DOI:
10.1039/C9SC00399A
(Edge Article)
Chem. Sci., 2019,
10, 4236-4245
Supramolecular chirality transformation driven by monodentate ligand binding to a coordinatively unsaturated self-assembly based on C3-symmetric ligands†
Received
24th January 2019
, Accepted 1st March 2019
First published on 5th March 2019
Abstract
Monodentate ligand binding is facilitated by supramolecular chirality transformations from propeller-shaped chirality into single-twist chirality by altering the self-assembly of C3-symmetric chiral ligands. The C3-symmetric chiral ligands (ImR3Bz and ImS3Bz) contain three chiral imidazole side arms (ImR and ImS) at the 1,3,5-positions of a central benzene ring. Upon coordination to zinc ions (Zn2+), which have a tetrahedral coordination preference, the C3-symmetric chiral ligands assemble, in a stepwise manner, into a propeller-shaped assembly with a general formula (Im(RorS)3Bz)4(Zn2+)3. In this structure each Zn2+ ion coordinates to the three individual imidazole side arms. The resulting assembly is formally coordinatively unsaturated (coordination number, n = 3) and capable of accepting monodentate co-ligands (imidazole: ImH2) to afford a coordinatively saturated assembly [(ImH2)3(ImR3Bz)4(Zn2+)3]. The preformed propeller-shaped chirality is preserved during this transformation. However, an excess of the monodentate co-ligand (ImH2/Zn2+ molar ratio of ∼1.7) alters the propeller-shaped assembly into a stacked dimer assembly [(ImH2)m(ImR3Bz)2(Zn2+)3] (m = 4–6) with single-twist chirality. This switch alters the degree of enhancement and the circular dichroism (CD) pattern, suggesting a structural transition into a chiral object with a different shape. This architectural chirality transformation presents a new approach to forming dynamic coordination-assemblies, which have transformable geometric chiral structures.
Introduction
Topology plays an important role in determining the properties of self-assembled structures at various hierarchical levels from the molecular to supramolecular scales. Among features that determine structure and function relationships, chirality mainly determines physical and morphological properties.1 Nature's topology is dynamic and transforms under the influence of certain stimuli.2–4 However, in the field of coordination assembly, supramolecular chirality is considered to be a static feature. In most cases, building blocks are rigid and inflexible, fixing the directionality of metal–ligand interactions, which is a requirement for predetermined chirality in discrete metallosupramolecular architectures.1,5 Conversely, the concept of dynamic assembly has drawn attention to the benefits of dynamic structures that can react to certain stimuli and change their behavior.6–23 For example, dynamic assembly has made it possible to design supramolecular architectures with “chirality inversion” through changes to an explicit chiral shape (e.g., helices), which is successfully reflected by chiroptical inversion, such as circular dichroism (CD) and circularly polarized luminescence (CPL) spectra.7–14 Besides these successful achievements, few cases of dynamic topology alteration have been reported and they have mostly concerned achiral to achiral transformations in two-component systems consisting of only one ligand component and one metal ion component.18–21 In such cases, an appropriate ratio of the building components drives alternative shapes in the output architecture. Stimuli-responsive or “smart” coordination assemblies22,23 have two competing features that must be balanced: the assembly should have metastability to enable acceptance of a third component (e.g., co-ligand), which induces the stimuli-responsive transformations. The system should also have sufficient thermodynamic stability to selectively form the target assembly in solution prior to any stimuli. It remains challenging to identify systems where both these properties are achieved.
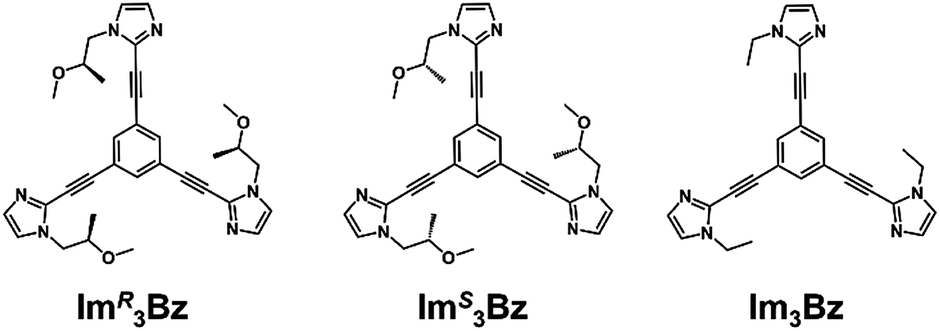
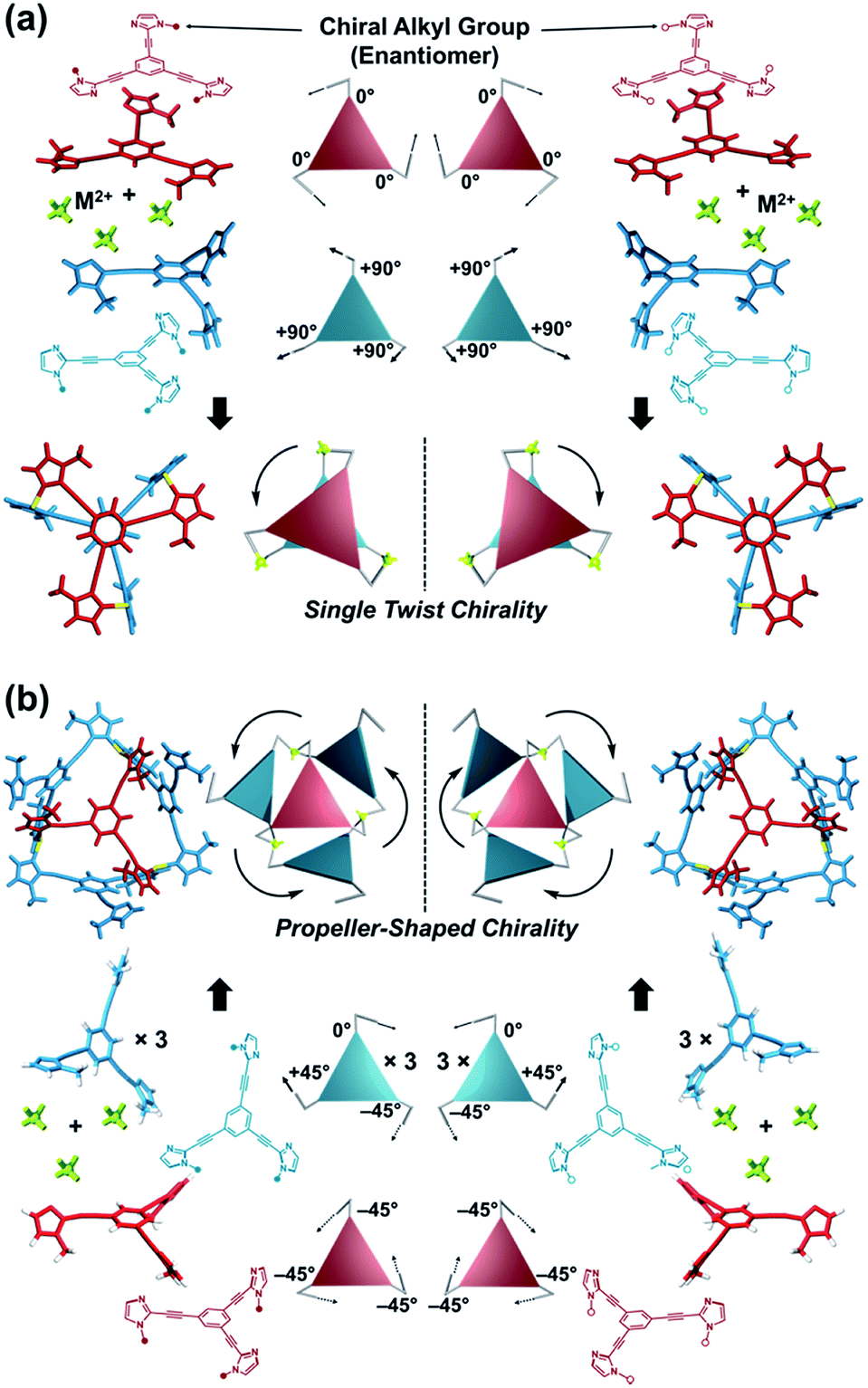
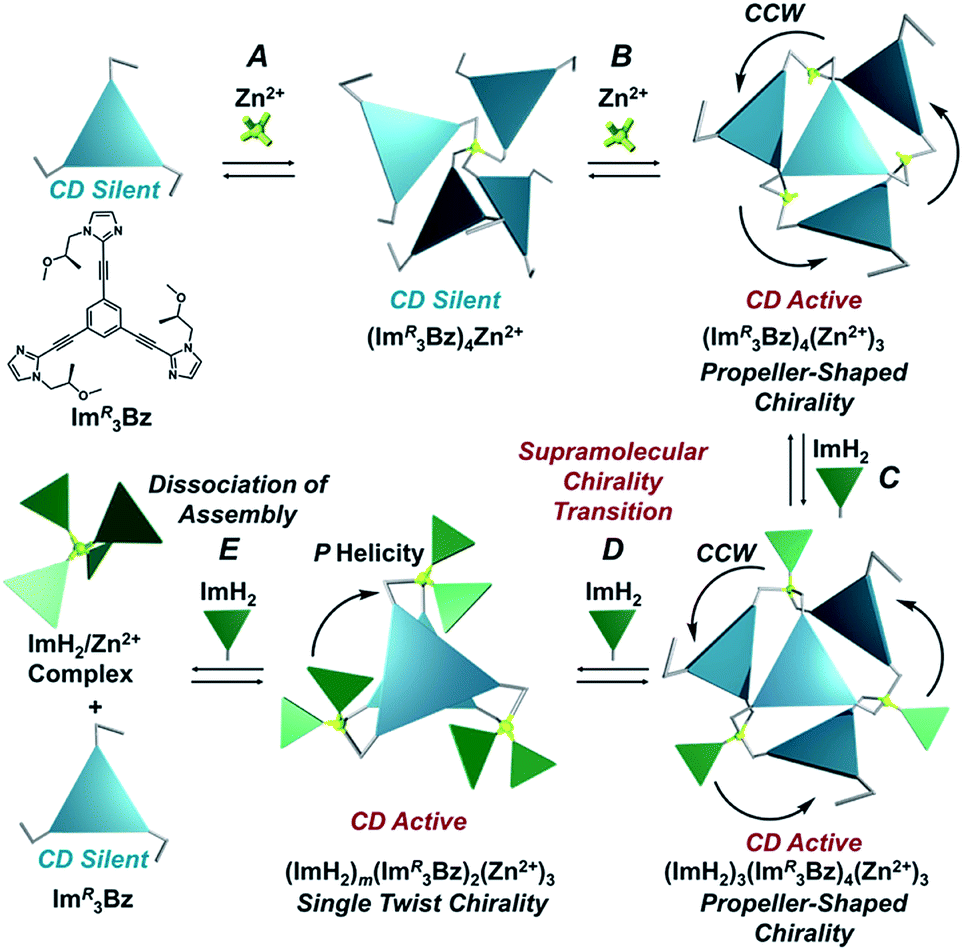
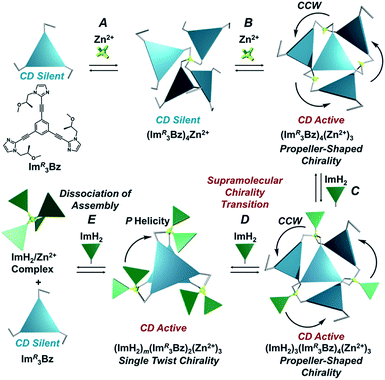
