
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning ligand field strength with pendent Lewis acids: access to high spin iron hydrides†
John J.
Kiernicki
a,
James P.
Shanahan
a,
Matthias
Zeller
b and
Nathaniel K.
Szymczak
 *a
*a
aUniversity of Michigan, 930 N. University, Ann Arbor, MI 48109, USA. E-mail: nszym@umich.edu
bH. C. Brown Laboratory, Purdue University, 560 Oval Dr, West Lafayette, IN 47907, USA
First published on 7th May 2019
Abstract
Geometrically flexible 9-borabicyclo[3.3.1]nonyl units within the secondary coordination sphere enable isolation of high-spin Fe(II)-dihydrides stabilized by boron–hydride interactions and a rare example of an isolable S = 3/2 reduction product. The borane-capped Fe(II)-dihydride: (1) rapidly deprotonates E–H (E = N, O, P, S) bonds to afford borane-stabilized Fe adducts and (2) releases H2 upon exposure to π-acids. The Lewis acids provide an avenue for redox-leveling in analogy to the near constant operating potential for N2 reduction in nitrogenase.
Iron-dihydrides are intermediates in myriad homogenous catalytic reactions and the recent push to develop earth-abundant transition metal catalysts has fueled many research groups to explore their reactivity.1 Beyond relevance to homogenous catalysis, iron-dihydrides have been implicated in biological reduction sequences. Whereas strong-field hydride ligands typically enforce low-spin configurations, metallocofactors including nitrogenase contain high-spin hydrides.2 To account for the modest conditions used by nitrogenase enzymes for N2 reduction, one proposal to accumulate reducing power at a near constant potential is to store reducing equivalents as Fe–μ–H–Fe intermediates.2,3 For example, the E4 state of the FeMoco center of nitrogenase is proposed to eliminate H2 from accumulated bridging hydrides concomitant with N2 binding/reduction (Fig. 1).
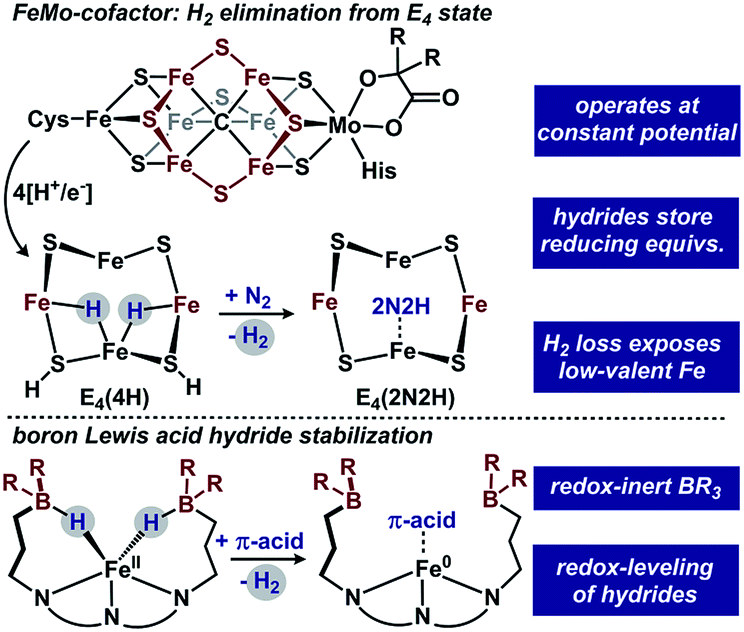 | ||
| Fig. 1 FeMo-cofactor and our approach to hydride accumulation with boron Lewis acids. | ||
Well characterized synthetic examples of open-shell Fe–μ–H–Fe complexes are rare,4 and thus, despite their relevance in biology, the synthesis and reactivity of such species remain largely unknown. This disparity is likely due to the mismatched requirements of the ligand/metal combination – strong-field hydride donor ligands rarely afford high-spin electronic configurations.
Our group is working to evaluate how the precise structural, electronic, and cooperative modes in the secondary coordination sphere can be used to regulate reactivity.5 One way in which the ligand-field strength of otherwise strong-field hydride ligands can be attenuated (accommodating high-spin states) is by introducing acidic groups to form bridging hydrides. The multiple Fe-centers in the E4 state may serve this role.6 To model these intermediates with redox-inactive acids, we targeted the synthesis of ferrous-dihydride compounds in the presence of appended boron Lewis acids. Herein, we report Lewis acid enabled isolation of a high-spin Fe(II)-di(boro)hydride as well as its reduction product, an S = 3/2 Fe-di(boro)hydride, and subsequent reactivity.
The complex, (BBNPDPtBu)FeBr2, contains a pair of moderately acidic 9-borabicyclo[3.3.1]nonyl (9-BBN) substituents that are capable of interacting with nitrogenous substrates independent of the metal center (N2H4) or cooperatively with the metal center (NH2−).7 Treatment of a freshly-thawed orange THF solution of (BBNPDPtBu)FeBr2 with two equiv. KBHEt3 affords an olive-tan powder, assigned as (BBNPDPtBu)FeH2 (1) (70%, Fig. 2). 1 is modestly stable with a half-life of ∼24 h in THF at room temperature. Investigation by 1H-NMR spectroscopy (THF) revealed a paramagnetically shifted spectrum with resonances ranging −16.9–63.6 ppm. The decrease in solution symmetry from C2v to C2 is consistent with the trialkylboranes interacting with the hydride ligands at the Fe-center. Infrared spectroscopy (KBr) supported this formulation with a broad Fe–H–B stretch at ∼1839 cm−1.8 Notably, the energy of this absorption is at significantly lower energy than bis(tris(mercaptoimidazolyl)hydroborato)-Fe(II) species that display a bridging borohydride.9 Solution magnetic susceptibly studies (25 °C, THF) establish 1 as high-spin Fe(II) (μeff = 4.6 ± 0.2 μB).
