
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayered Zn2[Co(CN)6](CH3COO) double metal cyanide: a two-dimensional DMC phase with excellent catalytic performance†
Carlos
Marquez
 a,
Arkadiy
Simonov
b,
Michael T.
Wharmby
c,
Cédric
Van Goethem
a,
Ivo
Vankelecom
a,
Bart
Bueken
a,
Andraž
Krajnc
d,
Gregor
Mali
d,
Dirk
De Vos
*a and
Trees
De Baerdemaeker
*a
a,
Arkadiy
Simonov
b,
Michael T.
Wharmby
c,
Cédric
Van Goethem
a,
Ivo
Vankelecom
a,
Bart
Bueken
a,
Andraž
Krajnc
d,
Gregor
Mali
d,
Dirk
De Vos
*a and
Trees
De Baerdemaeker
*a
aCentre for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be; trees.debaerdemaeker@kuleuven.be
bInorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, UK
cDeutsches Elektronen-Synchrotron (DESY), Notkestraße 85, 22607 Hamburg, Germany
dDepartment of Inorganic Chemistry and Technology, National Institute of Chemistry, Hajdrihova 19, 1001 Ljubljana, Slovenia
First published on 4th April 2019
Abstract
Double metal cyanides (DMCs) are well known, industrially applied catalysts for ring opening polymerization reactions. In recent years, they have been studied for a variety of catalytic reactions, as well as other applications, such as energy storage and Cs sorption. Herein, a new, layered DMC phase (L-DMC), Zn2[Co(CN)6](CH3COO)·4H2O, was synthesized. The structure, which crystallizes in the monoclinic space group P21/m, consists of positively charged {Zn2Co(CN)6}+ DMC layers linked through acetate groups and presents a new layered structure to the family of double metal cyanides. L-DMC proved to be a reusable and stable catalyst that exhibited a higher activity than the benchmark DMC catalyst in two important applications: hydroamination of phenylacetylene with 4-isopropylaniline and polymerization of 1,2-epoxyhexane.
Introduction
Double metal cyanides (DMCs) are inorganic coordination polymers containing two metal atoms connected by cyanide groups. They have been used industrially as solid catalysts for the ring opening polymerization of epoxides since the 1960s,1–3 and their potential as Lewis acid catalysts for other organic transformations such as co-polymerization of epoxides and CO2,4,5 hydroamination,6–8 transesterification9 and coupling reactions10,11 has recently been uncovered. DMCs can generally be described as polynuclear transition metal cyanides with the structural formula Mu[M′(CN)n]v·xH2O (M and M′ are two metal cations), which often crystallize with a cubic unit cell.12 The common u–v combination 3–2 can be described as a rock salt type structure consisting of alternating M2+ and [M′(CN)6]3− ions. The charge imbalance between these ions produces vacancies that occur at one in three [M′(CN)6]3− sites.13–15 Even though the cubic phase is the most common, the occurrence of other phases, for example a monoclinic and a rhombohedral phase for Zn3[Co(CN)6]2, has also been observed.16,17In all the aforementioned phases, however, the pores are too narrow to allow the diffusion of the majority of aromatic compounds, which for DMC-catalyzed reactions of these molecules signifies that the reaction takes place on the outer surface.7 In the light of this, many strategies have been suggested in order to increase the accessibility and availability of the catalytically active sites. For example, we recently showed that by dispersing the catalytically active Zn3[Co(CN)6]2 DMC on silica, its activity can be vastly improved in hydroamination and epoxide polymerization reactions because of the increased accessibility of the active sites.8 Furthermore, Robertson et al.18 proposed layered Co(H2O)2[M′(CN)4] DMCs (M′ = Ni, Pd or Pt) as high surface area catalysts for the propylene oxide (PO) homopolymerization and PO/CO2 copolymerization. In this case, the activity of the Co[M′(CN)4] system still remained lower than that of the Zn3[Co(CN)6]2 DMCs. Layered materials, such as layered double hydroxides (LDHs), layered metal–organic frameworks (MOFs) and clay minerals, are generally of wide interest because of their highly tunable properties and wide range of potential applications, including in catalysis,19–22 energy storage,23,24 and CO2 capture.25,26 Additionally, their potential for modification through, for instance, exfoliation has been extensively investigated in the case of clays, layered zeolite precursors or layered double hydroxides.27–35
During our investigation of Zn3[Co(CN)6]2 DMCs, we discovered single crystals of a phase that was not cubic, but instead, consisted of positively charged DMC layers linked by acetate anions. To the best of our knowledge, such a cationic, layered DMC structure has not yet been reported in the literature. Given the known catalytic activity of other Zn3[Co(CN)6]2 DMCs – cubic,36 monoclinic,16 or (partially) amorphous37–39 – we were motivated to synthesize this layered DMC phase as a powder to investigate its catalytic properties. Herein, we report the synthesis, structure, physicochemical characterization and catalytic activity of this layered DMC (L-DMC). Moreover, the advantages of L-DMC over the benchmark DMC catalyst will be demonstrated for two important catalytic applications of DMCs: hydroamination of phenylacetylene and 4-isopropylaniline, and epoxide polymerization of 1,2-epoxyhexane.
Experimental section
Synthesis of single-crystal L-DMC
Single crystals of L-DMC were grown from silica gel using a modification of the technique reported in ref. 40 and 41. The silica gel was formed by mixing 2.5 mL of a 1.37 M aqueous solution of Na2SiO3 with 2.5 mL of a 0.0125 M aqueous solution of zinc acetate dihydrate and 5 mL of an aqueous acetic acid solution (2 M), and leaving the solution to stand overnight. Subsequently, a K3[Co(CN)6] solution (3 mL, 0.0833 M) was layered on top of the gel and the crystals were left to grow for three days.Optimized synthesis of L-DMC (powder)
Zinc acetate dihydrate (Zn(CH3COO)2·2H2O, 3 mmol) was dissolved in 20 mL of a 4 M solution of acetic acid. The solution was stirred and heated to 80 °C in an oil bath before dropwise addition of 10 mL of an aqueous solution containing 1.5 mmol of K3[Co(CN)6]. The reaction was immediately removed from the bath and the precipitate was recovered by centrifugation, washed three times with distilled water and dried at 80 °C under vacuum (587 mg, 82% yield).Synthesis of benchmark DMC (DMC-PTMEG)
The benchmark DMC catalyst (DMC-PTMEG) was synthesized with tert-butanol (tBuOH) as complexing agent and poly(tetramethylene ether)glycol (PTMEG) as co-complexing agent following literature procedures.6–8,42 This material has been reported to be the most active compared to other Zn3[Co(CN)6]2 DMCs prepared by co-precipitation for intermolecular hydroamination of amines and alkenes or alkynes and ring opening polymerization reactions.6,42 ZnCl2 (15 mmol) and PTMEG (1.5 mmol) were dissolved in 150 mL of water. To this, 15 mL of a 0.1 M aqueous solution of K3[Co(CN)6] was added dropwise. tBuOH (37.5 mL) was added to the reaction mixture, which was subsequently stirred for 3 h at room temperature. The final solid was recovered by centrifugation and washed three times with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of water:tBuOH, and then dried at 60 °C overnight.
