (2.153(2) and 2.139(2) Å) than the corresponding tBu(IMe)B2–Te distances (2.358(2) and 2.331(2) Å), likely owing to Te-to-B π-donation, which is not present in the case of the sp3-hybridized B2.
Borylene complex 3 was also reacted with the Se and S compounds 2a and 2b under the same conditions as for 1. Interestingly, in these cases the Se and S analogs of the four-membered B2Te2 compound 4 were only observed as minor products, which we were unable to isolate, however we were able to obtain a few crystals of the selenium-containing analog of 4, which allowed us to determine a solid-state structure and confirm its connectivity (see ESI†). In these reactions, after crystallization from a THF/pentane mixture, the five-membered rings cyclo-[(tBu)(IMe)B–EE–B(Tp)–E] (E = Se (5a) and S (5b)) were obtained (yield 22% in both cases). In the 1,2,4-triselena-3,5-diborolane 5a, the 11B NMR signals for the two boron atoms were found at 6.2 and 72.1 ppm. Similarly, in 5b, they are found at 7.7 and 66.2 ppm. The five-membered rings 5a and 5b are likely to arise from the initial formation of Se and S analogues of 4, which then undergo ring expansion using the starting materials or reaction intermediates as sacrificial chalcogen sources. While 11B NMR monitoring of the reaction showed the formation of other boron chalcogenide species – as is predicted from the stoichiometry of the reaction – these byproducts could not be identified. 5a and 5b can however be reliably crystallized from the reaction mixture and isolated.
In the solid state, 5a is structurally comparable to the three previously reported examples of crystallographically characterized BSeBSeSe non-cluster five-membered rings.15 Interestingly, it represents, to our knowledge, the first example of such a cycle with an unsymmetrical substitution pattern. The planar sp2-hybridized TpB site features B–Se distances (1.919(5) and 1.921(4) Å) that are close to those reported by Tokitoh and coworkers in a symmetrical five-membered ring of selenium and sp2 boron.15b In contrast, on the tBu(IMe)B side of 5a, the B–Se distances (2.115(4) and 2.106(4) Å) are analogous to those found in triselena-1,3-diborolanes with sp3 boron atoms.10,15c Unfortunately, while we were able to grow crystals of 5b, their poor quality, combined with the absence of heavy elements, did not yield crystallographic data of a precision that would allow us to discuss structural parameters. Instead, the single-crystal X-ray diffraction analysis only allows us to confirm the connectivity and the substitution pattern in the molecule.
Since the insertion of TpB fragments into 2a and 2b did not allow us to selectively synthesize unsymmetrical B–Se dichalcogenadiboretane rings, we elected to react these precursors with other borylene sources. Thus, the Group 6 aminoborylene complex [(OC)5MoBN(SiMe3)2] (6) was reacted with 2a and 2b in toluene. In both cases, four-membered cyclic products, cyclo-[(tBu)(IMe)B–E–B{N(SiMe3)2}–E] (E = Se (7a) and S (7b)), were obtained after recrystallization from a toluene/pentane mixture (yield: 52% (7a) and 38% (7b)). The possibility of ring expansion processes (vide supra), combined with the difficulties in quantitatively crystallizing these products, explains the moderate yields of the reactions.
 | ||
| Fig. 3 Reactions of boradichalcogeniranes 2a and 2b with borylene complexes 6 and 8 (top). POV-Ray depictions of the crystallographically determined structures of compounds 7a (bottom left) and 7b (bottom right). Atomic displacement ellipsoids depicted at 50% probability level. Hydrogen atoms and ellipsoids of peripheral groups omitted for clarity. Selected bond distances (Å) and angles (°): 7a: B1–C(tBu) 1.628(6), B1–C(IMe) 1.631(6), B1–Se1 2.116(5), B1–Se2 2.112(5), B2–N 1.442(7), B2–Se1 1.957(5), B2–Se2 1.958(5), B1–Se1–B2 81.7(2), Se1–B2–Se2 102.9(2), B2–Se2–B1 81.8(2), Se2–B1–Se1 92.8(2); 7b: B1–C(tBu) 1.632(5), B1–C(IMe) 1.633(5), B1–S1 1.998(4), B1–S2 1.990(3), B2–N 1.440(5), B2–S1 1.855(3), B2–S2 1.844(4), B1–S1–B2 80.8(2), S1–B2–S2 104.0(2), B2–S2–B1 81.2(2), S2–B1–S1 94.0(2). | ||
In 7a, 11B NMR signals at −13.4 and 46.7 ppm can be assigned to the sp3 and sp2 boron centers, respectively. In the solid state, the 1,3-diselena-2,4-diboretane cycle is nearly planar, as in the case of 4. A slight pyramidalization of the N(SiMe3)2 group, as well as dihedral angles of ca. 35° between this group and the B2Se2 and a B–N bond distance of 1.442(7) Å (longer than 1.3549(18) Å in 6), suggest only a small amount of N-to-B π-donation. Consistent with this observation, the (Me3Si)2NB–Se bond distances (1.957(5) and 1.958(5) Å), while being longer than in 5a, are short enough to suggest some Se-to-B π-donation competing with the N–B multiple bonding. On the other side of the molecule, the (tBu)(IMe)B–Se distances (2.112(5) and 2.116(5) Å) are considerably longer, consistent with the presence of an sp3 boron center. In the case of 7b, 11B NMR signals were found at −6.1 and 49.8 ppm. The solid-state structure of 7b is comparable to that of 7a, with (Me3Si)2NB–S bond distances of 1.844(4) and 1.855(3) Å, tBu(IMe)B–S distances of 1.990(3) and 1.998(4) Å and a B–N bond measuring 1.440(5) Å (Fig. 3).