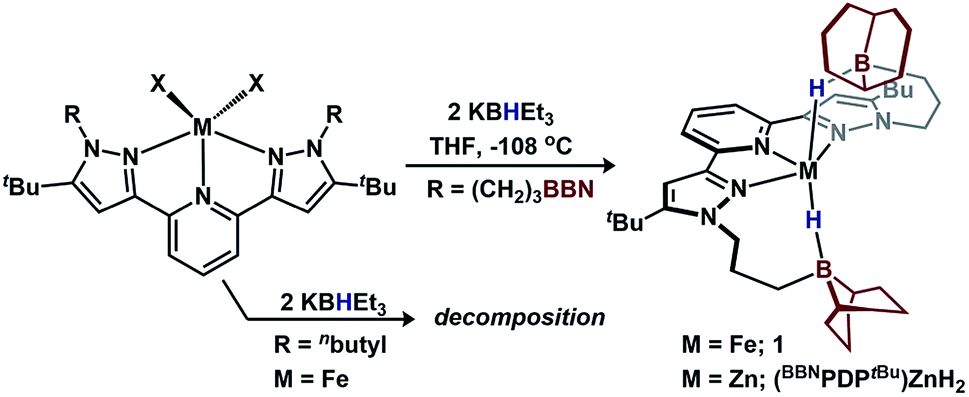 | ||
| Fig. 2 Synthesis of borane-capped metal–dihydrides. | ||
Single-crystal X-ray diffraction (XRD) experiments confirmed 1 as an iron-dihydride with each hydride capped by pyramidalized trialkylboranes (Fe–B = 2.970 Å; ∑Bα = 318.6°; Fig. 3B). The geometry about iron is best described as square pyramidal (τ5 = 0.31). Complex 1 is a rare example of a monometallic high-spin Fe(II)-dihydride (or Fe(II)-diborohydride).1a,6c,9
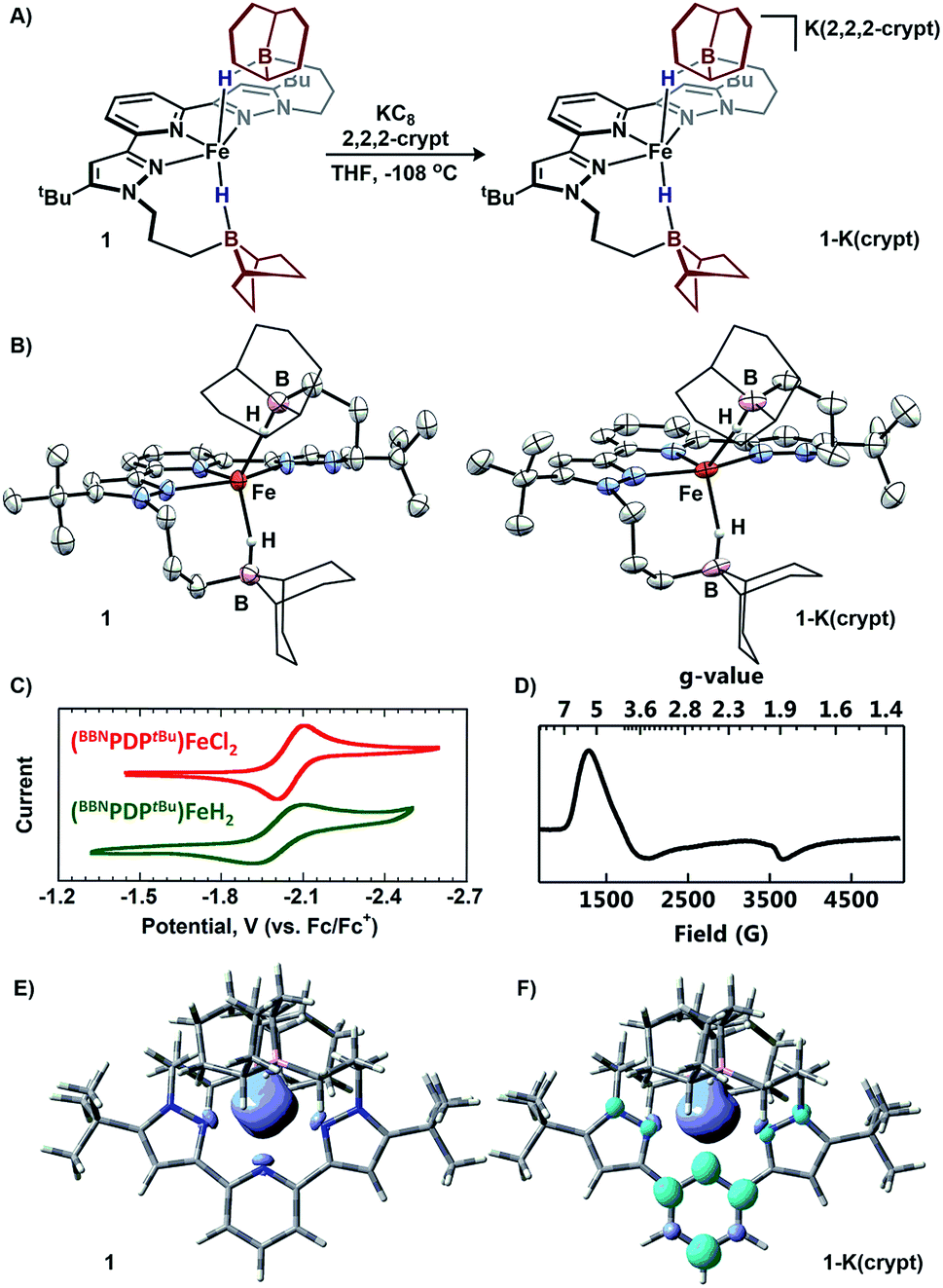 | ||
Fig. 3 (A) Reduction of 1. (B) Molecular structures of 1 and 1-K(crypt) (50% probability ellipsoids). For clarity, the 9-BBN substituents are displayed in wireframe. (C) Electrochemical comparison between 1 and (BBNPDPtBu)FeCl2. (D) X-band EPR of 1-K(crypt) at 10 K in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene/THF. (E and F) Spin-density isosurface plots (0.002 a.u.) of 1 and 1-K(crypt), respectively. :1 toluene/THF. (E and F) Spin-density isosurface plots (0.002 a.u.) of 1 and 1-K(crypt), respectively. | ||
The requirements of the intramolecular acids to stabilize 1 were assessed with a control ligand, BuPDPtBu, where the –(CH2)3BBN fragments are replaced with n-butyl groups (Fig. 2).7 Treating (BuPDPtBu)FeBr2 with 2 equiv. KBHEt3 afforded intractable mixtures, precluding characterization (see ESI†). We propose that the appended trialkylboranes in 1 serve two important roles: (1) to stabilize the hydride ligands, and (2) to decrease the entropic penalty for stabilization (intramolecular 9-BBN vs. exogenous BEt3). Attempts to install another donor ligand to 1 and enforce a low-spin configuration were unsuccessful: 1 was unreactive toward stoichiometric 1,4-diazabicyclo[2.2.2]octane, NMe3, and PMe3, but decomposed when treated with 4-dimethylaminopyridine.