:50 mixture of water:tBuOH, and then dried at 60 °C overnight.
Characterization
High-throughput powder X-ray diffraction (PXRD) data were recorded on a Malvern PANalytical Empyrean diffractometer equipped with a PIXcel3D solid state detector using a Cu anode (CuKα1,2 radiation) in transmission geometry (1.3–45° 2θ range; 0.013° step size), or on a STOE Stadi MP diffractometer (in transmission mode) using an image plate detector and focusing Ge(111) monochromator (CuKα1 radiation) over a range of 5–60° 2θ. All PXRD patterns were recorded at room temperature. Single-crystal X-ray diffraction measurements were performed at beamline I19 (Diamond Light Source, Oxfordshire, UK)43 using a wavelength of λ = 0.6889 Å with the crystal cooled to a temperature of −173 °C. The structure was solved using SUPERFLIP and refined using tools available within the Crystals and Olex2 suites.44–46 High-resolution synchrotron powder X-ray diffraction data were collected at beamline P02.1, PETRA III (DESY, Hamburg, Germany) at a wavelength of λ = 0.20738 Å using a PerkinElmer area detector.47 The as-prepared L-DMC powder sample was loaded into a 0.5 mm borosilicate glass capillary tube and data were collected for 60 s in Debye–Scherrer geometry. Raw two-dimensional detector images were integrated using the DAWN suite, using a bin size of 0.005° 2θ.48 Indexing, Pawley fitting and Rietveld refinement of the powder diffraction data were performed using the routines of TOPAS-Academic V5.49–51 Chemically sensible restraints were applied to the C–N, C–C, C–O, Co–C, Zn–N and Zn–O distances. The metal ratio of the L-DMC was determined with ICP-OES using a Varian 720-ES equipped with a double-pass glass cyclonic spray chamber, a Sea Spray concentric glass nebulizer and a high solids torch. Samples were digested in a 7:3 (v:v) solution of HNO3–HCl and heated to 200 °C in a microwave oven for 2 h, as reported by Lee et al.52 Fourier-transform infrared (FTIR) spectra of KBr wafers (1 wt% of DMC) were collected on a Bruker IFS 66 v/S Vacuum FTIR spectrometer (resolution 4 cm−1). Solid state nuclear magnetic resonance experiments (SSNMR) were performed on a 600 MHz Varian spectrometer, equipped with a 1.6 mm triple-resonance magic-angle spinning (MAS) probe. Larmor frequencies were 599.44 MHz for 1H and 150.74 MHz for 13C nuclei, and the spectral axes of the recorded spectra were referenced with respect to the signals of tetramethylsilane. The sample was spun at MAS rate of 40 kHz. The 1H MAS NMR spectrum was collected using Hahn-echo sequence with 90° and 180° pulses of 1.5 μs and 3.0 μs, respectively, and inter-pulse delay of 25 μs. 16 scans were accumulated with a recycle delay of 5 s. The 1H–13C cross-polarization (CP) MAS NMR spectrum was recorded by first exciting protons and transferring polarization to carbon nuclei using the ramped-amplitude CP block with a duration of 5 ms. 10000 scans were accumulated with the recycle delay of 1 s. During the acquisition, high-power XiX heteronuclear decoupling was applied. N2 physisorption isotherms were collected on a Micromeritics 3Flex Surface Analyzer at −196 °C. Before the measurements, the samples were evacuated at 150 °C for 16 h. The specific surface area (SBET) was determined using the BET method in a p/p0 range from 0.004 to 0.03. The specific external surface area (Sext) and the micropore volume (Vmicro) were obtained using t-plot analysis. In order to estimate the micropore size distribution (MPSD), Ar physisorption isotherms were collected under similar conditions and the MPSD was estimated using the Horvath–Kawazoe (HK) method. Scanning electron microscopy (SEM) images were taken using a JEOL-6010LV SEM. Prior to imaging, samples were coated with a Pd/Au layer using a JEOL JFC-1300 autofine coater under Ar plasma for 10 s. Transmission electron microscopy (TEM; high angle annular dark field, HAADF) images and energy-dispersive X-ray spectroscopy (EDX) maps were recorded with a JEOL ARM-200F TEM with a probe Cs corrector operated at 200 kV. Before imaging, the samples were suspended in ethanol and dropped onto a Cu grid (300 Mesh, Pacific Grid Tech, USA) coated with a Lacey carbon layer. Thermogravimetric mass spectrometry (TG-MS) analysis measurements were carried out on a NETZSCH STA 449 F3 Jupiter® thermal analyzer coupled with a Hiden HPR-20 EGA gas analysis system with a heating rate of 10 °C min−1 under air atmosphere.
