
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting the trifluoroethyl group as a precatalyst ligand in nickel-catalyzed Suzuki-type alkylations†
Yi
Yang
 *a,
Qinghai
Zhou
b,
Junjie
Cai
a,
Teng
Xue
c,
Yingle
Liu
a,
Yan
Jiang
a,
Yumei
Su
a,
Lungwa
Chung
*b and
David A.
Vicic
*c
*a,
Qinghai
Zhou
b,
Junjie
Cai
a,
Teng
Xue
c,
Yingle
Liu
a,
Yan
Jiang
a,
Yumei
Su
a,
Lungwa
Chung
*b and
David A.
Vicic
*c
aKey Laboratory of Green Catalysis of Higher Education Institutes of Sichuan, School of Chemistry and Environmental Engineering, Sichuan University of Science & Engineering, 180 Xueyuan Street, Huixing Lu, Zigong, Sichuan 643000, China. E-mail: yangyiyoung@163.com
bShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: oscarchung@sustc.edu.cn
cDepartment of Chemistry, Lehigh University, 6 E. Packer Ave., Bethlehem, PA 18015, USA. E-mail: vicic@lehigh.edu
First published on 17th April 2019
Abstract
We report herein the exploitment of the partially fluorinated trifluoroethyl as precatalyst ligands in nickel-catalyzed Suzuki-type alkylation and fluoroalkylation coupling reactions. Compared with the [LnNiII(aryl)(X)] precatalysts, the unique characters of bis-trifluoroethyl ligands imparted precatalyst [(bipy)Ni(CH2CF3)2] with bench-top stability, good solubilities in organic media and interesting catalytic activities. Preliminary mechanistic studies reveal that an eliminative extrusion of a vinylidene difluoride (VDF, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) mask from [(bipy)Ni(CH2CF3)2] is a critical step for the initiation of a catalytic reaction.
CF2) mask from [(bipy)Ni(CH2CF3)2] is a critical step for the initiation of a catalytic reaction.
Introduction
Transition metal catalyzed cross-coupling reactions have advanced organic synthesis in the last few decades and have become powerful tools for the generation of molecular complexity.1 Substantial effort has been devoted to identifying general and robust transition metal catalytic systems for reaction methodology research and chemical production improvement. A prominent example is the development of Suzuki–Miyaura coupling systems, which now employ a diverse combinations of transition-metals, supporting ligands, and coupling partners to construct C(sp2)–C(sp2) bonds.2 Although Pd catalysts operate with much success in this arena,3 the development of Ni-catalyzed protocols has been of interest because of the cost efficiency and complementary reactivities.4 For instance, Ni-catalyzed couplings are particularly useful for constructing synthetically challenging C(sp2)–C(sp3) linkages,5–7 due to the facile oxidation of low-valent nickel by C(sp3)-centered electrophiles and the suppression of undesired β-hydrogen eliminations at nickel.4,8 One of the most successful catalysts for nickel-catalyzed coupling reactions is derived from the [(bipyridine)nickel] motif which has been widely employed for both traditional cross-coupling and photoredox catalysis.5–7 However, it should be noted that the conventional [(bipyridine)nickel] systems characterized by a combination of Ni0 catalysts or inorganic NiII salts with bipyridyl ligand still suffer from some unneglectable limitations: (i) commonly used Ni0 sources for catalysis are expensive and air-sensitive, thus hindering their use out of glovebox for large-scale synthesis; (ii) the low solubility of inorganic NiII salts complicates the heteroleptic coordination of exogenous supporting ligands which could have deleterious effects on reaction outcomes.In this context, the development of robust nickel-based precatalysts in which the metallic cores are preligated with privileged ligands is highly desirable and constitutes a viable solution to address the above-mentioned limitations.9 Recently, the carbon-bound nickel precatalysts have exerted powers in a variety of coupling reactions as alternatives to the conventional [LnNiX2] precatalysts (L = P or N ligands).10 Notably, many previously reported carbon-bound Ni precatalysts [LnNi(X)(R)] feature sterically bulky ligands (R = o-tolyl, mesityl, 1-naphthyl), or highly stabilizing motifs (R = η3–allyl, η5–Cp) for sheltering reactive organometallic nickel cores (Fig. 1-A).10 Considering that fluoroalkyl ligands are known to confer enhanced stability to metal complexes relative to their non-fluorinated alkyl counterparts owing to fluorine's unique electronic and steric properties,11–13 we wondered whether the incorporation of selected fluoroalkyl moieties could support novel nickel-based precatalysts and render new catalytic activities for use in synthetic methods development (Fig. 1-B). Herein, we describe the synthesis of such a fluoroalkyl-bound nickel precatalyst and demonstrate its use in C(sp2)–C(sp3) Suzuki-type coupling reactions.