2a and 2b also react with [Cp(OC)2MnBtBu] (Cp = cyclopentadienyl) (8),12a a manganese borylene complex that has a rich reactivity12 and that is a precursor of 1, 2a and 2b. While we were not able to obtain high-quality crystals from this reaction, HRMS of the isolated products revealed a formula consistent with cyclo-[(tBu)(IMe)B–E–B(tBu)–E] (E = Se (9a) and S (9b)), arising once again from the transfer of a borylene fragment (tBuB) from 8 to 2a and 2b. The solution 1H and 13C NMR spectra of 9a and 9b are consistent with the formula assignment from HRMS, revealing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio of IMe and two inequivalent tBu groups. The 11B NMR resonances of 9a (δ = −10.8 and 77.5) and 9b (δ = −3.6 and 72.9) reveal a similar environment for the sp3 boron atom as that in the corresponding four-membered rings 7a and 7b, which leads us to postulate that products 9a and 9b are indeed the cyclic compounds cyclo-[(tBu)(IMe)B–E–B(tBu)–E].
:1:1 ratio of IMe and two inequivalent tBu groups. The 11B NMR resonances of 9a (δ = −10.8 and 77.5) and 9b (δ = −3.6 and 72.9) reveal a similar environment for the sp3 boron atom as that in the corresponding four-membered rings 7a and 7b, which leads us to postulate that products 9a and 9b are indeed the cyclic compounds cyclo-[(tBu)(IMe)B–E–B(tBu)–E].
Finally, 2a was also found to react with the macrocyclic complex cyclo-[(CAAC)BCN]4 (10).16 This metal-free tetrameric borylene was previously shown to be a useful source of the corresponding monomer [(CAAC)BCN] in its reaction with Lewis bases and to generate of one of the few reported examples of cyclo-[BSe]2 complexes by reaction with elemental selenium. Thus, the reaction of 2 and 10 in toluene led to the isolation of the new 1,3-diselena-2,4-diborolane cyclo-[(tBu)(IMe)B–Se–B(CAAC)CN–Se] (11) in a 34% yield. While once again unsymmetrically substituted, this compound is the first of the series reported herein to feature two sp3-hybridized boron atoms. The 11B NMR spectrum of 11 shows two signals at −9.5 and −32.8 ppm. While both signals lie within the typical range for sp3 boron atoms, we can assign the former to the (tBu)(IMe)B group by comparison to 7a and 9a, while the latter is similar to the signals of both isomers of the previously reported cyclo-[(CAAC)(CN)B–S–B(CN)(CAAC)–S] (δ = −31.8 and −33.5 ppm).10
Rather surprisingly, and in contrast with other 1,3-diselena-2,4-diboretes,9b,1011 adopts a clearly bent butterfly structure in the solid state. This situation is plausibly a consequence of the unsymmetrical substitution pattern, which gives rise to a large difference in steric bulk on both faces of the cycle. The B–Se distances are slightly shorter on the (CAAC)(NC)B side (2.067(1) and 2.079(2) Å) than around the (tBu)(IMe)B moiety (2.114(1) and 2.114(2) Å) (Fig. 4).