Catalytic reactions
Results and discussion
Structure solution and refinement
The structure of the crystals grown using slow diffusion in a silica gel was solved by single crystal X-ray diffraction. While all non-H atoms could be found, the quality of the data was too low to allow satisfactory structure refinement. Through further synthesis optimization (vide infra), we were able to obtain a phase pure microcrystalline powder of the L-DMC. Synchrotron powder diffraction data were collected and successfully refined by the Rietveld method. Details of the crystallographic parameters of the final refinement are given in Table 1 and a Rietveld plot for the final cycles of refinement is presented in Fig. 1.| Composition | Co(CN)6Zn2(CH3COO)·4H2O |
| Space group (crystal system) | P21/m (monoclinic) |
| a/Å | 12.3804(3) |
| b/Å | 7.50396(16) |
| c/Å | 8.4883(2) |
| β/° | 85.3299(19) |
| Cell volume/Å3 | 785.97(4) |
| R wp/RBragg | 2.36/1.31 |
| χ 2 | 0.652 |
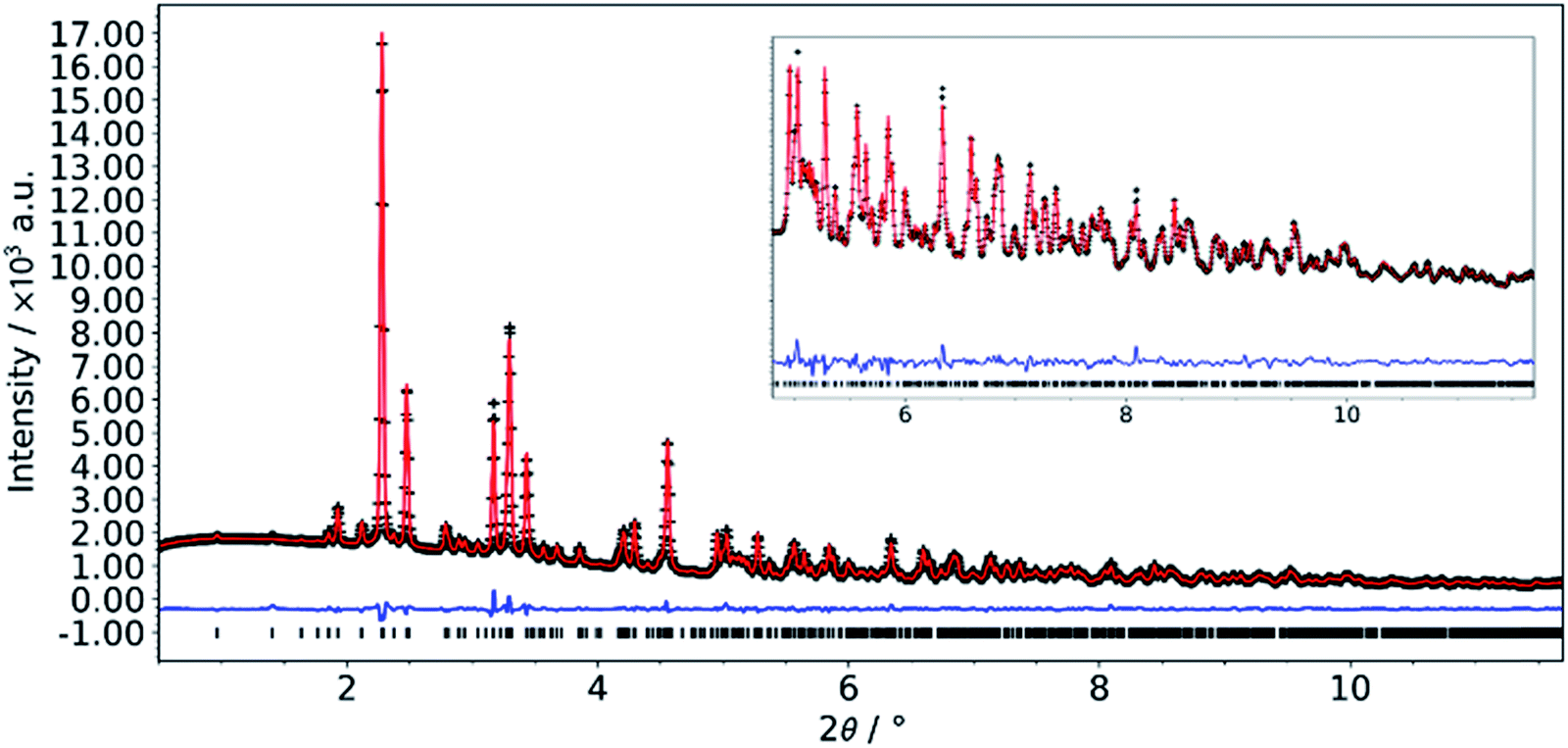 | ||
| Fig. 1 Final Rietveld plot for the refinement of L-DMC. Data collected at beamline P02.1, PETRA III (DESY, Hamburg, Germany); λ = 0.20738 Å. | ||
The structure crystallizes in the monoclinic space group P21/m and is shown in Fig. 2. The asymmetric unit consists of two symmetry independent Zn atoms and one Co atom, four cyanide groups (labelled CN20 to 23, with each C and N atom numbered accordingly), acetate and three water molecules (represented as isolated O atoms Ow1, Ow11 and Ow12). Zn1 is tetrahedrally coordinated to three N atoms from two symmetry independent cyanide groups (twice to CN20 and once to CN21) and one O atom of the acetate (O10). By contrast, Zn2 has an octahedral coordination environment consisting of three N atoms of two independent cyanide groups (twice CN22, once CN23), one O atom from an acetate group (O11) and two water molecules (Ow1). Co1 is also octahedrally coordinated by six C atoms, two from CN20 and CN22 and one from each of the other cyanide groups. The metal polyhedra are linked together through cyanide groups to form a puckered-layered structure in the ab-plane, in which two octahedrally coordinated Zn2 sites and one tetrahedral Zn1 site define a pocket, into which an octahedrally coordinated Zn2 from the underlying layer projects. The layers are coordinatively bonded together through an acetate group in the apical position of the tetrahedral Zn1, which bridges to the octahedral Zn2 in the layer below. Two capping water molecules (Ow1) project from this octahedral Zn2 site into the interstitial region where they form a H-bonding network with two further water molecules (Ow11 and Ow12), with O⋯O distances in the range 2.827(19)–3.054(16) Å.