 | ||
| Fig. 1 Strategy for the development of fluoroalkyl-bound nickel precatalyst. | ||
Result and discussion
At the outset, we began the rational design of precatalyst based on the principles of utilizing short fluoroalkyl and bipyridine as supporting ligands for atomic economy and C(sp2)–C(sp3) coupling reaction efficiency. Specifically, the short and partially fluorinated CF3CH2 group was selected as supporting ligand (analogue of ethyl group but end-capped with fluorines) which was anticipated to render distinctive thermostability and reactivities versus both the hydrocarbonated [(bipy)Ni(CH2CH3)2]14 and perfluorinated [(bipy)Ni(CF2CF3)2]12g counterparts (Fig. 1B, bipy = 2,2′-bipyridine). Gratifyingly, the reaction of [Ni(COD)2], CF3CH2I, and 2,2-bipyridine furnished [(bipy)Ni(CH2CF3)2] 2 in 41% isolated yield (eqn (1)) presumably via an interesting ligand redistribution12c,15 of the intermediate [(bipy)Ni(CH2CF3)(I)] 1. The solubility of complex 2 in benzene facilitated its isolation from the nickel halide co-products 3. The 19F NMR spectra of 2 exhibited a triplet at δ −47.98 ppm, demonstrating the presence of CH2CF3 groups and their bonding to Ni core. Dark red crystals of 2 can be grown from THF/pentane and are air-stable at room temperature for several weeks. | (1) |
X-ray diffraction analysis of 2 confirmed the ligation of two CH2CF3 groups at nickel (Fig. 2). Complex 2 featured a square planar arrangement at the NiII core with a rough linear trans N–Ni–C linkage (bond angle: 177.4(2) and 177.8(2)°). In contrast, more striking distortions were found in the previously reported and related complexes [(bipy)Ni(CF3)2] 4 (trans N–Ni–C bond angles: 165.1(2)° and 159.7(2)°) and [(bipy)Ni(CF2CF3)2] 5 (both at 152.2°),12g,12i indicating fewer steric and electronic repulsions of the CH2CF3 chains in 2 compared to the perfluorinated derivatives. Interestingly, Ni–C distances of 2 (1.944(5) and 1.942(4) Å) are substantially longer than those of 4 (1.872(6) and 1.883(6) Å) and 5 (1.910(6) and 1.911(6) Å). Besides, the value of C(2)–F(3) bond length [1.366(6) Å, trans coplanar to C(1)–Ni bond] was clearly larger than the others two carbon–fluorine bonds [C(2)–F(1) 1.346(5) Å; C(2)–F(2) 1.342(6) Å] which implied the possibile use of β-fluorine elimination for further coupling reaction development.
 | ||
| Fig. 2 ORTEP diagram of precatalyst [(bipy)Ni(CH2CF3)2] 2. | ||
Although we did not obtain the one fluoroalkyl accommodated nickel complex [(bipy)Ni(CH2CF3)(I)] 1 which showed more structural similarities to the reported [(bipy)Ni(o-tolyl)Cl] precatalyst,14b we presumed that β-fluorine elimination16 of [(bipy)Ni(CH2CF3)2] 2 hinted by the C–F bond length analysis could be leveraged for the in situ generation of [(bipy)Ni(F)(CH2CF3)] 2a with concurrent extrusion of vinylidenedifluoride (CH2CF2). Notably, the Ni–F structural motif of intermediate 2a was supposed to facilitate the transmetalation of arylboronic acids towards nickel according to a recent example of base-free Suzuki coupling.4g Furthermore, the bis-trifluoroethyl structural motifs of [(bipy)Ni(CH2CF3)2] 2 entails the bench-top stability and excellent solubility in organic solvents which is of vital importance for developing nickel-based precatalysts.9,10
With [(bipy)Ni(CH2CF3)2] 2 in hand, we initially assessed it as a precatalyst for the Suzuki-type coupling between CF3CH2I and arylboronic acids for C(sp2)–C(sp3) bonding. Based upon previously established Ni-catalyzed trifluoroethylation conditions,17 we were pleased to find that coupling products can be obtained in excellent yields at 80 °C with 5 mol% catalyst loading using K3PO4 as a base and DME as a solvent (Table 1, entry 1). Use of other solvents decreased the yields, and only polar non-protic DMSO solvent was comparatively effective. Furthermore, the use of K3PO4 was critical to the success of the coupling reaction and suppressing dehydrofluorination of the final products (for details, see ESI Tables S1 and S2†). The commercialized [(TMEDA)Ni(o-tolyl)Cl]10e,10f bearing modular TMEDA was found to be less efficient (yield 35%) with using the privileged bipyridine as the leading supporting ligand (Table 1, entry 2). In contrast, the bipyridine preligated [(bipy)Ni(o-tolyl)Cl]14b bearing the privileged o-tolyl ligand improved the coupling yield to 78% but was still inferior to that of [(bipy)Ni(CH2CF3)2]. These results demonstrated the importances of supporting ligation groups of both trifluoroethyl and bipyridine in the structural motif of [(bipy)Ni(CH2CF3)2] (Table 1, entries 2–3). Even the classical [(dppf)Ni(o-tolyl)Cl]10c or [Ni(COD)2]/bipyridine18 combined system gave unsatisfactory yield (25–40%) compared with the use of 2 (Table 1, entries 4–5). For further demonstrating the distinctive role of partially fluorinated trifluoroethyl ligand, we compared its catalytic performance with those surrogating [(bipy)NiEt2],14c [(bipy)Ni(CF2CF3)2]12i and [(bipy)Ni(CF3)2]12i (Table 1, entries 6–8). It was found that these fully hydrocarbonated and fluorinated counterpart complexes can not furnish comparable catalytic outcomes. Gratifyingly, the tests of decreasing the precatalyst loading and gram-scale synthesis also provided the coupling product in comparatively good yields (Table 1, entries 6–7). These results demonstrated proof-in-principle of the excellent catalytic efficiency of precatalyst 2 for the targeted Suzuki-type couplings.