To assess the requirements of the metal center and provide additional spectroscopic characterization, we synthesized the zinc analogue, (BBNPDPtBu)ZnH2, from (BBNPDPtBu)ZnI2 (Fig. 2). 11B-NMR spectroscopy (THF) of (BBNPDPtBu)ZnH2 revealed an upfield resonance at 5.43 ppm, consistent with a tetrahedral boron center.10 IR spectroscopy (KBr) revealed a broad Zn–H–B stretch at ∼1775 cm−1 that shifts upon deuterium labeling to ∼1300 cm−1.112H-NMR spectroscopy established the assignment of the hydride resonance at 0.65 ppm, which is comparable to other κ1-Zn(BH4)/Zn(BH3R) complexes.12 The molecular structure of (BBNPDPtBu)ZnH2 displays bonding metrics analogous to 1 (see ESI†).
Electrochemical investigation of 1 using cyclic voltammetry (0.2 M [Bu4N][PF6], THF) revealed a quasi-reversible reductive event at −2.06 V vs. Fc/Fc+ which is minimally shifted from (BBNPDPtBu)FeCl2 (Δ = +10 mV),13 despite their different X-type donors (Fig. 3C).14 The similar redox potentials suggest that, in analogy to the E4 state of nitrogenase, reducing equivalents can be delivered at a near constant potential to a Fe(II) state, when stored as bridging hydride equivalents.15 To prepare the reduced complex, 1 equiv. of KC8 was added to a freshly thawed THF solution of 1 in the presence of 2,2,2-cryptand, resulting in an immediate color change to a vibrant green species assigned as [K(2,2,2-cryptand)][(BBNPDPtBu)FeH2] (1-K(crypt); Fig. 3A).16 Investigation of 1-K(crypt) by 1H-NMR spectroscopy (THF) revealed a paramagnetically shifted spectrum with resonances ranging −70.6–95.0 ppm with solution C2 symmetry. Samples of 1-K(crypt) are less stable than 1 and have an approximate half-life of 12 h in THF at room temperature. Upon reduction, the Fe–H–B infrared absorption shifts to higher energy (∼1866 cm−1; KBr). Solution magnetic susceptibly studies (25 °C, THF) of 1-K(crypt) are consistent with an S = 3/2 complex (μeff = 4.1 ± 0.1 μB).17 X-band EPR spectroscopy was employed to confirm the spin-state. Regardless of coordination environment or geometry, S = 1/2 iron complexes, typically exhibit g values near the free electron value,18 while high-spin complexes exhibit gx-tensors > 3.5.2,191-K(crypt) displays a broad rhombic signal with g values of 5.6, 3.97, and 1.82 at 10 K in 1:1 THF/toluene glass (Fig. 3D), which suggests 1-K(crypt) is best described as high-spin.20 Reduced iron complexes with low coordination numbers (2, 3, occasionally 4) are often high-spin,19c,19e,21 while those stabilized by strong-field ligands (i.e. NHC, porphyrin, phosphines) are low-spin. 1-K(crypt) is a rare example of a high-spin complex with a coordination number ≥ 5.19b
For structural comparison, 1-K(crypt) was examined by XRD. Data refinement revealed the anionic portion of 1-K(crypt) to be geometrically similar (τ5 = 0.36) to 1 (Fig. 3B). The interacting trialkylboranes are equidistant from the metal center in 1 and 1-K(crypt). Upon reduction, the Fe–Npyridine bond distance decreases from 2.178(5) to 2.021(6) consistent with enhanced π-backbonding.22 DFT optimized geometries for the high-spin configuration of 1 and 1-K(crypt) are in agreement with their crystallographic structures.23 The spin-density for 1 is localized on iron, consistent with a high-spin Fe(II) description (Fig. 3E). For 1-K(crypt), significant spin-density is localized on the pyridyl-moiety of the chelate. The calculated β-SOMO for 1-K(crypt) corresponding to reduction is primarily comprised of ligand-π* orbitals on the pyridyl moiety with minimal Fe contribution (19%). This analysis is consistent with reduction of 1 affording an S = 3/2 system through antiferromagnetic coupling of high-spin Fe(II) with [BBNPDPtBu]1−.24 The calculated electronic structure is consistent with crystallographic bond metrics.25 Pyridyl-π* population is reflected by C3–C4 bond elongation (1: 1.377(6); 1-K(crypt): 1.433(12)ave Å; Fig. S85†).
We sought to examine the generality of the trialkylborane Lewis acids to stabilize both the accumulated hydrides and other small molecules. Addition of E–H substrates (E = NH2, NHMe, NHPh, OH, PHPh, SPh) to freshly thawed THF solutions of 1 afford 2-E as orange-tan powders with production of H2 (Fig. 4).262-E are high-spin Fe(II)-species with solution magnetic susceptibilities ranging μeff = 4.5–5.5 μB (THF, 25 °C). 2-OH and 2-PHPh provide diagnostic infrared handles with sharp υ(OH)/υ(PH) absorptions observed at 3630 and 2340 cm−1 (KBr), respectively—each consistent with previously reported M–OH–BR327 and Fe-phosphides.281H-NMR spectroscopy revealed 2-NHMe, 2-NHPh, 2-OH, and 2-PHPh are C2 symmetric in solution—consistent with Fe–E–B interactions remaining intact in solution. In contrast, 2-SPh exhibits a solution C2V symmetric spectrum that broadens upon cooling to −80 °C, consistent with reversible B–S binding; likely a consequence of a weaker B–S interaction.29 This observation was supported by DFT analyses. In contrast to the strong B–O interaction in 2-OH (favored by ∼10 kcal per mol per interaction), the calculated B–S interaction is thermodynamically disfavored by ∼9 kcal per mol per interaction. In solution, the B–SPh binding equilibria in 2-SPh were arrested by treating 2-SPh with two equiv. NH3. Competitive binding with NH3 afforded the ammonia-borane species, (BBNPDPtBu)Fe(SPh)2(NH3)2 (3), highlighting the utility of moderately acidic groups to reversibly interact with small molecule substrates (Fig. 5).