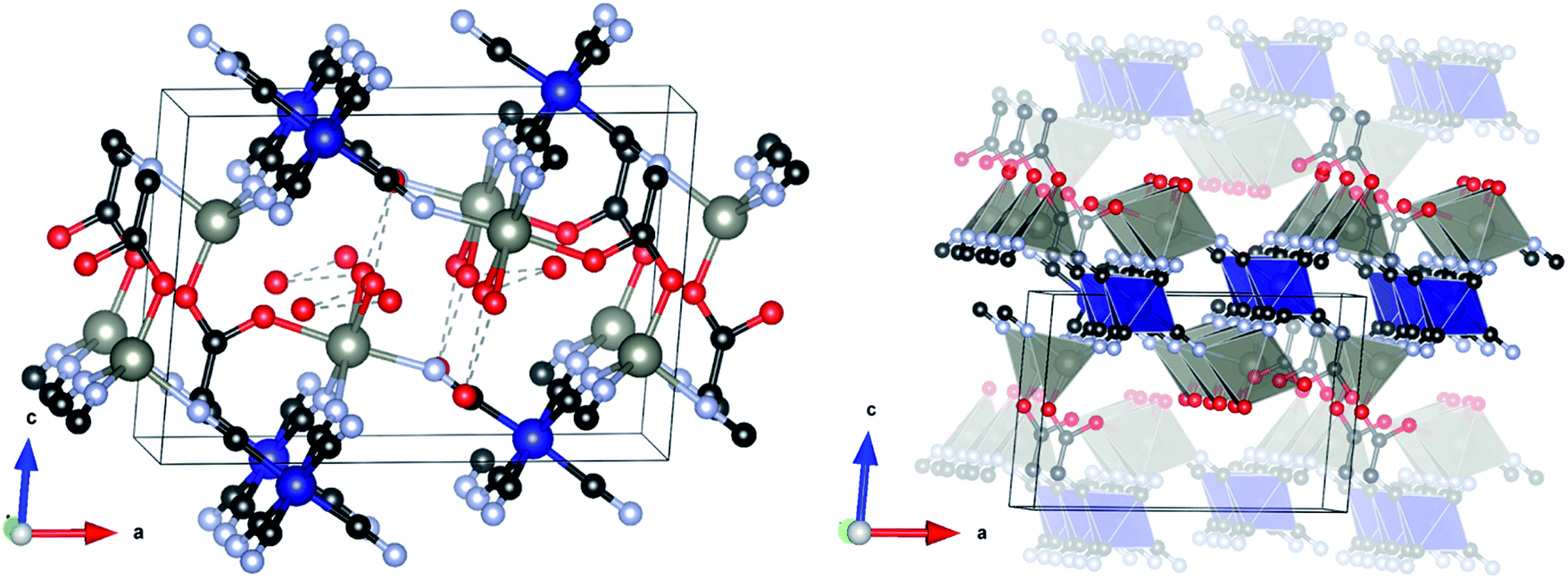 | ||
| Fig. 2 Crystal structure of L-DMC (left) and polyhedral view showing the stacking of the layers in L-DMC (right). Red, blue, light grey, dark grey and black spheres represent O, N, C, Zn and Co atoms, respectively. H atoms were omitted for clarity. | ||
It should be noted that the powder pattern reported by Zhang et al.53 of a DMC obtained by ball milling K3[Fe(CN)6] and zinc acetate (complex 2 in ref. 53) is qualitatively similar to the diffraction pattern of L-DMC. We therefore suggest that the structure they observed during ball milling is an Fe(CN)6 analogue of our layered phase. Furthermore, from the FTIR spectrum of their complex 2, they found that acetate should be coordinated to Zn, which is in agreement with our structure and FTIR analysis (vide infra).
Synthesis optimization
To understand the formation mechanism of L-DMC, the single crystal synthesis conditions were modified and the structural changes were followed by high-throughput PXRD and both SEM and TEM (Table 2 and Fig. S1–S4†). To begin with, ZnCl2 - the most common zinc source in the synthesis of Zn3[Co(CN)6]2 DMCs36 - was replaced by Zn(CH3COO)2·2H2O. To favor slow crystal formation, the Zn(CH3COO)2·2H2O aqueous solution was cooled down using an ice bath. The resulting solid, DMC-1, exhibited a PXRD pattern (Fig. S1†) that resembled that of L-DMC. However, SEM images revealed the presence of two phases with different morphologies (Fig. S2†), which suggests that DMC-1 actually consists of a mixture of the cubic (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and the layered phase (monoclinic, P21/m). In an attempt to ensure the incorporation of acetate ions, the synthesis was repeated with Zn(CH3COO)2·2H2O dissolved in a 2 M solution of acetic acid instead. This yielded a solid, DMC-2, that also consisted of a mixture of phases, as evidenced by its PXRD pattern and SEM images (Fig. S1 and S3†). When the concentration of the acetic acid solution was increased from 2 M to 4 M, we obtained a solid (DMC-3) with a PXRD pattern very similar to that of L-DMC. Nevertheless, TEM images revealed that the synthesized solid was not phase pure (Fig. S4†), as two different morphologies (with two different metal ratios) were discerned. Only after increasing the crystallization temperature to 80 °C, phase pure L-DMC was finally obtained. This synthesis temperature has also been used to crystallize a rhombohedral Zn3[Co(CN)6]2 phase, in which all Zn atoms adopt a tetrahedral coordination.36,54 Additionally, we studied the effect of the presence of acetate ions during the synthesis. To that end, we replaced Zn(CH3COO)2·2H2O by other zinc salts, such as ZnCl2 and Zn(CF3COO)2 while keeping the acetic acid concentration at 4 M (DMC-4 and DMC-5, respectively). The pattern of DMC-4 exhibits reflections that are readily assigned to the rhombohedral phase, while the pattern of DMC-5 shows reflections typical of both a rhombohedral and a cubic structure. Substitution of acetic acid by trifluoroacetic acid (DMC-6) also produced a cubic DMC. These results indicate the importance of both the presence of acetic acid in the synthesis solution and the use of Zn(CH3COO)2·2H2O as Zn source to form the desired layered structure, since the acetate ions are needed to balance the positive charge of the {Zn2[Co(CN)6]}+ layers and ensure their linkage. The other studied anions most likely do not have the appropriate geometry to form the desired 2D structure and incomplete hydrolysis of Zn acetate might be necessary to avoid formation of the cubic structure. Furthermore, the electronic properties of the anions, in all likelihood, also play a crucial role in the synthesis of the layered phase. In this sense, CF3COO− may bind too poorly to Zn, as it offers low electron density, and thus it can be easily displaced by water, forming a cubic structure.