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Isolated yield |
| a General conditions: 4-biphenylboronic acid (0.3 mmol), CF3CH2I (0.2 mmol), K3PO4 (0.4 mmol), 5.0 mol% precatalyst loading of 2, DME (1.0 mL). b Gram-scale synthesis. | ||
| 1 | None | 93% |
| 2 | [(TMEDA)Ni(o-tolyl)Cl] (5.0 mol%) and 2,2-bipyridine (5.0 mol%) instead of 2 | 35% |
| 3 | [(bipy)Ni(o-tolyl)Cl] (5.0 mol%) instead of 2 | 78% |
| 4 | [(dppf)Ni(o-tolyl)Cl] (5.0 mol%) instead of 2 | 25% |
| 5 | [Ni(COD)2] (5.0 mol%) and 2,2-bipyridine (5.0 mol%) instead of 2 | 40% |
| 6 | [(bipy)NiEt2] (5.0 mol%) instead of 2 | 13% |
| 7 | [(MeCN)2Ni(CF2CF3)2] (5.0 mol%) and 2,2-bipyridine (5.0 mol%) instead of 2 | 55% |
| 8 | [(MeCN)2Ni(CF3)2] (5.0 mol%) and 2,2-bipyridine (5.0 mol%) instead of 2 | 51% |
| 6 | 2.5 mol% loading of precatalyst 2 | 91% (83%b) |
| 7 | 1.0 mol% loading of precatalyst 2 | 79% |
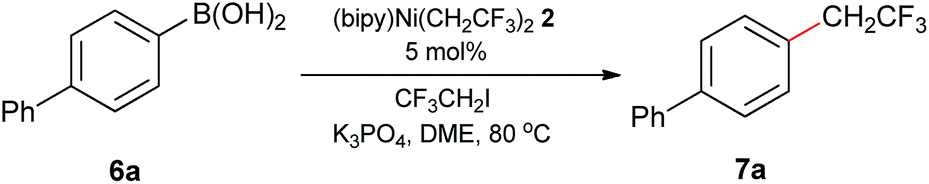
Under the optimized conditions, a wide array of arylboronic acid coupling partners were found to successfully participate in the Suzuki-type trifluoroethylation catalyzed by 2 (Table 2). Both the electron-donating and electron-withdrawing groups substituted arylboronic acids were competent substrates and gave the desired product in moderate to good yield. Broad functional groups were well tolerated, including ethers (7c–7e), aldehydes (7f, 7j), enolizable ketones (7g, 7k), esters (7h, 7l) and nitriles (7i, 7m). Notably, the nitrogen-containing heterocyclic boronic acids (7o, 7p) proceeded smoothly with good yields despite the potential strong binding affinity of the nitrogen atoms with Ni. To further exhibit the synthetic practicality of our precatalyst and trifluoroethylation protocol, the late-stage modifications of fenofibrate and clofibrate (drugs against cardiovascular disease) were accomplished (7s, 7t). Therefore, this synthetic strategy should provide important opportunities for making more diverse biologically active molecules.
|
|
|---|
| a General conditions: (hetero)arylboronic acid (0.6 mmol), CF3CH2I (0.4 mmol), base (0.8 mmol), 2.5 mol% precatalyst loading, DME (2.0 mL), 80 °C. b Isolated yield. c Yield determined by 19F NMR spectroscopy using PhCF3 as an internal standard due to the volatility of naphthalene products. Data in parentheses refer to yields of isolated products. d 5.0 mol% precatalyst loading. |
|
Further demonstration of the privileged catalytic utilities of precatalyst [(bipy)Ni(CH2CF3)2] 2 was showcased by several types of C(sp2)–C(sp3) Suzuki-type alkylations. Iodoethane (8), 3-iodooxetane (9), ethyl bromoacetate (10), allyl bromide (11), (4,4,4-trifluoro-3-iodobutyl)benzene (12a), HCF2CH2I (12b) and FCH2CH2I (12c) were found to successfully couple with a series of arylboronic acids ranging from electron-poor and electron-rich types (Table 3). The encouraging results showed that these primary and secondary alkyl halides were readily compatible with this catalytic alkylation, regardless of the possible β-H or β-F elimination problems.8a Furthermore, the active allyl group can be coupled with the aromatic groups without detection of the migration of double bond (13d).7
|
|
|---|
| a General conditions: (hetero)arylboronic acid (0.6 mmol), the indicated R–X (0.4 mmol), base (0.8 mmol), 2.5 mol% precatalyst loading, DME (2.0 mL), 80 °C. b Isolated yield. c Yield determined by 1H NMR spectroscopy using Cl2CHCHCl2 as an internal standard due to the difficulties in separation of product from deboronative byproduct. d Using 5.0 mol% precatalyst loading and DMSO as the solvent instead of DME. e Using 5.0 mol% precatalyst loading. |

|