Here, we propose a flexible ligand approach to address supramolecular chirality transformations driven by monodentate ligand binding. We have designed new C3-symmetric ligands (ImR3Bz, ImS3Bz, and Im3Bz) containing three imidazole side arms connected to a central benzene ring at the 1,3,5-positions through ethynyl spacers (Scheme 1). The present C3-symmetric ligands show intrinsic rotational flexibility around the ethynyl axes but maintain conjugated aromatic units, resulting in structural diversity in terms of the dihedral angle (θ) between the imidazole and the central benzene rings (Scheme 2). Most C3-symmetric ligands containing ethynyl spacers have been designed with pyridine rings to make the direction of the metal–ligand interactions constant irrespective of the ligand conformation prior to self-assembly.24,25 This diversity in ligand chirality suggests potential to access a range of topologies with different chiral shapes through a coordination process we term “metal-ion clipping”.21 Among the types of supramolecular chirality accessible in this coordination assembly, the simplest should be a stacked dimer with single-twist chirality (Scheme 2 from top to middle).21c,26,27 This structure forms from two different C3-symmetric conformers (θ = +90°, +90°, +90°; θ = 0°, 0°, 0°) clipped by three metal ions with a tetrahedral coordination preference. Propeller-shaped chirality is another probable architecture,28 which forms from a C3-symmetric conformer (θ = −45°, −45°, −45°) with three non-symmetrical conformers (θ = −45°, +45°, 0°) clipped by three metal ions (Scheme 2 from bottom to middle). We reveal that the C3-symmetric ligands selectively assemble into a propeller-shaped chiral assembly upon coordination to zinc ions (Zn2+). The resulting propeller-shaped assembly [(Im(RorS)3Bz)4(Zn2+)3] is formally coordinatively unsaturated (coordination number, n = 3), and undergoes coordination of monodentate co-ligands (imidazole: ImH2) to afford a coordinatively saturated assembly [(ImH2)3(Im(RorS)3Bz)4(Zn2+)3]. The resulting coordinatively saturated assembly maintains the preformed propeller-shaped chirality but transforms into a stacked dimer assembly [(ImH2)m(Im(RorS)3Bz)2(Zn2+)3] (m = 4–6) with single-twist chirality at a certain excess of monodentate co-ligands (ImH2/Zn2+ ratio at approximately 1.7). This change considerably enhances and modifies the CD signals, suggesting that the structure transitions into different chiral objects with different shapes. The present system seems to be completely different from the past examples of the structural conversion associated with chiroptical inversion by external stimuli. Our findings will pave the way for rational design of dynamic and flexible coordination chirality, which can transform between different chiral shapes under stimuli.
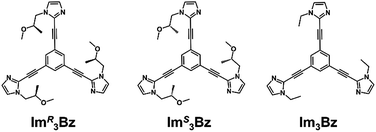 |
| | Scheme 1 Chemical structures of C3-symmetric ligands studied here. | |
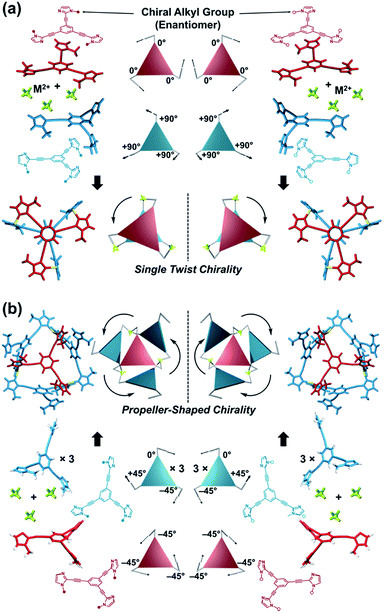 |
| | Scheme 2 (a) Supramolecular architecture with single twist chirality formed from the two C3-symmetric ligands clipped by three metal ions with a tetrahedral coordination preference. It should be noted that the suggested L2M3 complex was not formed in the present system in the absence of a monodentate co-ligand. (b) Supramolecular architecture with propeller-shaped chirality formed from the three C3-symmetric ligands clipped by three metal ions with a tetrahedral coordination preference. | |
Results and discussion
Chiral assembly formation of C3-symmetric chiral ligands upon coordination to zinc ions
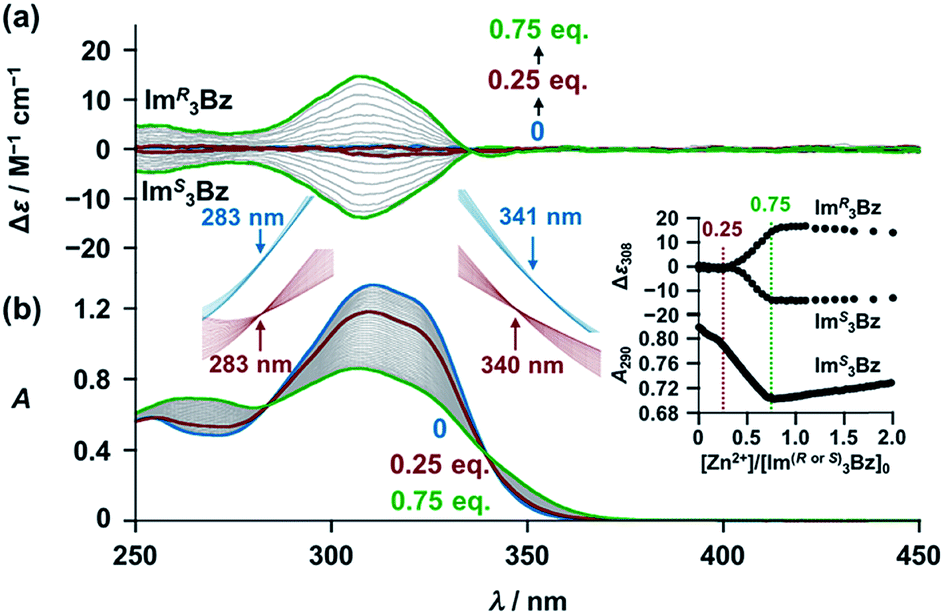
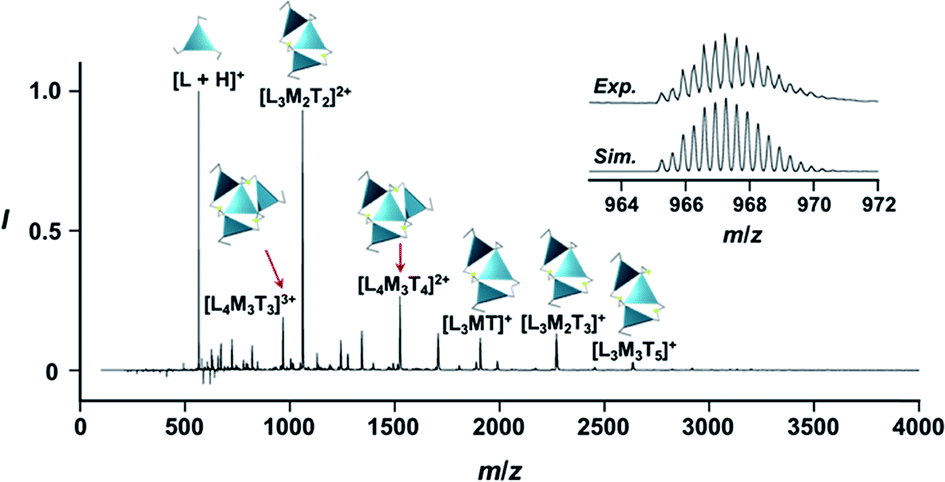