m) and the layered phase (monoclinic, P21/m). In an attempt to ensure the incorporation of acetate ions, the synthesis was repeated with Zn(CH3COO)2·2H2O dissolved in a 2 M solution of acetic acid instead. This yielded a solid, DMC-2, that also consisted of a mixture of phases, as evidenced by its PXRD pattern and SEM images (Fig. S1 and S3†). When the concentration of the acetic acid solution was increased from 2 M to 4 M, we obtained a solid (DMC-3) with a PXRD pattern very similar to that of L-DMC. Nevertheless, TEM images revealed that the synthesized solid was not phase pure (Fig. S4†), as two different morphologies (with two different metal ratios) were discerned. Only after increasing the crystallization temperature to 80 °C, phase pure L-DMC was finally obtained. This synthesis temperature has also been used to crystallize a rhombohedral Zn3[Co(CN)6]2 phase, in which all Zn atoms adopt a tetrahedral coordination.36,54 Additionally, we studied the effect of the presence of acetate ions during the synthesis. To that end, we replaced Zn(CH3COO)2·2H2O by other zinc salts, such as ZnCl2 and Zn(CF3COO)2 while keeping the acetic acid concentration at 4 M (DMC-4 and DMC-5, respectively). The pattern of DMC-4 exhibits reflections that are readily assigned to the rhombohedral phase, while the pattern of DMC-5 shows reflections typical of both a rhombohedral and a cubic structure. Substitution of acetic acid by trifluoroacetic acid (DMC-6) also produced a cubic DMC. These results indicate the importance of both the presence of acetic acid in the synthesis solution and the use of Zn(CH3COO)2·2H2O as Zn source to form the desired layered structure, since the acetate ions are needed to balance the positive charge of the {Zn2[Co(CN)6]}+ layers and ensure their linkage. The other studied anions most likely do not have the appropriate geometry to form the desired 2D structure and incomplete hydrolysis of Zn acetate might be necessary to avoid formation of the cubic structure. Furthermore, the electronic properties of the anions, in all likelihood, also play a crucial role in the synthesis of the layered phase. In this sense, CF3COO− may bind too poorly to Zn, as it offers low electron density, and thus it can be easily displaced by water, forming a cubic structure.
| Sample name | Synthesis conditions | ||
|---|---|---|---|
| Zn source | Crystallization temperature | Acid concentration | |
| a Acetic acid. b Trifluoroacetic acid. | |||
| L-DMC | Zn(CH3COO)2·2H2O | 80 °C | 4 Ma |
| DMC-1 | Zn(CH3COO)2·2H2O | Ice bath | — |
| DMC-2 | Zn(CH3COO)2·2H2O | Ice bath | 2 Ma |
| DMC-3 | Zn(CH3COO)2·2H2O | Ice bath | 4 Ma |
| DMC-4 | ZnCl2 | 80 °C | 4 Ma |
| DMC-5 | Zn(CF3COO)2 | 80 °C | 4 Ma |
| DMC-6 | Zn(CH3COO)2·2H2O | Ice bath | 4 Mb |
In an attempt to delaminate the layered structure, the optimized synthesis of L-DMC was slightly modified. Before final isolation (final washing step), the solid was dispersed in an aqueous miscible organic solvent (AMOS) for 16 h.55 The structural changes that occurred during the AMOS dispersion procedure were followed by high throughput powder X-ray diffraction (Fig. S5†). No significant changes were observed in the patterns of L-DMC after washing with the solvents (acetone, methanol, ethanol, acetonitrile and acetaldehyde). This suggests that no exfoliation took place and that the L-DMC structure was maintain, confirming its chemical stability.
Characterization
From ICP-OES analysis of L-DMC, a Zn:Co molar ratio of 2.0 was obtained, which corresponds with the theoretical composition of L-DMC - Zn2[Co(CN)6](CH3COO)·4H2O. TEM images were collected at different regions to corroborate the phase purity of L-DMC (Fig. 3). All images show a flake-like morphology, typical of a two-dimensional (2D) geometry, with many of the flakes randomly aggregated. Moreover, HAADF-STEM images and EDX mapping of L-DMC (Fig. S6†) confirm a homogeneous distribution of Zn and Co throughout the structure with a Zn:Co molar ratio of 2.0. The FTIR spectra of L-DMC (Fig. S7†) exhibits all the bands associated with DMCs,54 as well as two additional bands around 1550 cm−1 and 1450 cm−1 attributed to the asymmetric and symmetric vibrations of –COO−, respectively.56–58 The presence of such bands in the FTIR spectra is in agreement with the solved crystal structure. Moreover, the band attributed to the stretching vibrations of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond is observed at 2190 cm−1, a red shift compared to the ν(CN) frequency in the spectrum of cubic Zn3[Co(CN)6]2 DMC complexes (2200 cm−1), indicating the influence of the acetate anions on the electronic environment of Zn. 1H–13C CPMAS NMR spectrum of L-DMC (Fig. 4) exhibits two well resolved signals of acetate at about 184 and 22 ppm (belonging to carboxyl and methyl groups, respectively), and four partly overlapped signals between 125 and 140 ppm, which can be assigned to carbon nuclei of the four crystallographically distinct cyanide groups. This is in excellent agreement with the proposed structure of L-DMC. Note that signal intensities in the 1H–13C CPMAS NMR spectrum do not properly reflect the abundancies of individual species in the crystals, because the efficiency of polarization transfer from protons is very different to carbon nuclei within –CH3, –COO− or –CN− groups. 1H MAS NMR measurement (Fig. S8†) resolves a stronger signal of water molecules at 4.2 ppm and a weaker signal of the –CH3 groups of acetate at 1.7 ppm, and thus additionally confirms that L-DMC comprises coordinating water molecules. TG-MS analysis of L-DMC (Fig. S9†) showed two marked mass decays: a first decay before 100 °C (∼10% mass loss) attributed to the elimination of water, and a second decay between 320 and 390 °C (∼35% mass loss) corresponding to the decomposition of CN. Since the TG-MS analysis was performed under air flow, the cyanide ligands were removed as (CN)2, and the observed CO2 evolution indicates the decomposition of acetate as no acetic acid desorption was observed before this temperature. After the analysis, the recovered powder exhibited an intense green color, which is indicative of the formation of Zn1−xCoxO oxides (cobalt green). The N2 adsorption isotherm at −196 °C of cubic Zn3[Co(CN)6]2 DMCs is a Type I isotherm with no hysteresis loop, typical of purely microporous materials. In contrast, the isotherm of L-DMC, presented in Fig. S10,† shows a small hysteresis loop.59,60 The high initial N2 uptake at low p/p0 is attributed to the filling of micropores, while the slope in the 0.25 to 0.35 range indicates the presence of a considerable external surface area (Sext) in agreement with the small crystallite size and plate-like morphology.61 Analysis of the N2 sorption isotherm of L-DMC results in a BET surface area (SBET) of 784 m2 g−1 and a Sext of 123 m2 g−1, which are both higher than those obtained for DMC-PTMEG (Table S1†). Furthermore, the MPSD was estimated from the Ar adsorption–desorption isotherms. Horvath–Kawazoe (HK) pore size analyses disclose differences in the microporous nature of both materials (Fig. S11†). While DMC-PTMEG exhibits a relatively narrow MPSD, centered around 6.7 Å, the MPSD of L-DMC shows the presence of three micropore systems with maxima around 6, 10 and 11 Å. This evidences the different geometries between L-DMC and the common, cubic DMC structure.