The success of precatalyst 2 for the Suzuki-type alkylations further encouraged us to investigate the reaction mechanism. At the start, we intended to determine whether the activation mode of [(bipy)Ni(CH2CF3)2] was consistent with our β-fluorine elimination hypothesis. The precatalyst could undergo β-fluorine elimination to afford [(bipy)Ni(F)(CH2CF3)] 2a with extrusion of CH2CF2, or alternatively undergo reductive elimination like the analogous [(bipy)Ni(CH2CH3)2]14 to generate a [(bipy)Ni(0)] species and CF3CH2CH2CF3. Heating precatalyst 2 at elevated temperature indicated the clear formation of CH2CF2 rather than CF3CH2CH2CF3 through continuous 1H and 19F NMR monitoring (for details, see ESI Fig. S122 and S123†) which illustrated the direct formation of low-valent nickel species like [(bipy)Ni(0)] from [(bipy)Ni(CH2CF3)2] 2 is less likely (Scheme 1-A).19 Additionally, the identification of CH2CF2 and Ar–CH2CF3 (Ar = 4-biphenyl) in GC-MS and NMR analysis of the reactions in Table 3 (e.g.14b, coupling between 4-biphenylboronic acid and 3-iodooxetane in Scheme 1B) revealed the important roles of the trifluoroethyl groups bound to nickel core (for details, see ESI Fig. S124, S125 and Table S5†). These results suggested that the first trifluoroethyl group functioned as the mask of the suggested active species [(bipy)Ni(F)(CH2CF3)] 2avia a CH2CF2 extrusion and the second trifluoroethyl moiety contributed as coupling partner for the formation of Ar–CH2CF3. Interestingly, the finding of byproduct CH3OCH(Ar)CH2OCH3 and ArCH2OCH2CH2OCH3 (Scheme 1-B) illustrated plausible radical activation of DME through abstraction of ethereal α-hydrogens by solvent-caged alkyl radicals.20
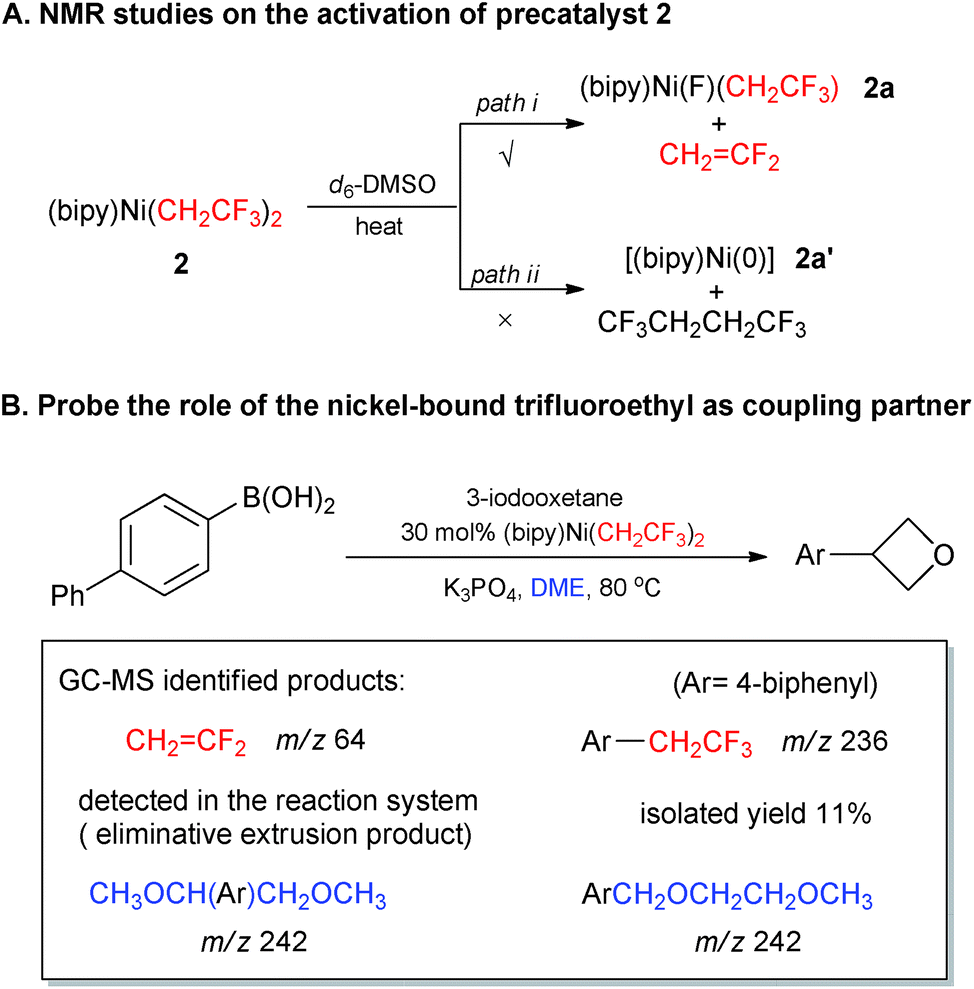 | ||
| Scheme 1 Control experiments for identifying the role of trifluoroethyl ligands in precatalyst 2. | ||
Next, a series of radical inhibition experiments were conducted to verify the possibilities of radical intermediacy (for details, see ESI Tables S8–S10†). It was found that the radical scavenger TEMPO shut down the coupling reactions completely when using the 3-iodooxetane or CF3CH2I as the alkyl electrophiles. Instead, TEMPO-alkyl (alkyl = 3-oxetanyl or trifluoroethyl) adducts 17 and 18 were observed in the GC-MS analysis, respectively. Also, when a radical-clock cyclopropane-based substrate was used, a few ring-opening products like the CF3CH2-merged product 20 and aryl-incorporated product 21 were identified. These experimental results suggested the involvement of CF3CH2˙ radicals (or R radicals) as well as aryl-bound nickel intermediates in the reaction profile.
With the above clues of reaction scenarios in hand, we conducted further interrogations on whether the reactions proceeded via a Ni0/NiII or NiI/NiIII redox shuttle. The important findings of bis-trifluoroethyl ligands of 2 serving as CH2CF2 mask and operational ligand for producing Ar–CH2CF3 inspired us to devise a stoichiometric reaction of complex 2 with 4-biphenylboronic acid as control experiment (Scheme 2-A). The intermediate [(bipy)Ni(F)(CH2CF3)] 2a could be generated in situ under the reaction conditions which was supposed to further undergo a facile Ni–B transmetalation4g to deliver [(bipy)Ni(Ar)(CH2CF3)] 2b (Ar = 4-biphenyl). However, the putative [(bipy)Ni(Ar)(CH2CF3)] intermediate did not proceed through a Ni(II)/Ni(0) reductive elimination10e to furnish Ar–CH2CF3. In addition, CF3CH2F and CF3CH2CH2CF3 were also not found which disfavored the scenario of Ni0 formation from the reductive elimination of 2a and 2. Taken together, these divalent organonickel intermediates (2, 2a and 2b) were not productive for the corresponding NiII/Ni0 reductive elimination under this current reaction system. Interestingly, the product Ar–CH2CF3 was almost comparably efficiently obtained when the precatalyst 2 was replaced by the putative [(bipy)NiI(Br)] complex21 for the coupling between ArB(OH)2 and CF3CH2I (Scheme 2-B). These experimental results suggested that a NiI/NiIII catalytic cycle was highly likely to be superior to Ni0/NiII counterpart in the current reaction systems.