New C3-symmetric ligands (ImR3Bz, ImS3Bz, and Im3Bz) were synthesized by Sonogashira coupling between the corresponding 2-iodoimidazoles and the 1,3,5-tris(ethynylbenzene) linker (see the ESI†). We used circular dichroism (CD) to investigate the chiral assembly of the C3-symmetric ligands upon coordination to zinc ions (Zn2+). Although the C3-symmetric chiral ligands contain chiral alkyl side chains, neither ImR3Bz nor ImS3Bz show a CD signal in the absence of Zn2+ (Fig. 1a, blue lines), because their chiral alkyl side chains are separated from the central chromophore unit (Scheme 1). However, the C3-symmetric chiral ligands exhibit a unique CD signal pattern once they assemble into geometrically chiral objects with Zn2+ (Fig. 1a, green lines, vide infra).21c Hence, the CD signals act as a probe for geometric changes occurring in the solution assembly process. These findings are further supported by analysis of NMR titration and ESI mass spectrometry results (Fig. 2 and 3, vide infra). The CD titration experiments were performed in acetonitrile solutions containing a constant concentration of ImR3Bz or ImS3Bz ([Im(RorS)3Bz]0 = 2.0 × 10−5 M) by changing the concentration of the Zn2+ ions (counter anion: OSO2CF3−). These results were compared with those from UV/vis titration experiments conducted under the same conditions (Fig. 1b). At a molar ratio of [Zn2+]/[Im(RorS)3Bz]0 = 0.25, Zn2+ induced no appreciable CD signal in the region of the π–π* transition (λ < 360 nm) of the C3-symmetric ligands (Fig. 1a, red lines); however, the absorption changed noticeably with isosbestic points at 283 and 341 nm (Fig. 1b, blue line to red line). The absorption spectral changes suggest conversion of Im(RorS)3Bz into a Zn2+ complex, which has no CD activity (Scheme 3A). However, as the molar ratio of [Zn2+]/[Im(RorS)3Bz]0 increased beyond 0.25, ImR3Bz and ImS3Bz exhibited positive and negative Cotton effects, respectively. Almost complete mirror-image CD signals can be seen between the enantiomers (Fig. 1a, green lines). Together with the considerable changes in the CD spectra, ImS3Bz showed changes in its absorption spectra with isosbestic points at 283 and 340 nm (Fig. 1b, red to green line).30 These results suggest a change of the CD-silent Zn2+-complex into a geometrically chiral Zn2+-assembly with CD activity (Scheme 3B). The CD intensity (Δε) at 308 nm and the absorbance at 290 nm are plotted against the molar ratio ([Zn2+]/[Im(RorS)3Bz]0) (inset of Fig. 1). This plot illustrates the stoichiometry of the initially formed CD-silent Zn2+-complex and the subsequently formed Zn2+-assembly with CD activity. A clear break can be seen at [Zn2+]/[Im(RorS)3Bz]0 = 0.25 in each case, indicating a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of Im(RorS)3Bz and Zn2+ [(Im(RorS)3Bz)4Zn2+], which conforms to the most common coordination number of Zn2+-complexes (n = 4). The 4:1 complex was assigned by high-resolution electrospray injection mass spectrometry (HR ESI-MS) (positive): m/z calcd. [(ImS3Bz)4(Zn2+)1]2+, 1160.53435; found 1160.53384.29 Saturation of the titration curves occurred at [Zn2+]/[Im(RorS)3Bz]0 = 0.75 (inset of Fig. 1), which corresponds to 4:3 binding stoichiometry for Im(RorS)3Bz and Zn2+. The most probable assembly is a 4:3 assembly [(Im(R or S)3Bz)4(Zn2+)3] with the lowest global complexity (GC = 4 + 3 = 7). To confirm the stoichiometry and the global complexity of the Zn2+-assembly in solution, we measured the ESI mass of ImS3Bz in the presence of 0.8 equivalents of Zn(OSO2CF3)2 in acetonitrile (Fig. 2). The proposed (ImS3Bz)4(Zn2+)3 was successfully detected as [(ImS3Bz)4(Zn2+)3(OSO2CF3)3]3+ and [(ImS3Bz)4(Zn2+)3(OSO2CF3)4]2+, in which the isotopic distribution of [(ImS3Bz)4(Zn2+)3(OSO2CF3)3]3+ agreed well with the calculated spectrum (Fig. 2 inset). We identified several fragment ion peaks, including [(ImS3Bz)3(Zn2+)3(OSO2CF3−)5]+ (m/z = 2629.4) and [(ImS3Bz)3(Zn2+)2(OSO2CF3−)2]2+ (m/z = 2118.6). Thus (ImS3Bz)4(Zn2+)3 contains labile ligand units (vide infra).
:1 complex of Im(RorS)3Bz and Zn2+ [(Im(RorS)3Bz)4Zn2+], which conforms to the most common coordination number of Zn2+-complexes (n = 4). The 4:1 complex was assigned by high-resolution electrospray injection mass spectrometry (HR ESI-MS) (positive): m/z calcd. [(ImS3Bz)4(Zn2+)1]2+, 1160.53435; found 1160.53384.29 Saturation of the titration curves occurred at [Zn2+]/[Im(RorS)3Bz]0 = 0.75 (inset of Fig. 1), which corresponds to 4:3 binding stoichiometry for Im(RorS)3Bz and Zn2+. The most probable assembly is a 4:3 assembly [(Im(R or S)3Bz)4(Zn2+)3] with the lowest global complexity (GC = 4 + 3 = 7). To confirm the stoichiometry and the global complexity of the Zn2+-assembly in solution, we measured the ESI mass of ImS3Bz in the presence of 0.8 equivalents of Zn(OSO2CF3)2 in acetonitrile (Fig. 2). The proposed (ImS3Bz)4(Zn2+)3 was successfully detected as [(ImS3Bz)4(Zn2+)3(OSO2CF3)3]3+ and [(ImS3Bz)4(Zn2+)3(OSO2CF3)4]2+, in which the isotopic distribution of [(ImS3Bz)4(Zn2+)3(OSO2CF3)3]3+ agreed well with the calculated spectrum (Fig. 2 inset). We identified several fragment ion peaks, including [(ImS3Bz)3(Zn2+)3(OSO2CF3−)5]+ (m/z = 2629.4) and [(ImS3Bz)3(Zn2+)2(OSO2CF3−)2]2+ (m/z = 2118.6). Thus (ImS3Bz)4(Zn2+)3 contains labile ligand units (vide infra).