N bond is observed at 2190 cm−1, a red shift compared to the ν(CN) frequency in the spectrum of cubic Zn3[Co(CN)6]2 DMC complexes (2200 cm−1), indicating the influence of the acetate anions on the electronic environment of Zn. 1H–13C CPMAS NMR spectrum of L-DMC (Fig. 4) exhibits two well resolved signals of acetate at about 184 and 22 ppm (belonging to carboxyl and methyl groups, respectively), and four partly overlapped signals between 125 and 140 ppm, which can be assigned to carbon nuclei of the four crystallographically distinct cyanide groups. This is in excellent agreement with the proposed structure of L-DMC. Note that signal intensities in the 1H–13C CPMAS NMR spectrum do not properly reflect the abundancies of individual species in the crystals, because the efficiency of polarization transfer from protons is very different to carbon nuclei within –CH3, –COO− or –CN− groups. 1H MAS NMR measurement (Fig. S8†) resolves a stronger signal of water molecules at 4.2 ppm and a weaker signal of the –CH3 groups of acetate at 1.7 ppm, and thus additionally confirms that L-DMC comprises coordinating water molecules. TG-MS analysis of L-DMC (Fig. S9†) showed two marked mass decays: a first decay before 100 °C (∼10% mass loss) attributed to the elimination of water, and a second decay between 320 and 390 °C (∼35% mass loss) corresponding to the decomposition of CN. Since the TG-MS analysis was performed under air flow, the cyanide ligands were removed as (CN)2, and the observed CO2 evolution indicates the decomposition of acetate as no acetic acid desorption was observed before this temperature. After the analysis, the recovered powder exhibited an intense green color, which is indicative of the formation of Zn1−xCoxO oxides (cobalt green). The N2 adsorption isotherm at −196 °C of cubic Zn3[Co(CN)6]2 DMCs is a Type I isotherm with no hysteresis loop, typical of purely microporous materials. In contrast, the isotherm of L-DMC, presented in Fig. S10,† shows a small hysteresis loop.59,60 The high initial N2 uptake at low p/p0 is attributed to the filling of micropores, while the slope in the 0.25 to 0.35 range indicates the presence of a considerable external surface area (Sext) in agreement with the small crystallite size and plate-like morphology.61 Analysis of the N2 sorption isotherm of L-DMC results in a BET surface area (SBET) of 784 m2 g−1 and a Sext of 123 m2 g−1, which are both higher than those obtained for DMC-PTMEG (Table S1†). Furthermore, the MPSD was estimated from the Ar adsorption–desorption isotherms. Horvath–Kawazoe (HK) pore size analyses disclose differences in the microporous nature of both materials (Fig. S11†). While DMC-PTMEG exhibits a relatively narrow MPSD, centered around 6.7 Å, the MPSD of L-DMC shows the presence of three micropore systems with maxima around 6, 10 and 11 Å. This evidences the different geometries between L-DMC and the common, cubic DMC structure.
 | ||
| Fig. 3 TEM images of L-DMC. | ||
 | ||
| Fig. 4 1H–13C CPMAS NMR spectrum of L-DMC (thick black line – measured; thin red line – decomposition into individual contributions). | ||
Catalytic reactions
The advantages of L-DMC over the benchmark DMC catalyst are evident in two important DMC applications: ring opening polymerization of 1,2-epoxyhexane and intermolecular hydroamination of phenylacetylene with 4-isopropylaniline. For the ring opening polymerization - one of the most important applications of DMCs62,63 - a significant increase in the 1,2-epoxyhexane conversion is observed when L-DMC is employed as catalyst (Fig. 5, top). In fact, almost full conversion is achieved after only 0.5 h reaction time (92%), which represents a vast improvement over the 61% conversion obtained with DMC-PTMEG. Furthermore, the initial TOF obtained with L-DMC is 1.5 higher than that obtained with DMC-PTMEG (TOFL-DMC = 6.2 and TOFDMC-PTMEG = 4.1). | ||
| Fig. 5 Catalytic activity results for L-DMC and DMC-PTMEG. Top: Epoxide polymerization (reaction conditions: 2 mmol of 1,2-epoxyhexane, 0.2 mmol of DEG, 5 mol% Zn, 110 °C). Bottom: Intermolecular hydroamination. Circles represent phenylacetylene conversion and bars represent yield of the hydroamination product (reaction conditions: 0.5 mmol of phenylacetylene, 1 mmol of 4-isopropylaniline, 1 mL of toluene, 1 mmol of tetradecane, 50 mol% Zn, 110 °C). | ||
The second application, the hydroamination reaction, is a 100% atom efficient route towards the production of fine chemicals,64,65 thus representing a greener alternative over other important C–N bond forming reactions, like the Buchwald–Hartwig amination. Furthermore, the formation of undesired products is limited, as only acetophenone was formed as a side product under these reaction conditions. Zn-based Lewis acid DMCs have been found to be superior catalysts for this reaction, showing a higher selectivity to the hydroamination product than materials that contain Brønsted acid sites.6,7,65,66Fig. 5 (bottom) shows the conversion of phenylacetylene and the yield of the desired hydroamination product obtained with DMC-PTMEG and L-DMC, and the kinetic data for L-DMC are presented in Fig. S12.† The higher catalytic activity of L-DMC (TOFL-DMC = 0.87 compared to TOFDMC-PTMEG = 0.23) can be attributed to a combination of the easier accessibility of the catalytic sites and the changed coordination environment of the Zn atoms in the structure. Additionally, the effect of the presence of acetate ions in the structure of L-DMC on its high catalytic activity cannot be disregarded, since it has been found to enhance the performance of DMC catalysts for other organic reactions.11 Nevertheless, in order to fully disclose their involvement in the reaction mechanism, further studies are required.