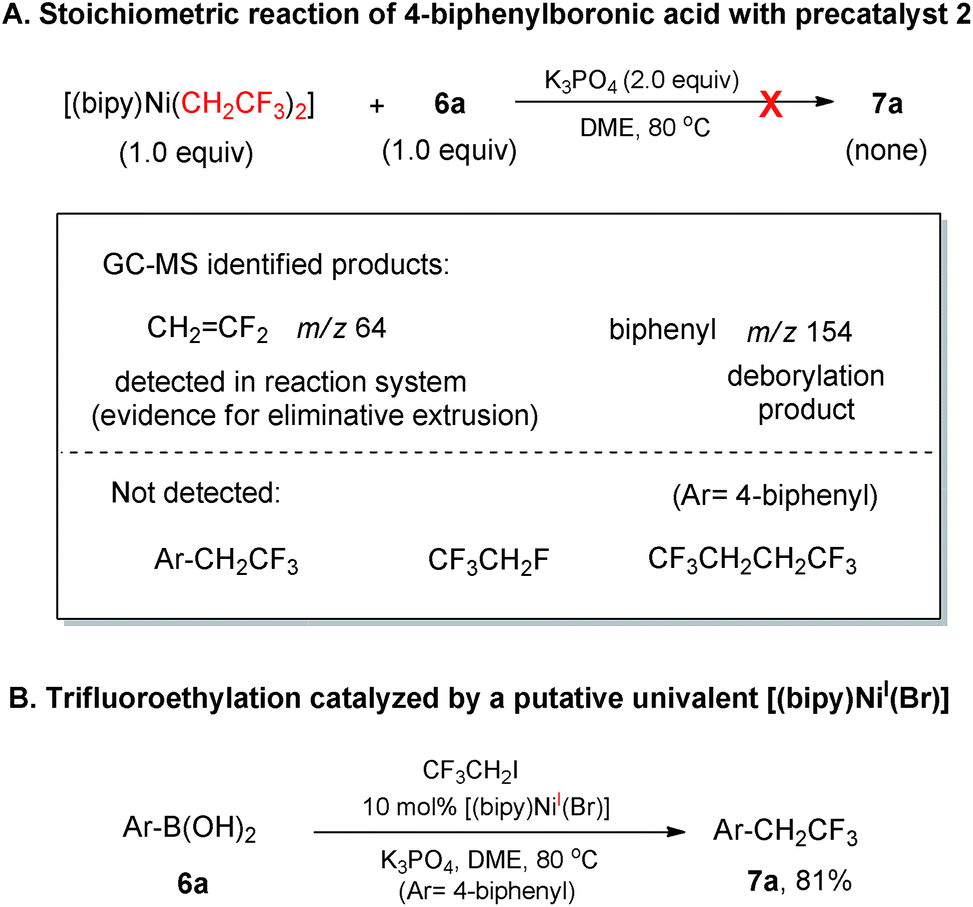 | ||
| Scheme 2 Control experiments to support NiI/NiIII redox shuttle in the catalytic cycle. | ||
Based on the above-mentioned experimental results and relevant previous reports,5,6,13,22 a plausible mechanism was proposed for these current cross-couplings (Scheme 3). The catalysis commences with an eliminative liberation of a CH2CF2 mask and 2a. Intermediate 2a is proposed to undergo transmetalation and subsequent abstraction of halogen atom from R–X to afford (bipy)NiIII(Ar)(CH2CF3)(X) A. Reductive elimination of A has been fingerprinted by the formation of Ar–CH2CF3 and delivered a key catalytic species (bipy)NiI(X) B. Upon the participation of B into the conventional NiI/NiII/NiIII catalytic cycle, the shuttles via transmetalation/oxidative addition22/reductive elimination provided efficient platform for the above described Suzuki-type C(sp2)–C(sp3) alkylation couplings.23
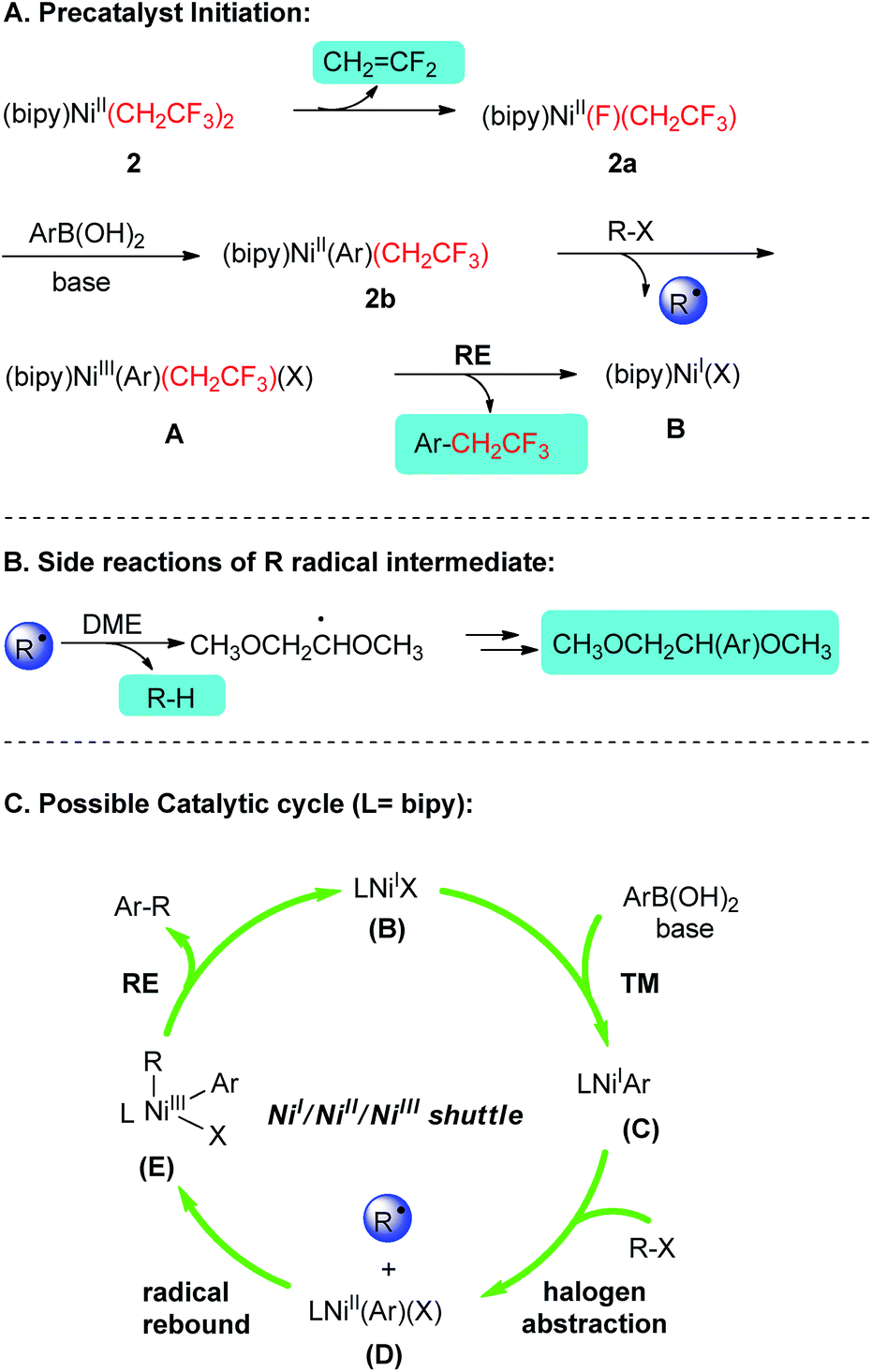 | ||
| Scheme 3 Proposed reaction mechanism for Suzuki-type alkylation couplings based on control experiments. The shadow depicted species in precatalyst initiation and radical-relay side reactions were fingerprinted by GC-MS. | ||
Conclusions
In conclusion, we demonstrated that the nickel-based precatalyst 2 [(bipy)Ni(CH2CF3)2] can be employed in Suzuki-type coupling reactions between (hetero)arylboronic acids and a variety of alkyl halides including several typical partially fluorinated alkyl halides bearing susceptible β-fluorine atoms (2-iodo-1,1,1-trifluoroethane and 12a–12c), leading to new C(sp2)–C(sp3) linkages. Catalytic performance tests demonstrated the advantages of the trifluoroethyl ligand motifs in the precatalyst [(bipy)Ni(CH2CF3)2] versus several sibling perfluorinated and hydrocarbonated counterparts.24 The critical roles of trifluoroethyl groups of precatalyst 2 as both CH2CF2 mask and triggering coupling-ligand in these nickel-catalyzed Suzuki-type alkylations were elucidated through mechanistic investigations. We believe that the initial success outlined here could prompt the utilization of more fluoroalkyl binding moieties for the development of new metal-based precatalysts with tailored activities. Further studies towards this endeavor and mechanistic details are underway in our laboratory, and the results will be reported in due course.
Conflicts of interest
We authors announce that there are no conflicts to declare.Acknowledgements
We thank National Natural Science Foundation of China (21502131, 21672096, 21702095), Science & Technology Department of Sichuan Province (2018JZ0061, 2018HH0128), Education Department of Sichuan Province (18CZ0024), Key Laboratory of Vanadium and Titanium of Sichuan Province (2018FTSZ03), Sichuan University of Science and Engineering (2017RCL03), Shenzhen Nobel Prize Scientists Laboratory Project (C17213101) and Office of Basic Energy Sciences of the U.S. Department of Energy (DE-SC0009363) for funding this work.Notes and references
- (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (b) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433 RSC; (c) J. Choi and G. C. Fu, Science, 2017, 356, 6334 CrossRef PubMed; (d) Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352 CrossRef CAS PubMed; (e) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (b) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed; (c) S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544 CrossRef CAS.