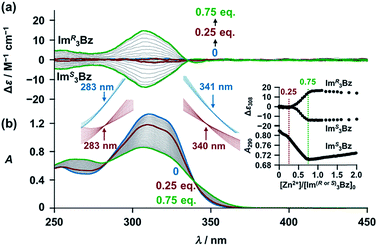 |
| | Fig. 1 (a) CD spectra of ImR3Bz and ImS3Bz (concentrations: 2.0 × 10−5 M) in the presence of Zn2+ [0 (blue), 5.0 × 10−6 (red) and 1.5 × 10−5 M (green)] in acetonitrile at 298 K, where Δε is calculated based on the ligand concentration (2.0 × 10−5 M). (b) Corresponding UV/vis absorption spectral changes observed in the titration of ImS3Bz by Zn2+. Inset: plots of Δε at 308 nm absorbance at 290 nm versus [Zn2+]/[Im(RorS)3Bz]0. The magnified spectra around the isosbestic points (blue lines: [Zn2+] = 0–5.0 × 10−6 M; red lines: 5.0 × 10−6 M to 1.5 × 10−5 M). For clarity reasons two step spectral changes are separated vertically. | |
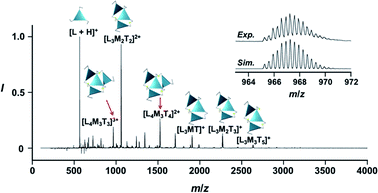 |
| | Fig. 2 Positive ESI-MS spectrum of ImS3Bz (normalized by its most intense fragment at m/z = 564.3 from the free ligand) in acetonitrile (2.0 × 10−3 M) in the presence of Zn2+ (1.6 × 10−3 M). Inset: isotopically resolved signals at m/z = 965.3 and the calculated isotopic distributions for [(ImS3Bz)4(Zn2+)3(OSO2CF3)3]3+. Objects correspond to the mass peak assignment (L: ImS3Bz, M: Zn2+, T: OSO2CF3−). | |
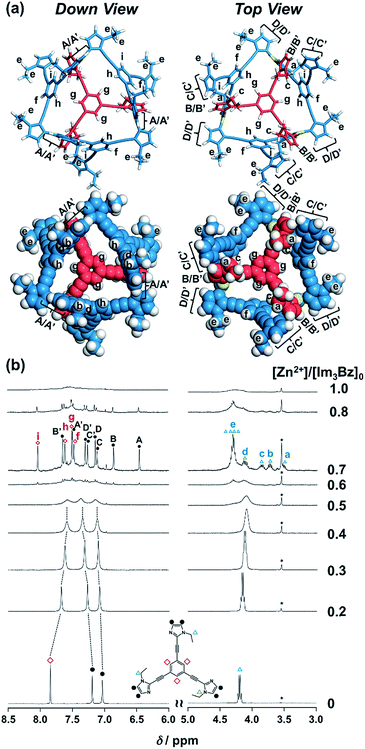 |
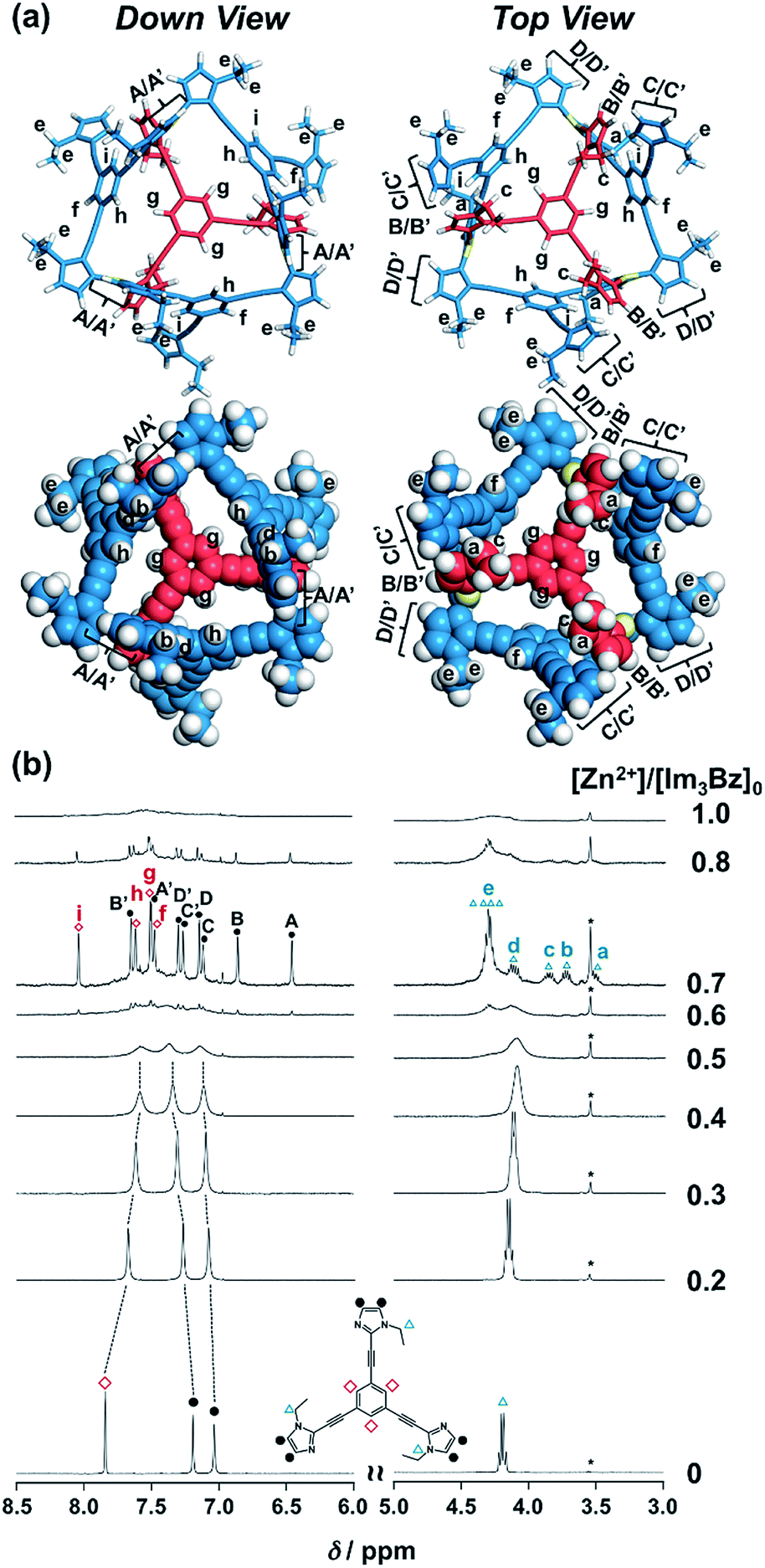
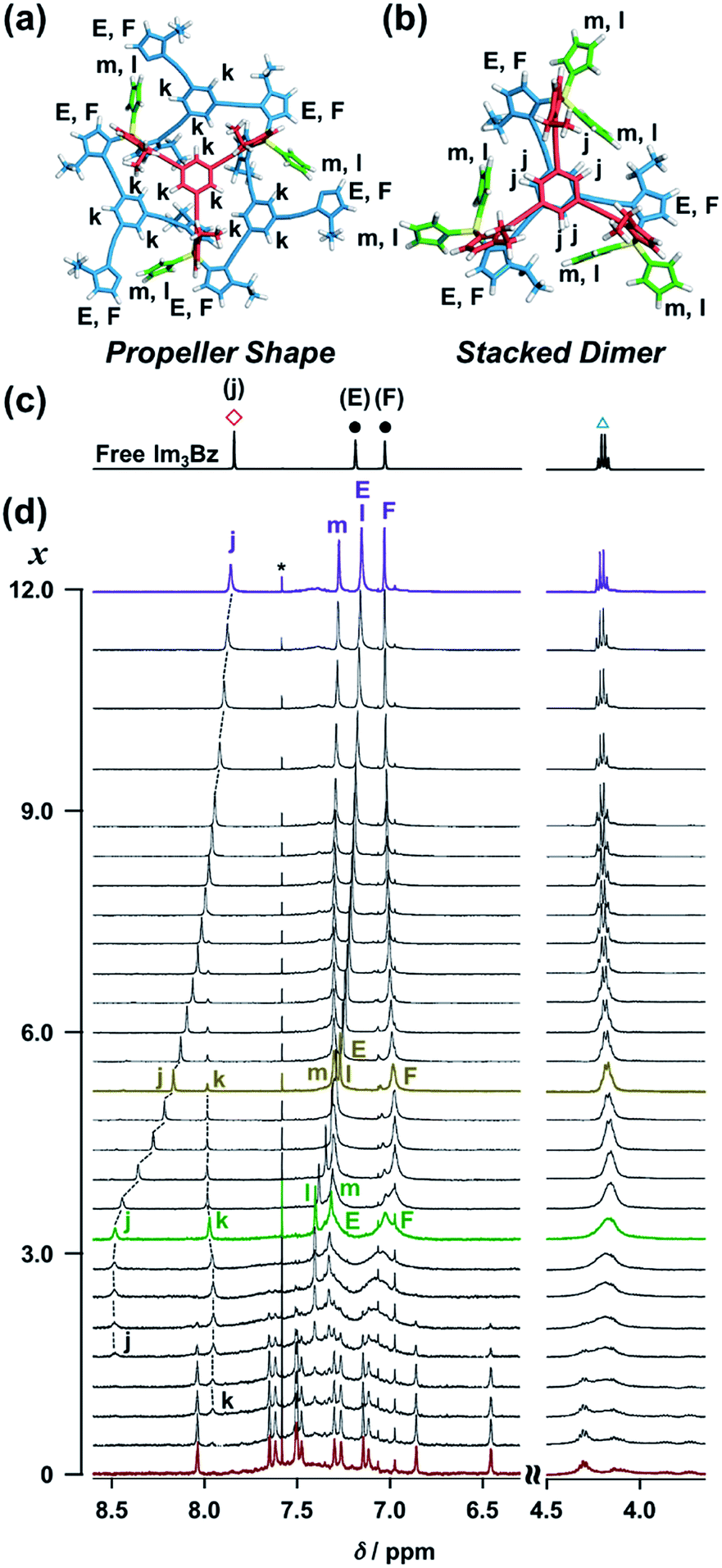
| | Fig. 3 (a) Solution structure and NMR signal assignment of (Im3Bz)4(Zn2+)3. (b) Stacked 1H NMR spectra of Im3Bz (3.3 × 10−3 M) in the presence of Zn2+ (0–3.3 × 10−3 M) in CD3CN at 298 K. The small peak annotated by an asterisk remains unchanged during the titration. Symbols correspond to those in the chemical structure of Im3Bz. Alphabet pairs (A/A′, B/B′, C/C′, and D/D′) labeling closed circles indicate vicinal protons at the imidazole ring. | |
 |
| | Scheme 3 Assembly formation of ImR3Bz with Zn2+ and ImH2. | |
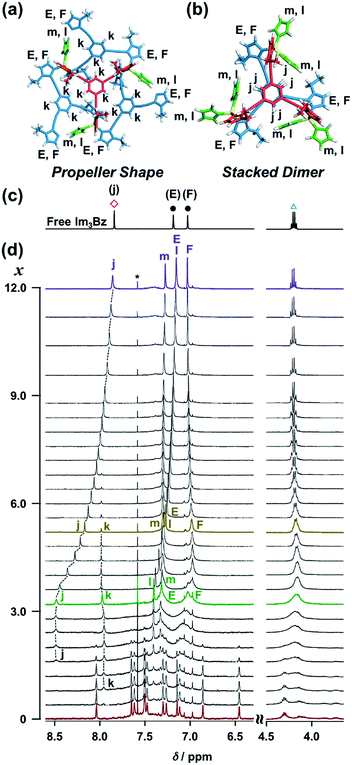
On the basis of the above analysis, we propose a propeller-shaped structure (Fig. 3a) for the (Im(RorS)3Bz)4(Zn2+)3 assembly, which is the most symmetrical geometry of the Zn2+-assemblies with GC = 7 and a stoichiometry of [Im(RorS)3Bz]:[Zn2+] = 4:3. Single-crystal X-ray diffraction analysis provides definitive structural information in the solid state. However, the propeller-shaped structure is formally coordinatively unsaturated (coordination number, n = 3) in solution, and unfortunately suitable crystals of the self-assembly could not be obtained. Hence, we verified the proposed structure by systematic NMR spectroscopic titration analysis.21,31 Upon titration of Zn(OSO2CF3)2 into ImS3Bz in acetonitrile-d3 (CD3CN) broadening of the 1H NMR signals occurred because of rapid exchange, i.e., dissociation and reassociation of the (ImS3Bz)4(Zn2+)3 assembly on the NMR timescale (Fig. S1†). We found that the NMR line broadening was prevented by modifying the side arms of the C3-symmetric ligands (Scheme 1), when the chiral chains were replaced by ethyl groups (vide infra, Fig. 3b). We note that the Im3Bz ligand exhibited absorption titration and ESI mass spectrometric results (Fig. S2 and S3†) similar to those for Im(RorS)3Bz (Fig. 1 and 2), which confirmed that Im3Bz also forms a 4:3 assembly [(Im3Bz)4(Zn2+)3] in a stepwise manner (as Scheme 3A and B). During the titration of Im3Bz with Zn2+ at [Zn2+]/[Im3Bz]0 molar ratios in the range of 0–0.6, the aromatic protons of the imidazole side arms and the central benzene rings gradually shifted and broadened (Fig. 3b). The initially observed NMR peak shift at [Zn2+]/[Im3Bz]0 molar ratios in the range of 0–0.4 is attributed to the formation of a 4:1 complex [(Im3Bz)4Zn2+]. Conversely, the NMR line broadening likely originated from rapid exchange between the resulting 4:1 complex [(Im3Bz)4Zn2+] and the subsequently formed 4:3 self-assembled [(Im3Bz)4(Zn2+)3]. However, clear NMR signals with a total of 12 aromatic protons were resolved upon addition of a certain amount (0.7 equivalents) of Zn2+ (Fig. 3b left).32 The preformed 4:1 complex was almost completely converted into the 4:3 self-assembly at a molar ratio close to [Zn2+]/[Im3Bz]0 = 0.75. On the basis of COSY correlations (Fig. S5†), we separated the 12 observed resonances into 4 resonances originating from the central benzene protons (Fig. 3b red squares, f, g, h, and i) and 8 other resonances corresponding to the imidazole (aromatic) protons (Fig. 3b closed circles, A–D and A′–D′). Among the central benzene protons (f, g, h, and i), we observed the 1H, 1H COSY correlations between the signals “f”, “h”, and “i” (Fig. S5†). Therefore, these signals come from neighboring protons that are directly bound to the same central benzene ring, but located in different chemical environments. Thus (Im3Bz)4(Zn2+)3 has Im3Bz ligands that lose the C3 symmetry of the original ligand upon self-assembly (blue colored molecules in Fig. 3a). Conversely, the benzene proton “g” has no 1H, 1H COSY correlation with other benzene protons (f, h, and i, see Fig. S5†), and therefore (Im3Bz)4(Zn2+)3 contains a central Im3Bz ligand that preserves the original ligand C3 symmetry (red colored molecule in Fig. 3a). On the basis of these results, we present the solution structure of (Im3Bz)4(Zn2+)3 (Fig. 3a), consisting of a central C3-symmetric ligand (red colored molecule) surrounded by three non-symmetrical conformers (blue colored molecules). Consequently, the proposed propeller-shaped assembly gives a total of four benzene protons with an integration ratio of 1:1:1:1 (3H:3H:3H:3H) in its NMR spectrum. One of these protons (g) comes from the central C3-symmetric ligand (red colored molecule) and the other three protons (f, h, and i) come from the three ligands (blue colored molecules). In this manner, NMR shows a total of 8 resonances for imidazole (aromatic) protons with the same integration ratio (3H amount for each signal), which is consistent with the experimentally observed NMR signals arising from the four pairs of vicinal protons at the imidazole rings (Fig. 3b, A/A′, B/B′, C/C′, and D/D′). The ethyl proton signals are likely caused by the imidazole side chains as much simpler probes, which support the proposed propeller-shaped structure. In the presence of 0.7 equivalents of Zn2+, the original quartet signal at 4.17 ppm splits into a double quartet signals with an integration ratio of 1:1:1:1 (Fig. 3b, blue triangles, a–d; 3H for each signal) and an intense overlapped signal consisting of 12 protons (e). The double quartet signals (a–d) suggest that the two CH2 protons (connected to the same CH2 carbon atom) are in different chemical environments owing to restricted rotation of the ethyl side chains upon formation of the propeller-shaped assembly. In fact, we observed 1H, 1H COSY correlations between “a” and “c” and between “b” and “d” (Fig. S6†). Therefore, these proton pairs must be connected to the same CH2 carbon atom. Among the four double quartet signals (a–d), the proton pairs “a” and “c” are assignable to the CH2 protons of the central C3-symmetric ligand (red colored molecule in Fig. 3a). These protons are shielded by the neighboring imidazole rings of the three propeller-shaped ligands (blue colored molecules). This steric configuration should restrict the rotation of the ethyl side chains in the central C3-symmetric ligand (red colored molecule). Conversely, the other proton pairs (b and d) are attributed to the CH2 protons of the side chains of the other three Im3Bz (blue colored molecules) at the bottom position. Consequently, the intense overlapping signal (e) corresponds to the other ethyl side chains of the three propeller-shaped ligands (blue colored molecules) at the top position in similar chemical environments (see the top view in Fig. 3a). The NMR line broadening above 0.8 equiv. of Zn2+ (Fig. 3b) indicates that an excess of Zn2+ ions promoted rapid dissociation and reassociation processes of (Im3Bz)4(Zn2+)3. On the basis of these findings, we conclude that the propeller-shaped structure (Fig. 3a) is the most probable main species in solution.33