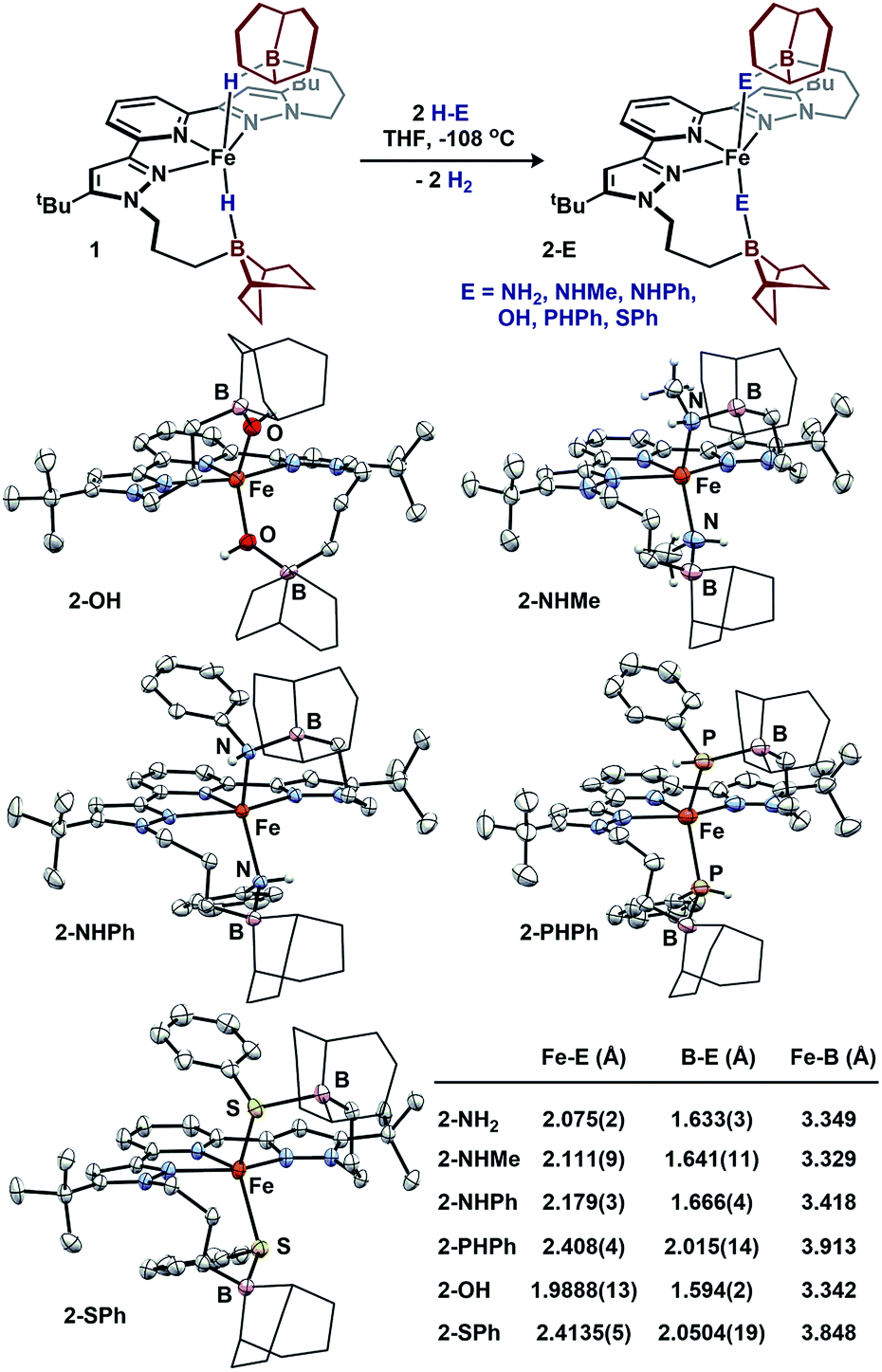 | ||
| Fig. 4 Formation of 2-E and molecular structures (50% probability ellipsoids) and averaged bond distances. For clarity, the 9-BBN substituents are displayed in wireframe. | ||
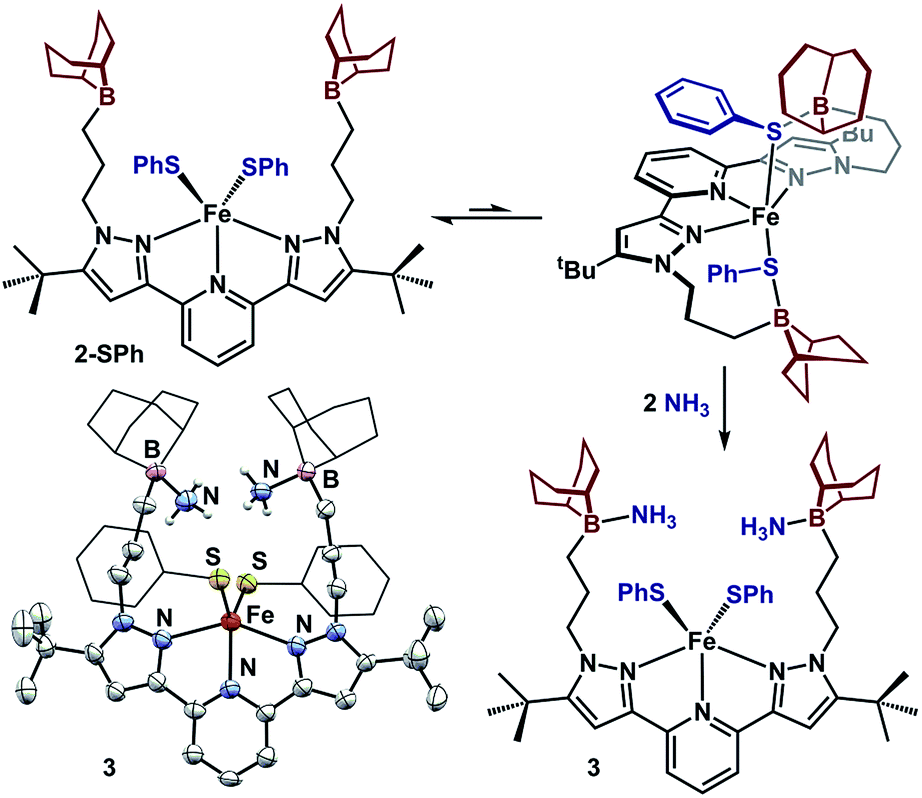 | ||
| Fig. 5 Reversible acid/base interaction in 2-SPh and molecular structure of 3 (50% probability ellipsoids). For clarity, the 9-BBN and phenyl substituents are displayed in wireframe. | ||
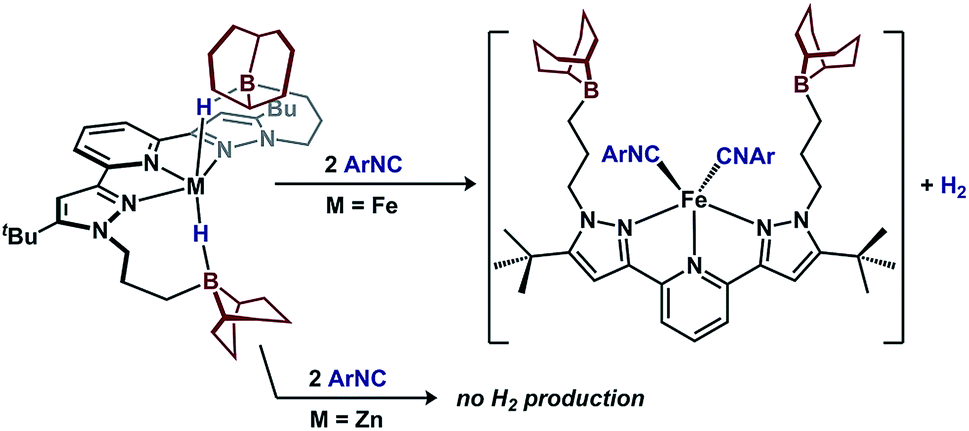 | ||
| Fig. 6 π-acid induced H2 extrusion from 1. | ||
To assess the structural similarities, 2-E were examined by XRD (Fig. 4). Each displays a pentacoordinate iron best described as distorted square-pyramidal with interacting trialkylboranes. The B-heteroatom distances range from 1.592(2)–2.0504(19) Å following the trend B–OH < B–NHMe < B–NHPh < B–PHPh = B–SPh. The same trend is observed for the Fe-heteroatom distance with 2-OH displaying the shortest bond (1.9812(13) Å) and 2-PHPh/2-SPh displaying the longest bond (2.408(4) and 2.4134(5) Å, respectively). The Fe–E bonds are longer than most reported examples, and we attribute this difference to due to quenching the ligand lone-pair by the trialkylborane.4d,19b,28,30 Upon substituting the B–S bond of 2-SPh with NH3 to afford 3, the Fe–S bond distance slightly decreases (0.02 Å) while the Fe–S–Ph angle expands (90.06(6) to 110.5(3)ave°). The ability of the appended trialkylborane to stabilize various sizes of small molecules at the metal center is highlighted by comparing the Fe–B distances of 1 and 2-PHPh which vary by nearly 0.9 Å (2.97 vs. 3.91 Å). The isolation of 2-E highlights the utility of secondary coordination sphere interactions to not only stabilize hydrides at a metal center, but also to stabilize intermediate species following H2 loss. Attempts to synthesize most compounds analogous to 2-E from (BuPDPtBu)FeBr2 through established methods were unsuccessful (see ESI†).19b,28,30b,31 The (BuPDPtBu)Fe(SPh)2 complex was the only isolable compound, which is consistent with the observed stability of 3 in the absence of Lewis acid/base interactions.
The acid/base interactions can be viewed as an avenue for redox-leveling: the reduction potentials vary by 340 mV across the series of compounds (1, 2-E, and (BBNPDPtBu)FeX2 (X = Cl, Br); Table S3†) despite their disparate ligand donor properties.32 Upon sequestering the Lewis acid of 2-SPh with NH3 to form 3, the redox potential shifts more negative by 80 mV (−2.21 V vs. Fc/Fc+). This modest shift is consistent with the weak or dynamic B–S interaction of 2-SPh. Further description of the Fe–E–B interactions were obtained from their DFT optimized structures.33 The LUMOs of the β manifold for 2-E provide insight into their reduction potentials.34 The boron Lewis acids temper the donor properties of the X-type donor ligand, lowering their energy and percent composition in the frontier orbitals (see ESI† for details). The LUMOs of 1 and 2-E span a modest range of 0.51 eV. In contrast, their computationally investigated truncated (MePDPtBu)Fe(E)2 counterparts range 1.72 eV. In analogy to the near constant operating potential for N2 reduction in nitrogenase where H−/NxHy components are present throughout the sequence, the [(BBNPDPtBu)Fe] system accommodates small molecule substrates whose donor properties are regulated through Lewis acid/base interactions.