- (a) T. Ben Halima, W. Zhang, I. Yalaoui, X. Hong, Y.-F. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311 CrossRef CAS PubMed; (b) G. Meng, S. Shi and M. Szostak, ACS Catal., 2016, 6, 7335 CrossRef CAS; (c) S. Handa, Y. Wang, F. Gallou and B. H. Lipshutz, Science, 2015, 349, 1087 CrossRef CAS PubMed; (d) Z. Feng, Q.-Q. Min, X.-P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918 CrossRef CAS PubMed.
- (a) C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347 RSC; (b) V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS; (c) J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk and G. C. Fu, Science, 2016, 354, 1265 CrossRef CAS PubMed; (d) G. C. Fu, ACS Cent. Sci., 2017, 3, 692 CrossRef CAS PubMed; (e) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (f) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC; (g) C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100 CrossRef CAS PubMed.
- (a) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920 CrossRef CAS PubMed; (b) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146 CrossRef CAS; (c) A. C. Wotal and D. J. Weix, Org. Lett., 2012, 14, 1476 CrossRef CAS; (d) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016 CrossRef CAS PubMed; (e) N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2015, 137, 10480 CrossRef CAS PubMed; (f) K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684 CrossRef CAS PubMed; (g) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (h) C. Xu, W.-H. Guo, X. He, Y.-L. Guo, X.-Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1170 CrossRef PubMed; (i) X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 1 CrossRef CAS PubMed; (j) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490 CrossRef CAS PubMed; (k) J. Sheng, H.-Q. Ni, H.-R. Zhang, K.-F. Zhang, Y.-N. Wang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 7634 CrossRef CAS PubMed.
- (a) Y.-L. Xiao, Q.-Q. Min, C. Xu, R.-W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837 CrossRef CAS PubMed; (b) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909 CrossRef CAS PubMed; (c) A. Joshi-Pangu, C.-Y. Wang and M. R. Biscoe, J. Am. Chem. Soc., 2011, 133, 8478 CrossRef CAS PubMed; (d) Z. Lu, A. Wilsily and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 8154 CrossRef CAS PubMed; (e) Z. Lu and G. C. Fu, Angew. Chem., Int. Ed., 2010, 49, 6676 CrossRef CAS PubMed; (f) N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 11908 CrossRef CAS PubMed; (g) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 624 CrossRef CAS PubMed; (h) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS PubMed; (i) J. Li, Y. Luo, H. W. Cheo, Y. Lan and J. Wu, Chem, 2019, 5, 192 CrossRef CAS; (j) H. Yi, W. Mao and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 3575 CrossRef CAS PubMed; (k) L. An, Y.-L. Xiao, Q.-Q. Min and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 9079 CrossRef CAS PubMed; (l) N. D. Schley and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 16588 CrossRef CAS PubMed; (m) Y. M. Su, G. S. Feng, Z. Y. Wang, Q. Lan and X. S. Wang, Angew. Chem., Int. Ed., 2015, 54, 6003 CrossRef CAS PubMed; (n) J. Sheng, H.-Q. Ni, K.-J. Bian, Y. Li, Y.-N. Wang and X.-S. Wang, Org. Chem. Front., 2018, 5, 606 RSC.