 | ||
| Fig. 4 Preparation and POV-Ray depiction of the crystallographically determined structure of 11. Atomic displacement ellipsoids depicted at 50% probability level. Hydrogen atoms and ellipsoids of peripheral groups omitted for clarity. Selected bond distances (Å) and angles (°): B1–C(tBu) 1.633(9), B1–C(IMe) 1.647(8), B1–Se1 2.113(5), B1–Se2 2.111(7), B2–C(CN) 1.591(9), B2–C(CAAC) 1.602(8), B2–Se1 2.079(7), B2–Se2 2.068(6), B1–Se1–B2 80.4(2), Se1–B2–Se2 96.1(3), B2–Se2–B1 80.7(2), Se2–B1–Se1 93.8(3), C(CN)–B2–B(CAAC) 117.9(5), C(tBu)–B1–C(IMe) 114.0(5). | ||
Conclusions
In conclusion, we demonstrate that borylene fragments can be inserted into the E–E bonds (E = Te, Se and S) of boradichalcogeniranes. This reaction gives unprecedented synthetic access to unsymmetrically substituted 1,3-dichalcogena-2,4-diboretes and 1,2,4-dichalcogena-3,5-diborolanes. We have thus reported the synthesis of heterocycles featuring unsymmetrical boron centers in sp2–sp3 and sp3–sp3 hybridization combinations. Given the lack of methods for the synthesis of such unsymmetrical compounds, this approach will provide us with a useful platform to study the properties and applications of these unusual heterocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial assistance from the Deutsche Forschungsgemeinschaft. S. Liu thanks the China Scholarship Council for a Ph.D. Scholarship. M.-A. L. acknowledges the Natural Science and Engineering Research Council of Canada for a postdoctoral fellowship.Notes and references
- (a) A. Behr, Chem. Ber., 1872, 5, 970 CrossRef; (b) A. M. Jeffrey, K. W. Jennette, S. H. Blobstein, I. B. Weinstein, F. A. Beland, R. G. Harvey, H. Kasai, I. Miura and K. Nakanishi, J. Am. Chem. Soc., 1976, 98, 5715–5717 CrossRef; (c) G. Frenking and S. Shaik, The Chemical Bond: Fundamental Aspects of Chemical Bonding, Wiley, 2014 CrossRef.
- (a) J. G. Contreras and S. T. Madariaga, Bioorg. Chem., 2001, 29, 57–64 CrossRef CAS PubMed; (b) N. Nagano, K. Nakano and R. Hara, J. Antibiot., 1991, 44, 415–421 CrossRef CAS PubMed; (c) S. G. Kim, Y. J. Suhr and J. A. Miller, Carcinogen, 1998, 19, 687–690 CrossRef CAS; (d) Y. J. Surh, M. Shlyankevich, J. W. Yoo and J. K. Yoo, Mutat. Res., 1996, 367, 219–224 CrossRef CAS; (e) R. W. Addor, J. Heterocycl. Chem., 1970, 7, 381–387 CrossRef CAS.
- (a) A. Haas, J. Fluorine Chem., 1986, 32, 415–439 CrossRef CAS; (b) R. J. Adrien, R. W. Gable, B. F. Hoskin and D. Dakternieks, J. Organomet. Chem., 1989, 359, 33–39 CrossRef CAS; (c) R. Boese, A. Haas and C. Limberg, J. Chem. Soc., Chem. Commun., 1991, 1378–1379 RSC.
- (a) C. Cui, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 1999, 18, 5120–5123 CrossRef CAS; (b) K. S. Klimek, J. Prust, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2001, 20, 2047–2051 CrossRef CAS; (c) W. J. Grigsby, C. L. Raston, V.-A. Tolhurst, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 2547–2556 RSC; (d) Y. Peng, H. Hao, V. Jancik, H. W. Roesky, R. Herbst-Irmer and J. Magull, Dalton Trans., 2004, 3548–3551 RSC; (e) H. Zhu, J. Chai, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt, D. Vidovic and J. Magull, Eur. J. Inorg. Chem., 2003, 3113–3119 CrossRef CAS; (f) V. Jancik, M. M. Moya Cabrera, H. W. Roesky, R. Herbst-Irmer, D. Neculai, A. M. Neculai, M. Noltemeyer and H.-G. Schmidt, Eur. J. Inorg. Chem., 2004, 3508–3512 CrossRef CAS; (g) C. Schnitter, A. Klemp, H. W. Roesky, H.-G. Schmidt, C. Röpken, R. Herbst-Irmer and M. Noltemeyer, Eur. J. Inorg. Chem., 1998, 2033–2039 CrossRef CAS; (h) R. J. Wehmschulte and P. P. Power, Chem. Commun., 1998, 335–336 RSC.
- S. González-Gallardo, A. S. Cruz-Zavala, V. Jancik, F. Cortés-Guzmán and M. M. Moya-Cabrera, Inorg. Chem., 2013, 52, 2793–2795 CrossRef PubMed.
- A. Ecker, R. Köppe, C. Üffing and H. Schnöckel, Z. Anorg. Allg. Chem., 1998, 624, 817–822 CrossRef CAS.
- N. J. Hardman and P. P. Power, Inorg. Chem., 2001, 40, 2474–2475 CrossRef CAS PubMed.