Given the unusual electronic and structural properties of 1, 1-K(crypt), and (BBNPDPtBu)ZnH2, Wiberg bond-indices for were investigated for each of these complexes to gauge the bonding contribution of the M–H–B interaction (i.e. metal–hydride or metal–borohydride character). For each, the interaction is predominantly borohydride in character with B–H indices ranging 0.65–0.78 and M–H indices ranging 0.14–0.22.35 (BBNPDPtBu)ZnH2 displays the most M–H character whereas 1-K(crypt) displays the most B–H character—consistent with IR spectroscopy. While the data suggest the electronic structures are best described as borohydride, the M–H character varies across the series, indicating M–H reactivity may be accessible.
A key difference between metal–hydrides and borohydrides is the ability of the former to undergo reductive elimination. To test the hydride accumulation hypothesis, i.e. whether the bridging hydrides in 1 provide access to Fe(0) by eliminating H2, 1 was treated with a π-acid (Fig. 6). Addition of two equiv. 2,4,6-tri-tert-butylphenylisocyanide to 1 expels H2 in 90(2)% yield and affords (BBNPDPtBu)Fe(CNAr)2.36 In contrast, when subjecting the redox-inactive zinc variant, (BBNPDPtBu)ZnH2, to analogous conditions, only 7(3)% H2 was detected. These results implicate that 1 contains accessible Fe–H character that is available for reactions. In contrast, the reactivity of (BBNPDPtBu)ZnH2 illustrates the inability to extrude H2 from a borohydride when access to a low-valent metal is not possible.
Conclusions
In summary, we have described a system capable of accumulating hydride equivalents at iron. Intramolecular borane Lewis acid serve a prominent role to not only impart stability to Fe-dihydrides, but also enable previously unobserved electronic structures at iron by regulating the Fe–X-type ligand interactions. Similarities can be drawn between our system and the E4 state of nitrogenase, where hydrides are accumulated at a near constant potential and addition of small molecule substrates induce H2 elimination concomitant with substrate stabilization. The geometric flexibility of the appended Lewis acids were previously shown to enable interactions both independent and cooperatively with a metal; here the geometric flexibility is highlighted by their ability to stabilize small molecules of varied sizes. Work is ongoing to interrogate how Lewis acids can be used to further regulate redox transformations at the metal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NIH (1R01GM111486-01A1). N. K. S. is a Camille Dreyfus Teacher-Scholar. JJK is supported by the NIH NIGMS F32GM126635. We thank Suzanne Bart for EPR assistance. X-ray diffractometers were funded by the NSF (CHE 1625543).Notes and references
- (a) H. Dai and H. Guan, Iron Dihydride Complexes: Synthesis, Reactivity, and Catalytic Applications, Isr. J. Chem., 2017, 57, 1170–1203 CrossRef CAS; (b) D. E. Prokopchuk, G. M. Chambers, E. D. Walter, M. T. Mock and R. M. Bullock, H2 Binding, Splitting, and Net Hydrogen Atom Transfer at a Paramagnetic Iron Complex, J. Am. Chem. Soc., 2019, 141, 1871–1876 CrossRef CAS.
- R. Y. Igarashi, M. Laryukhin, P. C. Dos Santos, H.-I. Lee, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, Trapping H-Bound to the Nitrogenase FeMo-Cofactor Active Site during H2 Evolution: Characterization by ENDOR Spectroscopy, J. Am. Chem. Soc., 2005, 127, 6231–6241 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Nitrogenase: A Draft Mechanism, Acc. Chem. Res., 2013, 46, 587–595 CrossRef CAS PubMed.