- (a) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (b) F. Chen, K. Chen, Y. Zhang, Y. He, Y.-M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929 CrossRef CAS PubMed; (c) L. Peng, Y. Li, Y. Li, W. Wang, H. Pang and G. Yin, ACS Catal., 2018, 8, 310 CrossRef CAS.
- (a) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed; (b) X. Hu, Chem. Sci., 2011, 2, 1867 RSC; (c) A. Kaga and S. Chiba, ACS Catal., 2017, 7, 4697 CrossRef CAS.
- (a) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 25 CrossRef CAS PubMed; (b) L. M. Guard, M. Mohadjer Beromi, G. W. Brudvig, N. Hazari and D. J. Vinyard, Angew. Chem., Int. Ed., 2015, 54, 13352 CrossRef CAS PubMed; (c) A. B. Dürr, H. C. Fisher, I. Kalvet, K.-N. Truong and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 13431 CrossRef PubMed.
- (a) E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 1585 CrossRef CAS PubMed; (b) C. M. Lavoie, P. M. MacQueen, N. L. Rotta-Loria, R. S. Sawatzky, A. Borzenko, A. J. Chisholm, B. K. V. Hargreaves, R. McDonald, M. J. Ferguson and M. Stradiotto, Nat. Commun., 2016, 7, 11073 CrossRef PubMed; (c) R. S. Sawatzky, M. J. Ferguson and M. Stradiotto, Synlett, 2017, 28, 1586 CrossRef CAS; (d) N. H. Park, G. Teverovskiy and S. L. Buchwald, Org. Lett., 2014, 16, 220 CrossRef CAS PubMed; (e) J. D. Shields, E. E. Gray and A. G. Doyle, Org. Lett., 2015, 17, 2166 CrossRef CAS PubMed; (f) J. Magano and S. Monfette, ACS Catal., 2015, 5, 3120 CrossRef CAS; (g) P. M. MacQueen, J. P. Tassone, C. Diaz and M. Stradiotto, J. Am. Chem. Soc., 2018, 140, 5023 CrossRef CAS PubMed; (h) A. J. Nett, S. Cañellas, Y. Higuchi, M. T. Robo, J. M. Kochkodan, M. T. Haynes, J. W. Kampf and J. Montgomery, ACS Catal., 2018, 8, 6606 CrossRef CAS PubMed; (i) M. Mohadjer Beromi, G. Banerjee, G. W. Brudvig, N. Hazari and B. Q. Mercado, ACS Catal., 2018, 8, 2526 CrossRef CAS PubMed.
- (a) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC; (b) B. Lantaño, M. R. Torviso, S. M. Bonesi, S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2015, 285, 76 CrossRef; (c) H. Wang and D. A. Vicic, Synlett, 2013, 24, 1887 CrossRef CAS; (d) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- (a) M. D. Kosobokov, A. Sandleben, N. Vogt, A. Klein and D. A. Vicic, Organometallics, 2018, 37, 521 CrossRef CAS; (b) K. R. McGarry and D. A. Vicic, J. Fluorine Chem., 2017, 203, 206 CrossRef CAS; (c) L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536 CrossRef CAS PubMed; (d) S. Yu, Y. Dudkina, H. Wang, K. V. Kholin, M. K. Kadi-rov, Y. H. Budnikova and D. A. Vicic, Dalton Trans., 2015, 44, 19443 RSC; (e) C.-P. Zhang, H. Wang, A. Klein, C. Biewer, K. Stirnat, Y. Yamaguchi, L. Xu, V. Gomez-Benitez and D. A. Vicic, J. Am. Chem. Soc., 2013, 135, 8141 CrossRef CAS PubMed; (f) P. T. Kaplan, L. Xu, B. Chen, K. R. McGarry, S. Yu, H. Wang and D. A. Vicic, Organometallics, 2013, 32, 7552 CrossRef CAS; (g) Y. Yamaguchi, H. Ichioka, A. Klein, W. W. Brennes-sel and D. A. Vicic, Organometallics, 2012, 31, 1477 CrossRef CAS; (h) A. Klein, D. A. Vicic, C. Biewer, I. Kieltsch, K. Stirnat and C. Hamacher, Organometallics, 2012, 31, 5334 CrossRef CAS; (i) Compounds 4 and 5 were theoretically modulated in ref. 12g. We used the combination of bipyridine and (MeCN)2Ni(Rf)2 as the surrogate. The complexes (MeCN)2Ni(Rf)2 (Rf = CF3 or C2F5) were synthesized according to our developed method (ref. 12e).
- (a) N. M. Camasso and M. S. Sanford, Science, 2015, 347, 1218 CrossRef CAS PubMed; (b) J. R. Bour, N. M. Camasso and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 8034 CrossRef CAS PubMed; (c) J. R. Bour, N. M. Camasso, E. A. Meucci, J. W. Kampf, A. J. Canty and M. S. Sanford, J. Am. Chem. Soc., 2016, 138, 16105 CrossRef CAS PubMed; (d) J. W. Schultz, K. Fuchigami, B. Zheng, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2016, 138, 12928 CrossRef CAS PubMed; (e) M. B. Watson, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 35 CrossRef CAS PubMed.
- Complex [(bipy)Ni(CH2CH3)2] are known to be air-sensitive and eliminate the alkyl ligands (reductive elimination and β-eliminations) at temperatures close to room temperature, please see references: (a) T. Yamamoto and M. Abla, J. Organomet. Chem., 1997, 535, 209 CrossRef CAS; (b) M. Uchino, K. Asagi, A. Yamamoto and S. Ikeda, J. Organomet. Chem., 1975, 84, 93 CrossRef CAS; (c) A. Yamamoto, K. Morifuji, S. Ikeda, T. Saito, Y. Uchida and A. Misono, J. Am. Chem. Soc., 1965, 87, 4652 CrossRef CAS.
- Ligand redistribution processes are common in nickel chemistry, please see references: (a) Y. Takakazu, K. Teiji and Y. Akio, Bull. Chem. Soc. Jpn., 1981, 54, 2010 CrossRef; (b) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef CAS PubMed; (c) T. Yamamoto, S. Wakabayashi and K. Osakada, J. Organomet. Chem., 1992, 428, 223 CrossRef CAS.