Supramolecular chirality transformation driven by monodentate ligand binding
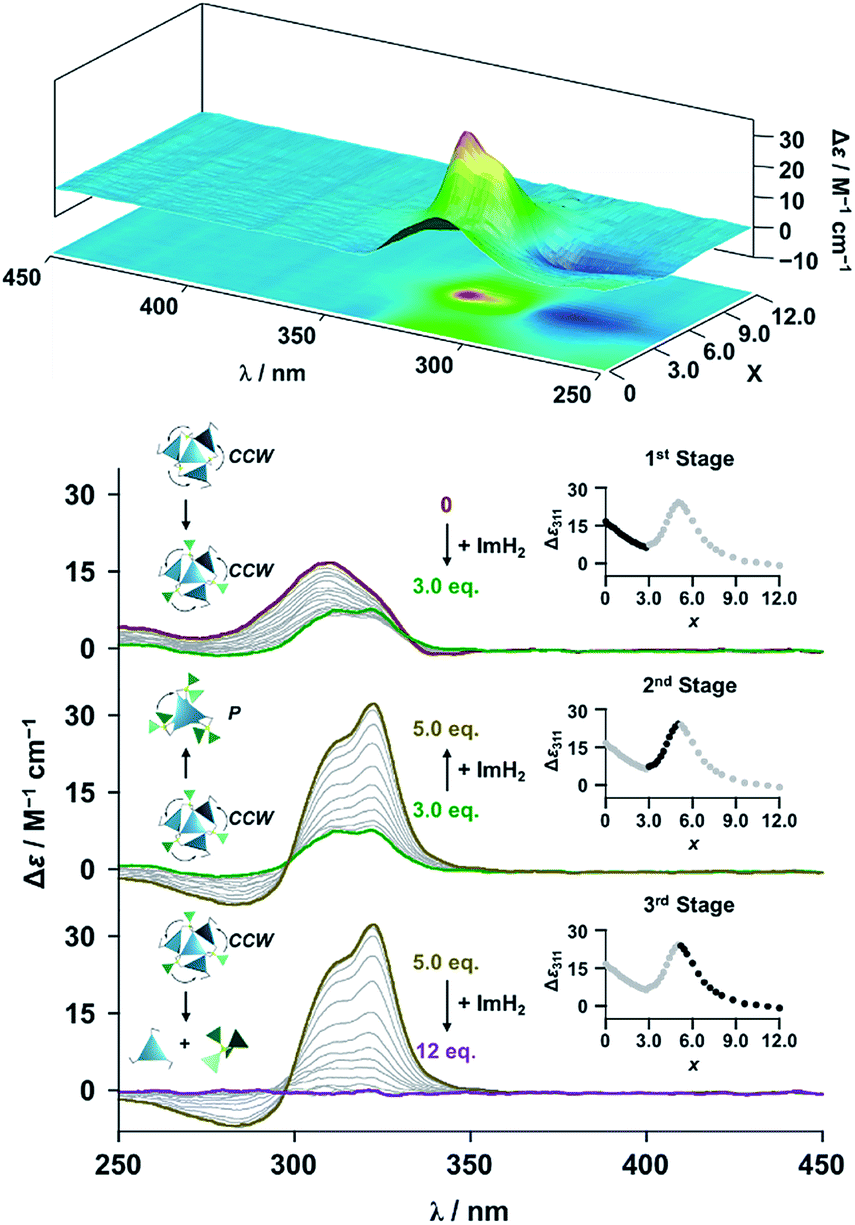
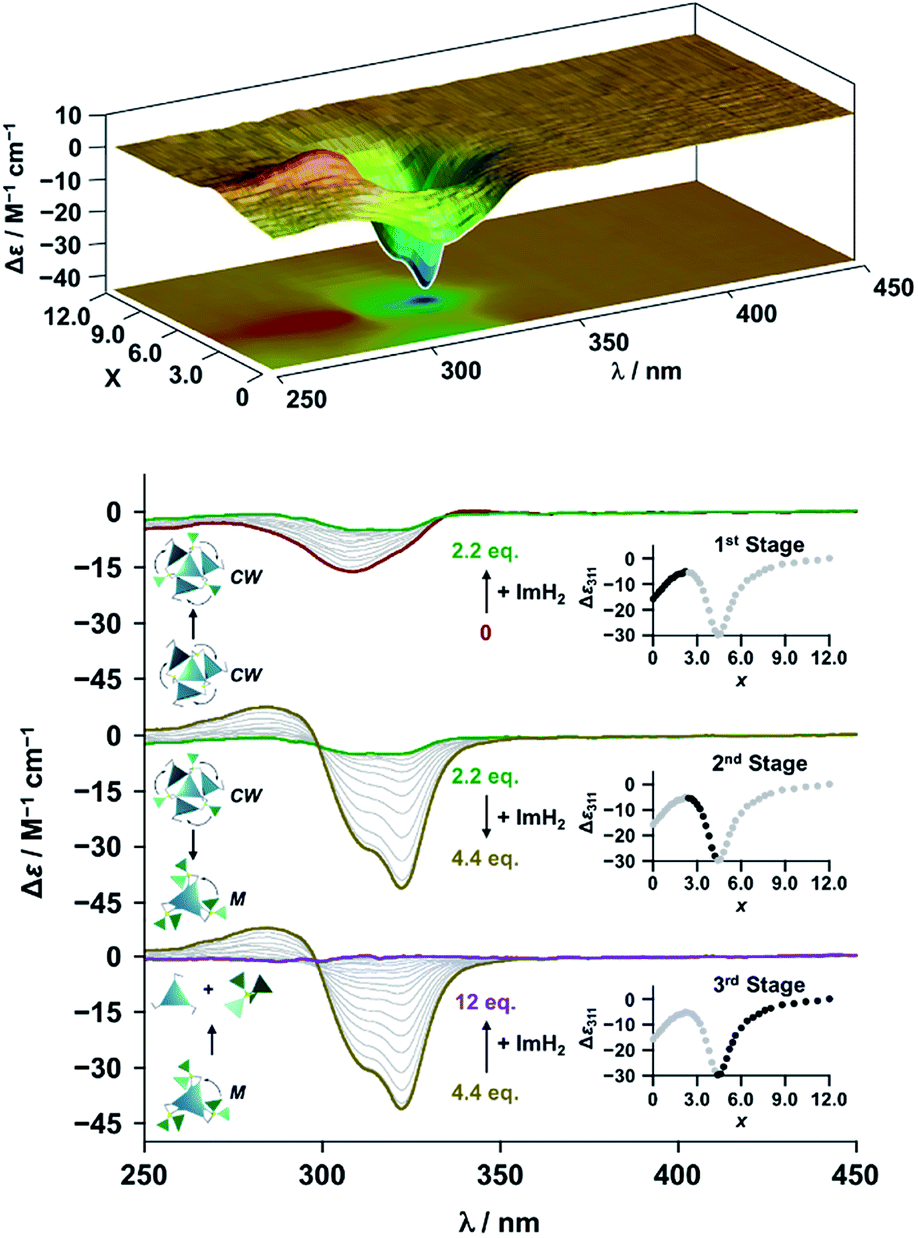
The propeller-shaped assembly is formally coordinatively unsaturated (Fig. 3a), where each Zn2+ ion coordinates to the three individual imidazole side arms (coordination number, n = 3). On the basis of ESI mass spectrometric analysis in solution (Fig. 2, vide supra), their vacant sites are likely occupied by solvent molecules or counter anions (OSO2CF3−), which can be easily replaced by an appropriate monodentate co-ligand. This motivated us to investigate monodentate ligand binding (imidazole: ImH2) to the resulting coordinatively unsaturated assembly (Scheme 3C and E). This process was again monitored by CD spectroscopy (Fig. 4 and 5; UV/vis titration in Fig. S8†). Upon addition of 0–3.0 equivalents of ImH2 to the resulting (Im(RorS)3Bz)4(Zn2+)3 assembly in acetonitrile, the CD spectrum of the propeller-shaped assembly [(Im(RorS)3Bz)4(Zn2+)3] gradually disappeared (Fig. 4 and 5, red line to green line). We then found a sharp change of the signal with an enhancement of the CD intensity in the presence of 5.0 equivalents (ImH2/Zn2+ molar ratio: ∼1.7) of ImH2 (Fig. 4 and 5, green line to yellow line). The resulting CD signals almost disappeared upon further addition of 12 equivalents of ImH2 (Fig. 4 and 5, purple line), owing to dissociation of self-assemblies in the presence of excess ImH2, which yielded ImH2/Zn2+ complexes (Scheme 3E). We analyzed the observed stepwise process (Scheme 3C and E) by using titration plots (insets of Fig. 4 and 5) of Δε at 311 nm vs. the molar ratio (x = [ImH2]/[(Im(RorS)3Bz)4(Zn2+)3]0), where inflection points occurred at [ImH2]/[(Im(RorS)3Bz)4(Zn2+)3]0 = 3.0 and around 5.0. The initial inflection is responsible for binding of the monodentate co-ligand (ImH2) to the vacant sites of (Im(RorS)3Bz)4(Zn2+)3, yielding a coordinatively saturated (ImH2)3(Im(RorS)3Bz)4(Zn2+)3 assembly (Scheme 3C). The resulting CD pattern of (ImH2)3(Im(RorS)3Bz)4(Zn2+)3 resembles the original CD profile owing to the propeller-shaped assembly despite the subtle red shift (Fig. 4 and 5, red line vs. green line), indicating that the resulting (ImH2)3(Im(RorS)3Bz)4(Zn2+)3 preserves the preformed propeller-shaped chirality (Scheme 3C). Notably, the suggested ternary complex was successfully identified by HR ESI MS, m/z calcd. as [(ImH−)3(ImR3Bz)4(Zn2+)3(OSO2CF3)1 + 2CH3CN]2+, 1440.51049; found 1440.51380 (Fig. 6). These results were obtained from an ESI mass spectrum of (ImR3Bz)4(Zn2+)3 in the presence of 3.0 equivalents of ImH2 in acetonitrile. The subsequently formed coordinatively saturated (ImH2)3(Im(RorS)3Bz)4(Zn2+)3 assembly contained no Zn2+ center capable of binding with the additional monodentate co-ligands (Fig. 7a). However, we observed a second inflection point in the CD titration plot (vide supra, Fig. 4 and 5 insets), which is associated with a sharp change of the CD spectrum (vide supra, Fig. 4 and 5, yellow lines). We posit that this feature arises from a supramolecular chirality transition driven by monodentate ligand binding (Scheme 3D). The observed intense CD signals (Fig. 4 and 5, yellow line) are characteristic of an exciton-coupled biphasic (splitting) CD pattern caused by twisting of the chromophore molecules.21c,26,34–36 In this context, Katoono et al. have recently developed stacked dimer assemblies of C3-symmetric benzene rings containing ethynyl spacers in the twisting positions.26 They provided CD profiles, which resemble those in our study (yellow lines in Fig. 4 and 5), suggesting structural similarities to our proposed stacked dimer assemblies here.26 From this analysis, we estimated the stacked dimer assemblies in the twisting position [(ImH2)m(Im3Bz)2(Zn2+)3] (m = 4–6) to be the most probable assembly in solution in the second stage of the titration (corresponding to yellow lines in Fig. 4 and 5). It should be noted that we have measured the ESI mass spectrum of ImS3Bz in the presence of Zn2+ and 1.7 equivalents (toward Zn2+) of ImH2, where the fragment mass of [(ImH2)m(ImS3Bz)2(Zn2+)3] (m = 4–6) was found (Fig. S10†).