- (a) A. Jablonskytė, J. A. Wright, S. A. Fairhurst, J. N. T. Peck, S. K. Ibrahim, V. S. Oganesyan and C. J. Pickett, Paramagnetic Bridging Hydrides of Relevance to Catalytic Hydrogen Evolution at Metallosulfur Centers, J. Am. Chem. Soc., 2011, 133, 18606–18609 CrossRef PubMed; (b) W. Wang, M. J. Nilges, T. B. Rauchfuss and M. Stein, Isolation of a Mixed Valence Diiron Hydride: Evidence for a Spectator Hydride in Hydrogen Evolution Catalysis, J. Am. Chem. Soc., 2013, 135, 3633–3639 CrossRef CAS PubMed; (c) R. A. Kinney, C. T. Saouma, J. C. Peters and B. M. Hoffman, Modeling the Signatures of Hydrides in Metalloenzymes: ENDOR Analysis of a Di-iron Fe(μ-NH)(μ-H)Fe Core, J. Am. Chem. Soc., 2012, 134, 12637–12647 CrossRef CAS PubMed; (d) J. M. Smith, R. J. Lachicotte and P. L. Holland, NN Bond Cleavage by a Low-Coordinate Iron(II) Hydride Complex, J. Am. Chem. Soc., 2003, 125, 15752–15753 CrossRef CAS PubMed; (e) J. Rittle, C. C. L. McCrory and J. C. Peters, A 106-Fold Enhancement in N2-Binding Affinity of an Fe2(μ-H)2 Core upon Reduction to a Mixed-Valence FeIIFeI State, J. Am. Chem. Soc., 2014, 136, 13853–13862 CrossRef CAS PubMed.
- (a) E. W. Dahl, J. J. Kiernicki, M. Zeller and N. K. Szymczak, Hydrogen Bonds Dictate O2 Capture and Release within a Zinc Tripod, J. Am. Chem. Soc., 2018, 140, 10075–10079 CrossRef CAS PubMed; (b) E. W. Dahl, H. T. Dong and N. K. Szymczak, Phenylamino derivatives of tris(2-pyridylmethyl)amine: hydrogen-bonded peroxodicopper complexes, Chem. Commun., 2018, 54, 892–895 RSC; (c) C. M. Moore and N. K. Szymczak, Redox-induced fluoride ligand dissociation stabilized by intramolecular hydrogen bonding, Chem. Commun., 2015, 51, 5490–5492 RSC; (d) O. Tutusaus, C. Ni and N. K. Szymczak, A Transition Metal Lewis Acid/Base Triad System for Cooperative Substrate Binding, J. Am. Chem. Soc., 2013, 135, 3403–3406 CrossRef CAS PubMed; (e) E. W. Dahl and N. K. Szymczak, Hydrogen Bonds Dictate the Coordination Geometry of Copper: Characterization of a Square-Planar Copper(I) Complex, Angew. Chem., Int. Ed., 2016, 55, 3101–3105 CrossRef CAS PubMed.
- (a) D.-H. Manz, P.-C. Duan, S. Dechert, S. Demeshko, R. Oswald, M. John, R. A. Mata and F. Meyer, Pairwise H2/D2 Exchange and H2 Substitution at a Bimetallic Dinickel(II) Complex Featuring Two Terminal Hydrides, J. Am. Chem. Soc., 2017, 139, 16720–16731 CrossRef CAS PubMed; (b) K. J. Anderton, B. J. Knight, A. L. Rheingold, K. A. Abboud, R. García-Serres and L. J. Murray, Reactivity of hydride bridges in a high-spin [Fe3(μ-H)3]3+ cluster: reversible H2/CO exchange and Fe–H/B–F bond metathesis, Chem. Sci., 2017, 8, 4123–4129 RSC; (c) Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, The Reactivity Patterns of Low-Coordinate Iron–Hydride Complexes, J. Am. Chem. Soc., 2008, 130, 6624–6638 CrossRef CAS PubMed.
- J. J. Kiernicki, M. Zeller and N. K. Szymczak, Hydrazine Capture and N–N Bond Cleavage at Iron Enabled by Flexible Appended Lewis Acids, J. Am. Chem. Soc., 2017, 139, 18194–18197 CrossRef CAS PubMed.
-
(a) M. A. Nesbit, D. L. M. Suess and J. C. Peters, E–H Bond Activations and Hydrosilylation Catalysis with Iron and Cobalt Metalloboranes, Organometallics, 2015, 34, 4741–4752 CrossRef CAS;
(b) D. L. M. Suess and J. C. Peters, H–H and Si–H Bond Addition to Fe
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NNR2 Intermediates Derived from N2, J. Am. Chem. Soc., 2013, 135, 4938–4941 CrossRef CAS PubMed;
(c) E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Selective Hydrogen Production from Methanol with a Defined Iron Pincer Catalyst under Mild Conditions, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed;
(d) R. Bau, H. S. H. Yuan, M. V. Baker and L. D. Field, An X-ray study of FeH(dmpe)2(BH4): a compound containing a singly-bridged BH4 ligand with a bent Fe–H–B linkage, Inorg. Chim. Acta, 1986, 114, L27–L28 CrossRef CAS;
(e) D. L. M. Suess and J. C. Peters, A CO-Derived Iron Dicarbyne That Releases Olefin upon Hydrogenation, J. Am. Chem. Soc., 2013, 135, 12580–12583 CrossRef CAS PubMed.
NNR2 Intermediates Derived from N2, J. Am. Chem. Soc., 2013, 135, 4938–4941 CrossRef CAS PubMed;
(c) E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Selective Hydrogen Production from Methanol with a Defined Iron Pincer Catalyst under Mild Conditions, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed;
(d) R. Bau, H. S. H. Yuan, M. V. Baker and L. D. Field, An X-ray study of FeH(dmpe)2(BH4): a compound containing a singly-bridged BH4 ligand with a bent Fe–H–B linkage, Inorg. Chim. Acta, 1986, 114, L27–L28 CrossRef CAS;
(e) D. L. M. Suess and J. C. Peters, A CO-Derived Iron Dicarbyne That Releases Olefin upon Hydrogenation, J. Am. Chem. Soc., 2013, 135, 12580–12583 CrossRef CAS PubMed. -
(a) J. S. Figueroa, J. G. Melnick and G. Parkin, Reactivity of the Metal → BX3 Dative σ-Bond: 1,2-Addition Reactions of the Fe → BX3 Moiety of the Ferraboratrane Complex [κ4-B(mimBut)3]Fe(CO)2, Inorg. Chem., 2006, 45, 7056–7058 CrossRef CAS PubMed;
(b) C. Kimblin, D. G. Churchill, B. M. Bridgewater, J. N. Girard, D. A. Quarless and G. Parkin, Tris(mercaptoimidazolyl)hydroborato complexes of cobalt and iron, [TmPh]2M (M
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Fe, Co): structural comparisons with their tris(pyrazolyl)hydroborato counterparts, Polyhedron, 2001, 20, 1891–1896 CrossRef CAS;
(c) See ESI Table S21† for further literature comparisons to Fe–B bond distances and υ(Fe–H–B) vibrational data.