As observed in both catalytic activity tests, the differences in activity exhibited by the DMCs highlight the advantages of L-DMC. The catalyst L-DMC also exhibits a higher hydroamination activity than the other synthesized DMCs and other Lewis acid catalysts (Tables S2 and S3†). Furthermore, recycling tests, PXRD and FTIR characterization after three hydroamination runs show the high stability and reusability of L-DMC as a solid catalyst (Fig. S13–S15†). The leaching of active species from the solid catalysts was studied by a hot filtration test (Fig. S16†). To this end, the catalyst was removed from the hot reaction mixture after 0.5 h reaction time (phenylacetylene conversion, ∼24%). The clear supernatant (in the absence of solid catalysts) was allowed to react for an additional 19.5 h reaction time, after which no increase in phenylacetylene conversion was detected. This indicates that no leaching of catalytically active species from the solid occurs during the reaction under the studied conditions.
Conclusions
In summary, we have successfully synthesized and characterized a new 2D layered DMC phase, L-DMC, consisting of positively charged {Zn2[Co(CN)6]}+ layers connected through acetate anions. L-DMC was more active than the benchmark DMC catalyst in both the ring opening polymerization of 1,2-epoxyhexane and in the intermolecular hydroamination of phenylacetylene and 4-isopropylaniline. Furthermore, recycling, PXRD, FTIR and hot filtration tests demonstrated the good reusability and stability of L-DMC. This represents, to the best of our knowledge, the first two-dimensional DMC based on the catalytically active Zn–Co metal combination. The successful synthesis and excellent catalytic properties of L-DMC may lead to new studies focused on exploring further modifications through, for instance, intercalation of other molecules or pillaring, in order to fine-tune its physicochemical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 641887 (project acronym: DEFNET). D. D. V. thanks KU Leuven for the Metusalem grant CASAS. F.W.O. Vlaanderen (Research Foundation Flanders) is thanked for project funding (D. D. V.: grant no. G.07811.18N; C. V. G. and I. V.: grant no. G.0256.14N) and for a Postdoctoral Fellowship (T. D. B. & B. B.). A. K. and G. M. acknowledge the financial support by the Slovenian Research Agency (research core funding No. P1-0021). Funding for the TEM through Hercules project AKUL/13/19 is kindly acknowledged. The authors are grateful to Prof. Jin Won Seo (KU Leuven) for TEM support, to Prof. Andrew L. Goodwin (Oxford) for the use of resources and discussions, to the beamline staff of I19 (Diamond Light Source, MT13639) for help and assistance, to Kwinten Janssens (KU Leuven) for the collection of additional FTIR data, and to Guangxia Fu and João Marreiros for Ar physisorption support.Notes and references
- J. Milgrom, US pat. 3278457A, 1966.
- R. J. Belner, US pat. 3278458, 1966.
- R. J. Herold, US pat. 3278459A, 1966.
- Y. Dienes, W. Leitner, M. G. J. Muller, W. K. Offermans, T. Reier, A. Reinholdt, T. E. Weirich and T. E. Muller, Green Chem., 2012, 14, 1168–1177 RSC.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- A. Peeters, P. Valvekens, F. Vermoortele, R. Ameloot, C. Kirschhock and D. De Vos, Chem. Commun., 2011, 47, 4114–4116 RSC.
- A. Peeters, P. Valvekens, R. Ameloot, G. Sankar, C. E. A. Kirschhock and D. E. De Vos, ACS Catal., 2013, 3, 597–607 CrossRef CAS.
- C. Marquez, M. Rivera-Torrente, P. P. Paalanen, B. M. Weckhuysen, F. G. Cirujano, D. De Vos and T. De Baerdemaeker, J. Catal., 2017, 354, 92–99 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2006, 241, 34–44 CrossRef CAS.
- C. Marquez, F. G. Cirujano, C. Van Goethem, I. Vankelecom, D. De Vos and T. De Baerdemaeker, Catal. Sci. Technol., 2018, 8, 2061–2065 RSC.
- C. Marquez, F. G. Cirujano, S. Smolders, C. Van Goethem, I. Vankelecom, D. De Vos and T. De Baerdemaeker, Dalton Trans., 2019, 48, 3946–3954 RSC.
- A. Ludi and H. U. Güdel, in Inorganic Chemistry. Structure and Bonding, Springer, Berlin, Heidelberg, 1973, pp. 1–21 Search PubMed.
- P. Valvekens and D. De Vos, in New Materials for Catalytic Applications, Elsevier, Amsterdam, 2016, pp. 1–12 Search PubMed.
- S. H. Byun, H. S. Seo, S. H. Lee, C.-S. Ha and I. Kim, Macromol. Res., 2007, 15, 393–395 CrossRef CAS.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef CAS PubMed.
- J. C. Wojdeł, S. T. Bromley, F. Illas and J. C. Jansen, J. Mol. Model., 2007, 13, 751–756 CrossRef PubMed.
- J. Roque, E. Reguera, J. Balmaseda, J. Rodríguez-Hernández, L. Reguera and L. F. del Castillo, Microporous Mesoporous Mater., 2007, 103, 57–71 CrossRef CAS.
- N. J. Robertson, Z. Qin, G. C. Dallinger, E. B. Lobkovsky, S. Lee and G. W. Coates, Dalton Trans., 2006, 5390–5395 RSC.
- C. G. Silva, Y. Bouizi, V. Fornes and H. Garcia, J. Am. Chem. Soc., 2009, 131, 13833–13839 CrossRef PubMed.