- For selected examples of nickel-catalyzed β-fluorine eliminations for synthetic methods development, see: (a) T. Ichitsuka, T. Fujita, T. Arita and J. Ichikawa, Angew. Chem., Int. Ed., 2014, 53, 7564 CrossRef CAS PubMed; (b) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947 CrossRef CAS; (c) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245 CrossRef CAS; (d) Z. Lin, Y. Lan and C. Wang, ACS Catal., 2019, 9, 775 CrossRef CAS.
- (a) X. Zhang and C. Yang, Adv. Synth. Catal., 2015, 357, 2721 CrossRef CAS; For some other related metal catalyzed trifluoroethylations: (b) Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 1033 CrossRef CAS PubMed; (c) A. Liang, X. Li, D. Liu, J. Li, D. Zou, Y. Wu and Y. Wu, Chem. Commun., 2012, 48, 8273 RSC; (d) S. Xu, H.-H. Chen, J.-J. Dai and H.-J. Xu, Org. Lett., 2014, 16, 2306 CrossRef CAS PubMed.
- Although [(bipy)Ni(CH2CF3)2] could be generated in situ within the [Ni(COD)2]/bipyridine trifluoroethylation systems, the deleterious effect of 1,5-cyclooctadiene (COD) on the current coupling can not be neglected. Please see related ref. 10a, e J. D. Shields, D. T. Ahneman, T. J. A. Graham and A. G. Doyle, Org. Lett., 2014, 16, 142 CrossRef CAS PubMed , and .
- The reductive eliminations of difluoroalkylnickel species were also proved to be challenging in the ref. 12e and 13c.
- When using CF3CH2I or BrCH2CO2Et as electrophiles, we also found the formation of by product R–H (R = CF3CH2– or EtOCOCH2–) which could be ascribed to the abstraction of ethereal α-hydrogen in the DME solvent cages (for details, see ESI Tables S6 and S7†). For related references for radical reactions in solvent cage: (a) D. A. Braden, E. E. Parrack and D. R. Tyler, Coord. Chem. Rev., 2001, 211, 279 CrossRef CAS; (b) K. Michio, G. Michiko and Y. Masato, Bull. Chem. Soc. Jpn., 1987, 60, 295 CrossRef; (c) R. A. Sheldon and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 4395 CrossRef CAS.
- [(bipy)NiI(Br)] was prepared according to the synthetic method of [(dppf)Ni(I)Cl]: (a) L. M. Guard, M. Mohadjer Beromi, G. W. Brudvig, N. Hazari and D. J. Vinyard, Angew. Chem., Int. Ed., 2015, 54, 13352 CrossRef CAS PubMed. It was characterized by IR and XPS due to its bad solubility for single crystal formation (for details, see ESI†). Very recently, Hazari group disclosed the synthesis of [(neocuproine)2NiI](Cl) which had a similar skeleton: (b) M. Mohadjer Beromi, G. W. Brudvig, N. Hazari, H. M. C. Lant and B. Q. Mercado, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201901866.
- For selected example of radical-rebound oxidative addition of alkyl halide: (a) Z. Ruan, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 3153 CrossRef CAS PubMed. If the radical-chain mechanism was operative, the cage-escaped radicals should dimerize via radical recombination. Also, we did not detect any radical dimerization products (R–R) in these experiments which could support a fast cage-rebound other than cage-escape process. For our recent report about the radical-rebound oxidative addition step in the nickel-catalyzed Suzuki-type fluoroethylation: (b) Y. Yang, J. Cai, G. Luo, Y. Jiang, Y. Su, Y. Su, C. Li, Y. Zheng, J. Zeng and Y. Liu, Org. Chem. Front., 2019 10.1039/c9qo00066f. For some seminal literatures for elucidating cage-rebound or cage-escape modes: ref. 15b and (c) J. Breitenfeld, J. Ruiz, M. D. Wodrich and X. Hu, J. Am. Chem. Soc., 2013, 135, 12004 CrossRef CAS PubMed.
- According to the recent reports about the catalytic activities of [LnNiIAr], the catalytic shuttle started from the species [LnNiIX] Bvia a transmetalation/oxidative addition/reductive elimination sequence other than oxidative addition/transmetalation/reductive elimination order should be plausible. The observed inertness of [(bipy)NiIBr] towards CF3CH2I seemed to rule out the oxidative addition/transmetalation/reductive elimination sequence. For some references about related [LnNiIAr] complexes: (a) J. B. Diccianni, J. Katigbak, C. Hu and T. Diao, J. Am. Chem. Soc., 2019, 141, 1788 CrossRef CAS PubMed; ref. 10i. More corroborative computational studies can be found in these references: (b) Z. Li, Y.-Y. Jiang and Y. Fu, Chem.–Eur. J., 2012, 18, 4345 CrossRef CAS PubMed; (c) X. Lin and D. L. Phillips, J. Org. Chem., 2008, 73, 3680 CrossRef CAS PubMed. For selected references that support NiI/NiIII catalytic shuttle: ref. 13c and d (d) G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175 CrossRef CAS PubMed; (e) B. Zheng, F. Tang, J. Luo, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2014, 136, 6499 CrossRef CAS PubMed.
- GC-MS analysis of the reaction mixtures (Table 1, entries 7–8) revealed no formation of Ar–CF2CF3 or Ar–CF3, respectively. These results demonstrated quite different catalytic profiles in which CF3CF2 and CF3 did not serve as coupling ligands. Unlike the CF3CH2 counterpart, their reluctance to contribute as triggering coupling-ligand might be explained by the challenging Ar–Rf (Rf = perfluoroalkyl) reductive elimination. For related reference: G. G. Dubinina, W. W. Brennessel, J. L. Miller and D. A. Vicic, Organometallics, 2008, 27, 3933 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1436475. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00554d |
| This journal is © The Royal Society of Chemistry 2019 |