Fe, Co): structural comparisons with their tris(pyrazolyl)hydroborato counterparts, Polyhedron, 2001, 20, 1891–1896 CrossRef CAS;
(c) See ESI Table S21† for further literature comparisons to Fe–B bond distances and υ(Fe–H–B) vibrational data. - B–H coupling is not observed presumably due to low symmetry at boron.
- Attempts at deuterium labeling 1 with LiBDEt3 were unsuccessful.
- S. Marks, R. Köppe, T. K. Panda and P. W. Roesky, Unprecedented Zinc–Borane Complexes, Chem.–Eur. J., 2010, 16, 7096–7100 CrossRef CAS PubMed.
- Potentials were assessed by square wave voltammetry.
- A typical shift of 200-300 mV is expected per Cl/H substitution, see: (a) T. Liu, D. L. DuBois and R. M. Bullock, An iron complex with pendent amines as a molecular electrocatalyst for oxidation of hydrogen, Nat. Chem., 2013, 5, 228 CrossRef CAS PubMed; (b) M. Tilset, I. Fjeldahl, J.-R. Hamon, P. Hamon, L. Toupet, J.-Y. Saillard, K. Costuas and A. Haynes, Theoretical, Thermodynamic, Spectroscopic, and Structural Studies of the Consequences of One-Electron Oxidation on the Fe–X Bonds in 17- and 18-Electron Cp*Fe(dppe)X Complexes (X = F, Cl, Br, I, H, CH3), J. Am. Chem. Soc., 2001, 123, 9984–10000 CrossRef CAS PubMed; (c) Y. Hu, L. Li, A. P. Shaw, J. R. Norton, W. Sattler and Y. Rong, Synthesis, Electrochemistry, and Reactivity of New Iridium(III) and Rhodium(III) Hydrides, Organometallics, 2012, 31, 5058–5064 CrossRef CAS; (d) A. P. Shaw, J. R. Norton, D. Buccella, L. A. Sites, S. S. Kleinbach, D. A. Jarem, K. M. Bocage and C. Nataro, Synthesis, Electrochemistry, and Reactivity of Half-Sandwich Ruthenium Complexes Bearing Metallocene-Based Bisphosphines, Organometallics, 2009, 28, 3804–3814 CrossRef CAS.
- B. J. McNicholas, R. H. Grubbs, J. R. Winkler, H. B. Gray and E. Despagnet-Ayoub, Tuning the formal potential of ferrocyanide over a 2.1 V range, Chem. Sci., 2019, 10, 3623–3626 RSC.
- Crown ethers are not required to isolate the reduced Fe-di(boro)hydride, see ESI.†.
- Y. Nakajima, Y. Nakao, S. Sakaki, Y. Tamada, T. Ono and F. Ozawa, Electronic Structure of Four-Coordinate Iron(I) Complex Supported by a Bis(phosphaethenyl)pyridine Ligand, J. Am. Chem. Soc., 2010, 132, 9934–9936 CrossRef CAS PubMed.
- (a) A. V. Polezhaev, C. J. Liss, J. Telser, C.-H. Chen and K. G. Caulton, A PNNH Pincer Ligand Allows Access to Monovalent Iron, Chem.–Eur. J., 2018, 24, 1330–1341 CrossRef CAS PubMed; (b) C. E. MacBeth, S. B. Harkins and J. C. Peters, Synthesis and characterization of cationic iron complexes supported by the neutral ligands NPi-Pr3, NArPi-Pr3, and NSt-Bu3, Can. J. Chem., 2005, 83, 332–340 CrossRef CAS; (c) C. V. Thompson, I. Davis, J. A. DeGayner, H. D. Arman and Z. J. Tonzetich, Iron Pincer Complexes Incorporating Bipyridine: A Strategy for Stabilization of Reactive Species, Organometallics, 2017, 36, 4928–4935 CrossRef CAS; (d) N. Ehrlich, M. Kreye, D. Baabe, P. Schweyen, M. Freytag, P. G. Jones and M. D. Walter, Synthesis and Electronic Ground-State Properties of Pyrrolyl-Based Iron Pincer Complexes: Revisited, Inorg. Chem., 2017, 56, 8415–8422 CrossRef CAS PubMed.
- (a) A. McSkimming and W. H. Harman, A Terminal N2 Complex of High-Spin Iron(I) in a Weak, Trigonal Ligand Field, J. Am. Chem. Soc., 2015, 137, 8940–8943 CrossRef CAS PubMed; (b) J. S. Anderson, M.-E. Moret and J. C. Peters, Conversion of Fe–NH2 to Fe–N2 with release of NH3, J. Am. Chem. Soc., 2013, 135, 534–537 CrossRef CAS PubMed; (c) P. P. Samuel, K. C. Mondal, N. Amin Sk, H. W. Roesky, E. Carl, R. Neufeld, D. Stalke, S. Demeshko, F. Meyer, L. Ungur, L. F. Chibotaru, J. Christian, V. Ramachandran, J. van Tol and N. S. Dalal, Electronic Structure and Slow Magnetic Relaxation of Low-Coordinate Cyclic Alkyl(amino) Carbene Stabilized Iron(I) Complexes, J. Am. Chem. Soc., 2014, 136, 11964–11971 CrossRef CAS PubMed; (d) S. A. Stoian, Y. Yu, J. M. Smith, P. L. Holland, E. L. Bominaar and E. Münck, Mössbauer, Electron Paramagnetic Resonance, and Crystallographic Characterization of a High-Spin Fe(I) Diketiminate Complex with Orbital Degeneracy, Inorg. Chem., 2005, 44, 4915–4922 CrossRef CAS PubMed; (e) K. P. Chiang, C. C. Scarborough, M. Horitani, N. S. Lees, K. Ding, T. R. Dugan, W. W. Brennessel, E. Bill, B. M. Hoffman and P. L. Holland, Characterization of the Fe–H Bond in a Three-Coordinate Terminal Hydride Complex of Iron(I), Angew. Chem., Int. Ed., 2012, 51, 3658–3662 CrossRef CAS PubMed.