- F. Gándara, B. Gomez-Lor, E. Gutiérrez-Puebla, M. Iglesias, M. A. Monge, D. M. Proserpio and N. Snejko, Chem. Mater., 2008, 20, 72–76 CrossRef.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- J. Helsen, J. Chem. Educ., 1982, 59, 1063–1065 CrossRef CAS.
- T. An, Y. Wang, J. Tang, Y. Wang, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2015, 445, 320–325 CrossRef CAS PubMed.
- X. Liu, C. Shi, C. Zhai, M. Cheng, Q. Liu and G. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 4585–4591 CrossRef CAS PubMed.
- Z.-H. Xuan, D.-S. Zhang, Z. Chang, T.-L. Hu and X.-H. Bu, Inorg. Chem., 2014, 53, 8985–8990 CrossRef CAS PubMed.
- M. K. Ram Reddy, Z. P. Xu, G. Q. (Max) Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504–7509 CrossRef CAS.
- T. J. Pinnavaia, M.-S. Tzou, S. D. Landau and R. H. Raythatha, J. Mol. Catal., 1984, 27, 195–212 CrossRef CAS.
- K. Suzuki, T. Mori, K. Kawase, H. Sakami and S. Iida, Clays Clay Miner., 1988, 36, 147–152 CrossRef CAS.
- U. Díaz and A. Corma, Dalton Trans., 2014, 43, 10292–10316 RSC.
- K. Varoon, X. Zhang, B. Elyassi, D. D. Brewer, M. Gettel, S. Kumar, J. A. Lee, S. Maheshwari, A. Mittal, C.-Y. Sung, M. Cococcioni, L. F. Francis, A. V. McCormick, K. A. Mkhoyan and M. Tsapatsis, Science, 2011, 334, 72–75 CrossRef CAS PubMed.
- A. Corma, V. Fornes, S. B. Pergher, Th. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353–356 CrossRef CAS.
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, Angew. Chem., 2000, 112, 1559–1561 CrossRef.
- A. Corma, V. Fornés and U. Díaz, Chem. Commun., 2001, 2642–2643 RSC.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- L. Li, R. Ma, Y. Ebina, N. Iyi and T. Sasaki, Chem. Mater., 2005, 17, 4386–4391 CrossRef CAS.
- J. Kuyper and G. Boxhoorn, J. Catal., 1987, 105, 163–174 CrossRef CAS.
- Y.-J. Huang, G. R. Qi and L. S. Chen, Appl. Catal., A, 2003, 240, 263–271 CrossRef CAS.
- I. Kim, K. Anas, S. Lee, C.-S. Ha and D.-W. Park, Catal. Today, 2008, 131, 541–547 CrossRef CAS.
- X.-H. Zhang, Z.-J. Hua, S. Chen, F. Liu, X.-K. Sun and G.-R. Qi, Appl. Catal., A, 2007, 325, 91–98 CrossRef CAS.
- Z. Blank, D. M. Speyer, W. Brenner and Y. Okamoto, Nature, 1967, 216, 1103–1104 CrossRef CAS.
- R. Z. LeGeros and J. P. LeGeros, J. Cryst. Growth, 1972, 13/14, 476–480 CrossRef.
- I. Kim, J. T. Ahn, C.-S. Ha, C. S. Yang and I. Park, Polymer, 2003, 44, 3417–3428 CrossRef CAS.
- D. R. Allan, H. Nowell, S. A. Barnett, M. R. Warren, A. Wilcox, J. Christensen, L. K. Saunders, A. Peach, M. T. Hooper, L. Zaja, S. Patel, L. Cahill, R. Marshall, S. Trimnell, A. J. Foster, T. Bates, S. Lay, M. A. Williams, P. V. Hathaway, G. Winter, M. Gerstel and R. W. Wooley, Crystals, 2017, 7, 336 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A.-C. Dippel, H.-P. Liermann, J. T. Delitz, P. Walter, H. Schulte-Schrepping, O. H. Seeck and H. Franz, J. Synchrotron Radiat., 2015, 22, 675–687 CrossRef CAS PubMed.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Naray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 CrossRef PubMed.
- G. S. Pawley, Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- W. Zhang, Q. Lin, Y. Cheng, L. Lu, B. Lin, L. Pan and N. Xu, J. Appl. Polym. Sci., 2012, 2, 977–985 CrossRef.
- C. P. Krap, B. Zamora, L. Reguera and E. Reguera, Microporous Mesoporous Mater., 2009, 120, 414–420 CrossRef CAS.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301–6303 RSC.
- E. A. Secco and G. G. Worth, Can. J. Chem., 1987, 65, 2504–2508 CrossRef CAS.
- N. Masciocchi, E. Corradi, A. Sironi, G. Moretti, G. Minelli and P. J. Porta, Solid State Chem., 1997, 131, 252–262 CrossRef CAS.
- D. C. Pereira, D. L. A. de Faria and V. R. L. Constantino, J. Braz. Chem. Soc., 2006, 17, 1651–1657 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- A. Gil, S. A. Korili and M. A. Vicente, Catal. Rev.: Sci. Eng., 2008, 50, 153–221 CrossRef CAS.
- W. J. Roth, B. Gil, W. Makowski, B. Marszalek and P. Eliasova, Chem. Soc. Rev., 2016, 45, 3400–3438 RSC.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- S. Lee, S. T. Baek, K. Anas, C.-S. Ha, D.-W. Park, J. W. Lee and I. Kim, Polymer, 2007, 48, 4361–4367 CrossRef CAS.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- J. Penzien, T. E. Müller and J. A. Lercher, Microporous Mesoporous Mater., 2001, 48, 285–291 CrossRef CAS.
- J. C. Penzien, C. Haessner, A. Jentys, K. Kohler, T. E. Muller and J. A. Lercher, J. Catal., 2004, 221, 302–312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization (XRD patterns, SEM and TEM images, N2 sorption isotherms, FTIR spectra, 1H MAS NMR spectra, TG-MS profiles) of the synthesized DMCs and L-DMC and catalytic data of the synthesized DMCs. Stability, recycling and hot filtration test of L-DMC. CCDC 1894386. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00527g |
| This journal is © The Royal Society of Chemistry 2019 |