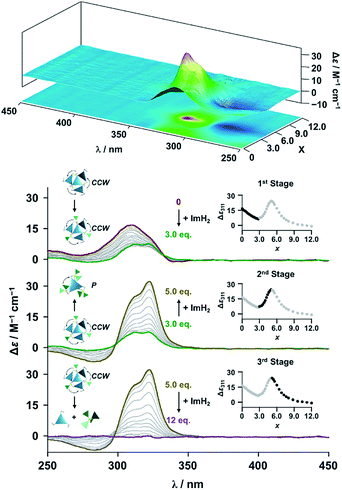 |
| | Fig. 4 CD spectral changes observed upon titration of an acetonitrile solution of ImR3Bz (2.0 × 10−5 M) containing Zn2+ (2.0 × 10−5 M) with ImH2 (0–6.0 × 10−5 M, 0: red line, 1.5 × 10−5 M: green line, 2.5 × 10−5 M: yellow line, and 6.0 × 10−5 M: purple line), where Δε is calculated based on the ligand concentration (2.0 × 10−5 M). Top panel shows the corresponding stacked plot for CD titration. Insets: plot of Δε at 311 nm vs. the molar ratio (x = [ImH2]/[(ImR3Bz)4(Zn2+)3]0). | |
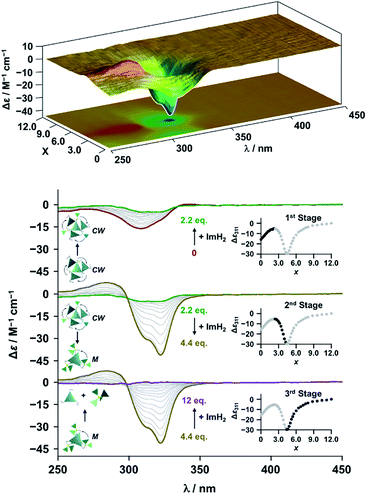 |
| | Fig. 5 CD spectral changes observed upon titration of an acetonitrile solution of ImS3Bz (2.0 × 10−5 M) containing Zn2+ (2.0 × 10−5 M) with ImH2 (0–6.0 × 10−5 M, 0: red line, 1.5 × 10−5 M: green line, 2.2 × 10−5 M: yellow line, and 6.0 × 10−5 M: purple line), where Δε is calculated based on the ligand concentration (2.0 × 10−5 M). Top panel shows the corresponding stacked plot for CD titration. Insets: plot of Δε at 311 nm vs. the molar ratio (x = [ImH2]/[(ImS3Bz)4(Zn2+)3]0). | |
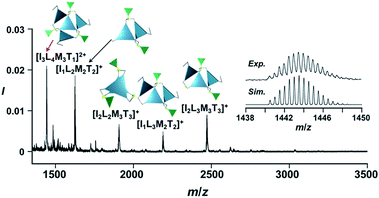 |
| | Fig. 6 Positive ESI-MS spectrum of ImS3Bz (normalized by its most intense fragment at m/z = 564.3 from the free ligand) in acetonitrile (2.2 × 10−3 M) in the presence of Zn2+ (1.7 × 10−3 M) and ImH2 (1.7 × 10−3 M). Inset: isotopically resolved signals at m/z = 1440.5 and calculated isotopic distributions for [(ImH−)3(ImS3Bz)4(Zn2+)3(OSO2CF3)(CH3CN)2]2+. Objects correspond to the mass peak assignment (I: ImH−, L: ImS3Bz, M: Zn2+, T: OSO2CF3−). Mass spectrum of the whole region is given in Fig. S9.† | |
 |
| | Fig. 7 Energy minimized structures and NMR signal assignment of (a) (ImH2)3(Im3Bz)4(Zn2+)3 and (b) (ImH2)6(Im3Bz)2(Zn2+)3. (c) 1H NMR spectrum of free Im3Bz (1.8 × 10−3 M) in CD3CN, where symbols correspond to those in the chemical structure Im3Bz (Fig. 3). (d) Stacked 1H NMR spectra of Im3Bz (1.8 × 10−3 M) with Zn2+ (1.4 × 10−3 M) in the presence of ImH2 (0–5.4 × 10−3 M, 0: red line, 1.4 × 10−3 M: green line, 2.3 × 10−3 M: yellow line, and 5.4 × 10−3 M: purple line) in CD3CN at 298 K (x = [ImH2]/[(Im3Bz)4(Zn2+)3]0). Asterisk denotes chloroform. | |
In order to investigate whether the reversible change between the (ImR3Bz)4(Zn2+)3 and (ImH2)m(ImR3Bz)2(Zn2+)3 complexes (Scheme 3C and D) is possible or not, we have measured CD spectra of (ImH2)m(ImR3Bz)2(Zn2+)3 upon addition of Zn2+ that acts as the sequester of ImH2 in (ImH2)m(ImR3Bz)2(Zn2+)3 (Fig. S11†). The CD intensity due to (ImH2)m(ImR3Bz)2(Zn2+)3 decreased with increasing Zn2+ concentration (Fig. S11†), where the CD spectrum of (ImH2)m(ImR3Bz)2(Zn2+)3 converted to that of (ImH2)3(ImR3Bz)4(Zn2+)3 upon addition of 2.5 × 10−5 M of Zn2+ (Fig. S11†). Hence, conversion of (ImH2)m(ImR3Bz)2(Zn2+)3 to (ImH2)3(ImR3Bz)4(Zn2+)3 (Scheme 3D) is reversible. Further addition of Zn2+ to the resulting solution caused a subsequent CD spectral change (Fig. S11†), indicating a structural transition of (ImH2)3(ImR3Bz)4(Zn2+)3. However, the resulting CD spectrum is slightly different from that of Zn3(ImR3Bz)4 (Fig. 4, red line). Thus, the reversible change between (ImH2)3(ImR3Bz)4(Zn2+)3 and (ImR3Bz)4(Zn2+)3 (Scheme 3C) is not possible under these conditions.
The ligand-driven architectural transformation is further discussed in detail by NMR titration experiments. We used Im3Bz instead of Im(RorS)3Bz in this case, because of the NMR broadening observed for (Im(RorS)3Bz)4(Zn2+)3 (vide supra, Fig. S1†). Note that the NMR titration experiments with ImR3Bz give almost the same results as those obtained with Im3Bz except for the initial NMR broadening (Fig. S12†). Upon addition of 0–3.0 equivalents of ImH2 to the resulting (Im3Bz)4(Zn2+)3 assembly in CD3CN (Fig. 7d, red line to green line), the well-resolved NMR signals from (Im3Bz)4(Zn2+)3 (red line) disappeared with a concomitant rise of sharp peaks at 8.48, 7.97, 7.40, and 7.32 ppm (j, k, l, and m, respectively) and broad signals at around 7.32 and 7.03 ppm (E and F, respectively). Among them, the sharp peaks (j and k) are considered to originate from two coordinated species in solution, and the peak “k” disappears with a concomitant increase of the relative intensity of the peak “j” with increasing molar ratio (x) [ImH2]/[(Im3Bz)4(Zn2+)3]0 (Fig. 7d, green line to yellow line). Upon further increasing ImH2 concentrations, the remaining peak “j” gradually shifted upfield and became the same NMR signal of free Im3Bz owing to the central benzene proton (Fig. 7c red square) at a molar ratio of [ImH2]/[(Im3Bz)4(Zn2+)3]0 = 12.0 (Fig. 7d, yellow line to purple line). This observation is consistent with dissociation of the self-assemblies to yield the ImH2–Zn2+ complex at higher concentrations of ImH2 (Scheme 3E). Taken together, we conclude that the peak “j” originates from the central benzene protons of Im3Bz in the subsequently formed stacked dimer assembly [(ImH2)m(Im3Bz)2(Zn2+)3], and the peak “k” derives from (ImH2)3(Im3Bz)4(Zn2+)3 prior to the stacked dimer assembly formation. Partial dissociation and reassociation likely occur on the NMR timescale in both assemblies, giving rise to a rather simple NMR signal pattern (Fig. 7c, green line). The two assemblies coexist at a molar ratio of [ImH2]/[(Im3Bz)4(Zn2+)3]0 = 3.0 (Fig. 7d, green line) under the conditions used for the NMR experiments.37 Imidazole protons (as well as CH2 protons from the side chains) of Im3Bz overlapped at similar chemical shifts for the two assemblies and showed rather broad signals (E and F on the green line).38 Conversely, the chemical shifts of the central benzene protons were markedly different between the two assemblies (peaks j and k), where the peak “j” shifted downfield compared with the peak “k” that is assigned to the propeller-shaped assembly (Fig. 7d). This differentiation may be the result of a difference in the relative positional relationship of the central benzene rings between the two assemblies. In the propeller-shaped assembly (Fig. 7a), the central C3-symmetric ligand (red colored molecule) is almost perpendicular to the three propeller-shaped ligands (blue colored molecules). Hence, their chemical environments are not markedly different from those of the free ligand. Conversely, in the stacked dimer assembly, clipping by the three Zn2+ ions forces the two benzene rings into a twisted face-to-face position (Fig. 7b). The observed downfield shift (peak j in Fig. 7d) is attributed to the deshielding effect of the stacked benzene rings, which gives experimental confirmation of the stacked dimer assembly.39,40
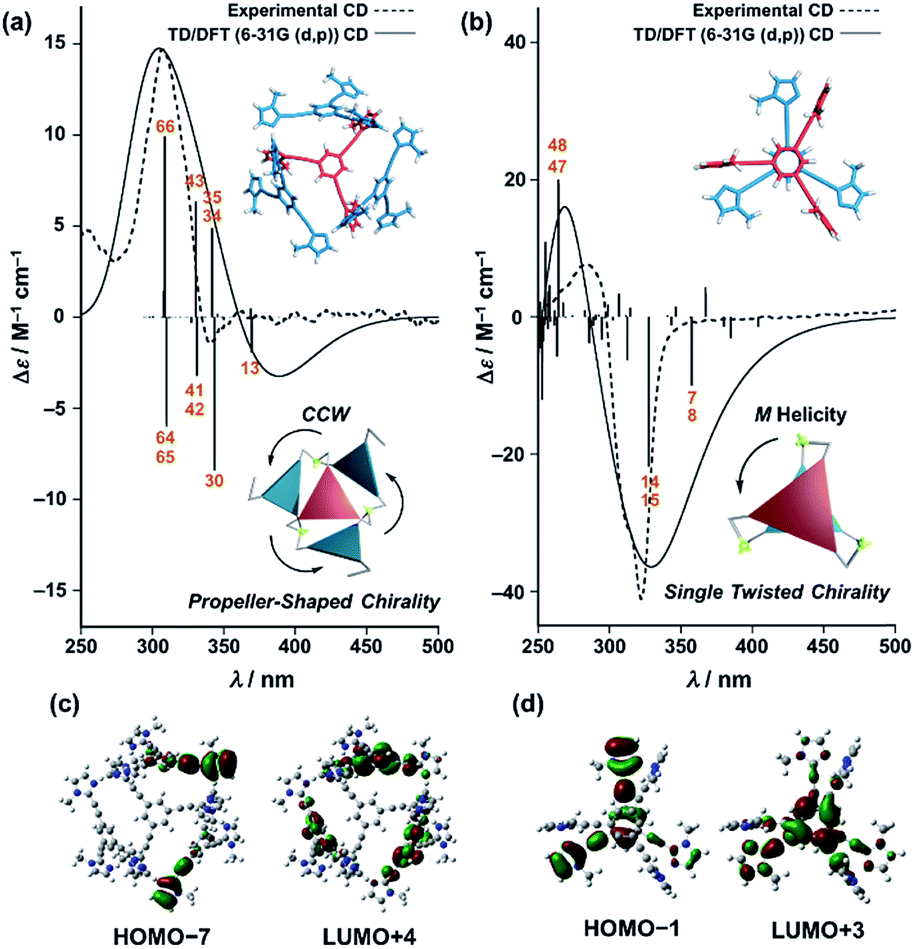