- g-Values were obtained by simulation, see ESI.†.
- (a) Z. Ouyang, J. Du, L. Wang, J. L. Kneebone, M. L. Neidig and L. Deng, Linear and T-Shaped Iron(I) Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis and Structure Characterization, Inorg. Chem., 2015, 54, 8808–8816 CrossRef CAS PubMed; (b) M. I. Lipschutz, T. Chantarojsiri, Y. Dong and T. D. Tilley, Synthesis, Characterization, and Alkyne Trimerization Catalysis of a Heteroleptic Two-Coordinate FeI Complex, J. Am. Chem. Soc., 2015, 137, 6366–6372 CrossRef CAS PubMed.
- The pyridine(dipyrazole) framework has not been observed to act as a redox-active ligand, see B. J. Cook, C.-H. Chen, M. Pink, R. L. Lord and K. G. Caulton, Inorg. Chim. Acta, 2016, 451, 82–91 CrossRef CAS . It has been proposed as an intermediate species before, see T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2017, 56, 8651–8660 CrossRef PubMed.
- See ESI† for comparison between alternative spin configurations.
- (a) A. M. Tondreau, S. C. E. Stieber, C. Milsmann, E. Lobkovsky, T. Weyhermüller, S. P. Semproni and P. J. Chirik, Oxidation and Reduction of Bis(imino)pyridine Iron Dinitrogen Complexes: Evidence for Formation of a Chelate Trianion, Inorg. Chem., 2013, 52, 635–646 CrossRef CAS PubMed; (b) S. K. Russell, A. C. Bowman, E. Lobkovsky, K. Wieghardt and P. J. Chirik, Synthesis and Electronic Structure of Reduced Bis(imino)pyridine Manganese Compounds, Eur. J. Inorg. Chem., 2012, 2012, 535–545 CrossRef CAS.
- Q. Knijnenburg, S. Gambarotta and P. H. M. Budzelaar, Ligand-centred reactivity in diiminepyridine complexes, Dalton Trans., 2006, 5442–5448 RSC.
- Protonolysis reactions with 1-K(crypt) resulted in disproportionation. Treating 1 with a diatom surrogate, Me3SiCN, affords dimeric [(BBNPDPtBu)Fe(NC)2]2, see ESI.†.
- (a) B. L. Conley and T. J. Williams, Thermochemistry and Molecular Structure of a Remarkable Agostic Interaction in a Heterobifunctional Ruthenium–Boron Complex, J. Am. Chem. Soc., 2010, 132, 1764–1765 CrossRef CAS PubMed; (b) R. Choukroun, C. Lorber, L. Vendier and C. Lepetit, Vanadocene-Mediated Ionization of Water in the Aqua Species [H2O·B(C6F5)3]: Structural Characterization of the Hydride and Hydroxide Complexes [Cp2V(μ-H)B(C6F5)3] and [Cp2V(μ-OH)B(C6F5)3], Organometallics, 2006, 25, 1551–1553 CrossRef CAS; (c) N. Tsoureas and A. A. Danopoulos, Pyridyl- and diphenylphosphinoethyl-functionalised N-heterocyclic carbene platinum methyl complexes, J. Organomet. Chem., 2015, 775, 178–187 CrossRef CAS.
- A. K. Hickey, S. B. Muñoz, S. A. Lutz, M. Pink, C.-H. Chen and J. M. Smith, Arrested α-hydride migration activates a phosphido ligand for C–H insertion, Chem. Commun., 2017, 53, 412–415 RSC.
- 2-SPh and the zinc analogue can be prepared from the reaction of (BBNPDPtBu)MH2 with PhSSPh. Both reactions extrude H2, see ESI.†.
- (a) S. E. Creutz and J. C. Peters, Exploring secondary-sphere interactions in Fe–NxHy complexes relevant to N2 fixation, Chem. Sci., 2017, 8, 2321–2328 RSC; (b) Y. M. Badiei, M. A. Siegler and D. P. Goldberg, O2 Activation by Bis(imino)pyridine Iron(II)–Thiolate Complexes, J. Am. Chem. Soc., 2011, 133, 1274–1277 CrossRef CAS PubMed.
- N. A. Eckert, J. M. Smith, R. J. Lachicotte and P. L. Holland, Low-Coordinate Iron(II) Amido Complexes of β-Diketiminates: Synthesis, Structure, and Reactivity, Inorg. Chem., 2004, 43, 3306–3321 CrossRef CAS PubMed.
- (a) R. D. Bemowski, A. K. Singh, B. J. Bajorek, Y. DePorre and A. L. Odom, Effective donor abilities of E-t-Bu and EPh (E = O, S, Se, Te) to a high valent transition metal, Dalton Trans., 2014, 43, 12299–12305 RSC; (b) S. A. DiFranco, N. A. Maciulis, R. J. Staples, R. J. Batrice and A. L. Odom, Evaluation of Donor and Steric Properties of Anionic Ligands on High Valent Transition Metals, Inorg. Chem., 2012, 51, 1187–1200 CrossRef CAS PubMed.
- Analysis of the LUMO's were used in lieu of the optimized reduced complexes of 2-E and truncated variants because (1) the reorganization of 1 upon reduction is minimal, (2) the high correlation between the β-LUMO of 1 and the β-SOMO of 1-K(crypt), see Fig. S88.† An analysis LUMO energies and calculated reduction potentials or a subset of 1 and 2-E is included in the ESI.†.
- (a) D. D. Méndez-Hernández, J. G. Gillmore, L. A. Montano, D. Gust, T. A. Moore, A. L. Moore and V. Mujica, Building and testing correlations for the estimation of one-electron reduction potentials of a diverse set of organic molecules, J. Phys. Org. Chem., 2015, 28, 320–328 CrossRef; (b) J. R. Levin, W. L. Dorfner, A. X. Dai, P. J. Carroll and E. J. Schelter, Density Functional Theory as a Predictive Tool for Cerium Redox Properties in Nonaqueous Solvents, Inorg. Chem., 2016, 55, 12651–12659 CrossRef CAS PubMed.
- (a) R. Hoffmann, P. von Ragué Schleyer and H. F. Schaefer III, Predicting Molecules—More Realism, Please!, Angew. Chem., Int. Ed., 2008, 47, 7164–7167 CrossRef PubMed; (b) The truncated analogue, (MePDPtBu)FeH2, is calculated to have a M–H bond index of 0.65.
- The assignment of (BBNPDPtBu)Fe(CNAr)2 is based on 1H NMR and IR spectroscopies as well as MALDI mass spectrometry. We have been unable to isolate analytically pure samples of this species due to its high solubility.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1884220–1884232 and 1903500. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00561g |
| This journal is © The Royal Society of Chemistry 2019 |