- (a) J. A. Forstner and E. L. Muetterties, Inorg. Chem., 1966, 5, 164 CrossRef CAS; (b) G. W. Bushnel and G. A. Rivett, Can. J. Chem., 1977, 55, 3294–3297 CrossRef.
- (a) D. Mannig, C. K. Narula, H. Nöth and U. Wietelmann, Chem. Ber., 1985, 118, 3748–3758 CrossRef; (b) E. Hanecker, H. Nöth and U. Wietelmann, Chem. Ber., 1986, 119, 1904–1910 CrossRef CAS; (c) J. M. Burke, M. A. Fox, A. E. Goeta, A. K. Hugues and T. D. Marder, Chem. Commun., 2000, 2217–2218 RSC; (d) T. Wang, S. Kohrt, C. G. Daniliuc, G. Kehr and G. Erker, Org. Biomol. Chem., 2017, 15, 6223–6232 RSC; (e) H. Borrmann, A. Simon and H. Vahrenkamp, Angew. Chem., Int. Ed. Engl., 1989, 28, 180–181 CrossRef; (f) H. Braunschweig, K. Radacki and A. Schneider, Angew. Chem., Int. Ed., 2010, 49, 5993–5996 CrossRef CAS PubMed.
- D. Auerhammer, M. Arrowsmith, R. D. Dewhurst, T. Kupfer and H. Braunschweig, Chem. Sci., 2018, 9, 2252–2260 RSC.
- S. Liu, M.-A. Légaré, A. Hofmann and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 11223–11226 CrossRef CAS PubMed.
- (a) H. Braunschweig, M. Burzler, T. Kupfer, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2007, 46, 7785–7787 CrossRef PubMed; (b) H. Braunschweig, W. C. Ewing, K. Ferkinghoff, A. Hermann, T. Kramer, R. Shang, E. Siedler and C. Werner, Chem. Commun., 2015, 51, 13032–13035 RSC; (c) H. Braunschweig, M. Burzler, T. Kupfer, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2007, 46, 8071–8073 CrossRef CAS PubMed.
- S. Liu, M.-A. Légaré, D. Auerhammer, A. Hofmann and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 15760–15762 CrossRef CAS PubMed.
- (a) B. Blank, M. Colling-Hendelkens, C. Kollann, K. Radacki, D. Rais, K. Uttinger, G. R. Whittell and H. Braunschweig, Chem.–Eur. J., 2007, 13, 4770–4781 CrossRef CAS PubMed; (b) H. Braunschweig, C. Kollann and U. Englert, Angew. Chem., Int. Ed., 1998, 37, 3179–3180 CrossRef CAS; (c) H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., 2006, 118, 5380–5400 CrossRef; (d) H. Braunschweig, K. Radacki, K. Kraft and S. Stellwag, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 1073–1076 CAS; (e) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS; (f) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC; (g) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS; (h) H. Braunschweig, Q. Ye, K. Radacki and A. Damme, Angew. Chem., Int. Ed., 2012, 51, 7839–7842 CrossRef CAS; (i) H. Braunschweig, Q. Ye, A. Vargas, R. D. Dewhurst, K. Radacki and A. Damme, Nat. Chem., 2012, 4, 563–567 CrossRef CAS; (j) M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schäffer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem.–Eur. J., 2018, 24, 6843–6847 CrossRef CAS; (k) M. Nutz, B. Borthakur, R. D. Dewhurst, A. Deißenberger, T. Dellermann, M. Schäfer, I. Krummenacher, A. K. Phukan and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef CAS PubMed; (l) H. Braunchweig, R. D. Dewhurst, T. Herbst and R. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5978–5980 CrossRef PubMed; (m) H. Braunchweig, T. Herbst, D. Rais and F. Seeler, Angew. Chem., Int. Ed., 2005, 44, 7461–7463 CrossRef PubMed; (n) S. Bertsch, H. Braunschweig, B. Christ, M. Forster, K. Schwab and K. Radacki, Angew. Chem., Int. Ed., 2010, 49, 9517–9520 CrossRef CAS PubMed; (o) H. Braunschweig, M. Colling, C. Hu and K. Radacki, Angew. Chem., Int. Ed., 2003, 42, 205–208 CrossRef CAS PubMed.
- (a) According to the Cambridge Crystallographic Database, Version 5.35, Cambridge Crystallographic Data Centre, November 2013, http://www.ccdc.cam.ac.uk/data_request/cif Search PubMed; (b) M. Ito, N. Tokitoh, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1999, 40, 5557–5560 CrossRef CAS; (c) M. Yalpani, R. Köster and R. Boese, Eur. J. Inorg. Chem., 1990, 123, 707–712 CAS.
- M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grüne, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2016, 55, 14464–14468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1886448–1886454. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00657e |
| This journal is © The Royal Society of Chemistry 2019 |