Finally, we performed time-dependent density functional theory (TD-DFT) calculations on the C3-symmetric ligands in a propeller-shaped arrangement and a twisting dimer arrangement (solid lines in Fig. 8a and b, respectively). Here, only electronic transitions of the C3-symmetric ligand part were calculated. Neither the Zn2+ (d10 configuration) nor the imidazole co-ligands have any appreciable absorption above 250 nm contributing to the observed CD spectra (dashed lines in Fig. 8). In addition, the chiral alkyl side chains of the C3-symmetric ligands were replaced by methyl groups to reduce the calculation complexity. These modifications enabled us to perform the TD-DFT calculations in a reasonable time frame. The theoretical CD spectrum for the propeller-shaped geometry agreed well with the experimental one for (ImR3Bz)4(Zn2+)3 (Fig. 8a, solid line vs. dashed line), suggesting a counter clockwise (ccw) rotation for (ImR3Bz)4(Zn2+)3, and a clockwise (cw) rotation for (ImS3Bz)4(Zn2+)3. According to the calculated rotatory strength (see Fig. S13†), most of the high rotatory strength arises from electronic transitions within the three molecules that represent the propeller shape (blue colored molecules in the inset of Fig. 8a). For example, the dominant contribution (17%) of excitation 43 (Fig. 8a) is a transition from the HOMO−7 to LUMO+4, which features molecular orbitals delocalized over the two and three molecules in the propeller-shaped arrangement, respectively (Fig. 8c). Conversely, the HOMO, HOMO−1, and HOMO−2 are localized on the central C3-symmetric ligand (red colored molecule in the inset of Fig. 8a, see Fig. S13†). Those higher energy levels do not contribute to a high rotatory strength in Fig. 8a (see Fig. S13†). Consequently, the propeller-shaped assembly (ImR3Bz)4(Zn2+)3 has a positive Cotton effect at shorter wavelengths and the extremely weak negative Cotton effect at the longer wavelength is negligible (Fig. 8a). In contrast, splitting of the CD pattern is found in both the experimental and theoretical CD spectra for the stacked dimer in the twisting position (Fig. 8b), where both spectra show a strong negative Cotton effect in the first Cotton band and a weak positive Cotton effect in the second Cotton band. These calculations suggest that most of the high rotatory strength arises from electronic transitions between the two molecules in the twisting position (Fig. S14†). For example, a transition from HOMO−1 to LUMO+3 is the dominant contribution (47%) of excitation 14 in Fig. 8b, in which the molecular orbital of LUMO+3 is essentially delocalized over the two central benzene rings (Fig. 8d). This calculation indicates an M helix (excess) for (ImH2)m(ImS3Bz)2(Zn2+)3 and a P helix (excess) for (ImH2)m(ImR3Bz)2(Zn2+)3.
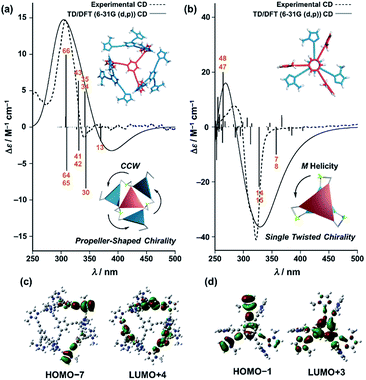 |
| | Fig. 8 Dashed lines show experimental CD spectra of (a) (ImR3Bz)4(Zn2+)3 (corresponding to the red line in Fig. 4) and (b) (ImH2)m(ImS3Bz)2(Zn2+)3 (corresponding to the yellow line in Fig. 5). Solid lines show theoretical CD spectra [TD-DFT/B3LYP-6-31G(d,p)] of the C3-symmetric ligands in (a) the propeller-shaped arrangement and (b) twisting dimer arrangement. Calculated spectrum in (a) is vertically scaled (scaling factor = 0.85). The numbered excitations correspond to those with high rotatory strength. Two molecular orbitals involved in (c) excitation 43 (17%) in (a) and (d) excitation 14 (47%) in (b). Insets: schematic representation of (a) propeller-shaped chirality and (b) single twisted chirality. | |
Conclusions
We have successfully demonstrated a supramolecular chirality transition driven by monodentate ligand binding. Newly synthesized C3-symmetric ligands (Im(RorS)3Bz) containing three chiral imidazole side arms (Im(R or S)) with ethynyl spacers assemble into a propeller-shaped assembly [(Im(RorS)3Bz)4(Zn2+)3] with 0.75 equivalents of Zn2+ in acetonitrile in the absence of monodentate co-ligands (imidazole: ImH2). The resulting (Im(RorS)3Bz)4(Zn2+)3 assembly is formally coordinatively unsaturated (coordination number: n = 3) and therefore capable of accepting a total of three equivalents of monodentate co-ligands (ImH2) to yield a coordinatively saturated assembly. The resulting coordinatively saturated assembly [(ImH2)3(Im(RorS)3Bz)4(Zn2+)3] preserves the preformed propeller-shaped chirality of (Im(RorS)3Bz)4(Zn2+)3. However, addition of monodentate co-ligands transforms the supramolecular chirality from propeller-shaped chirality into single-twist chirality, when (ImH2)3(Im(RorS)3Bz)4(Zn2+)3 transforms into a stacked dimer assembly (ImH2)m(Im(RorS)3Bz)2(Zn2+)3 (m = 4–6). The present strategy shows promise for the rational design of dynamic coordination chirality capable of alternating between chiral objects of different shapes driven by a specific external stimulus.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by JSPS Grant-in-Aid for JSPS Research Fellow Grant Numbers 16J11554, and KAKENHI Grant Numbers JP17H05386 in Scientific Research on Innovative Areas “Coordination Asymmetry”, Grant-in-Aid for Takahashi Industrial and Economic Research Foundation, and ASAHI Glass Foundation. We thank Andrew Jackson, PhD, from the Edanz Group (http://www.edanzediting.com/ac) for editing a draft of this manuscript.
Notes and references
-
(a) L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555 RSC;
(b) J. Crassous, Chem. Soc. Rev., 2009, 38, 830 RSC;
(c) J. Crassous, Chem. Commun., 2012, 48, 9684 RSC;
(d) R. W. Saalfrank, H. Maid and A. Scheurer, Angew. Chem., Int. Ed., 2008, 47, 8794 CrossRef CAS;
(e) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS;
(f) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1 CrossRef CAS.
- F. A. Samatey, K. Imada, S. Nagashima, F. Vonderviszt, T. Kumasaka, M. Yamamoto and K. Namba, Nature, 2001, 410, 331 CrossRef CAS.
- R. Kuroda, B. Endo, M. Abe and M. Shimizu, Nature, 2009, 462, 790 CrossRef CAS.
- C. Mao, W. Sun, Z. Shen and N. C. Seeman, Nature, 1999, 397, 144 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS.
-
(a) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656 RSC;
(b) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS;
(c) M. E. Carnes, M. S. Collins and D. W. Johnson, Chem. Soc. Rev., 2014, 43, 1825 RSC.
-
(a) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS;
(b) H. Miyake and H. Tsukube, Chem. Soc. Rev., 2012, 41, 6977 RSC;
(c) H. K. Bisoyi and Q. Li, Angew. Chem., Int. Ed., 2016, 55, 2994 CrossRef CAS.
-
(a) K. Miwa, Y. Furusho and E. Yashima, Nat. Chem., 2010, 2, 444 CrossRef CAS;
(b) K. Shimomura, T. Ikai, S. Kanoh, E. Yashima and K. Maeda, Nat. Chem., 2014, 6, 429 CrossRef CAS;
(c) R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853 CrossRef CAS;
(d) N. Ousaka, Y. Takeyama and E. Yashima, Chem. Sci., 2012, 3, 466 RSC;
(e) S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948 CrossRef CAS;
(f) S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868 CrossRef CAS;
(g) C. P. Montgomery, E. J. New, D. Parker and R. D. Peacock, Chem. Commun., 2008, 4261 RSC.
-
(a) J. Gregoliński, P. Starynowicz, K. T. Hua, J. L. Lunkley, G. Muller and J. Lisowski, J. Am. Chem. Soc., 2008, 130, 17761 CrossRef;
(b) J. Gregoliński and J. Lisowski, Angew. Chem., Int. Ed., 2006, 45, 6122 CrossRef;
(c) J. Gregoliński, T. Lis, M. Cyganik and J. Lisowski, Inorg. Chem., 2008, 47, 11527 CrossRef;
(d) J. Gregoliński, K. Ślepokura and J. Lisowski, Inorg. Chem., 2007, 46, 7923 CrossRef.
-
(a) J. Gregoliński, M. Hikita, T. Sakamoto, H. Sugimoto, H. Tsukube and H. Miyake, Inorg. Chem., 2016, 55, 633 CrossRef;
(b) H. Miyake, M. Ueda, S. Murota, H. Sugimoto and H. Tsukube, Chem. Commun., 2012, 48, 3721 RSC;
(c) H. Miyake, K. Yoshida, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2004, 126, 6524 CrossRef CAS;
(d) H. Miyake, H. Sugimoto, H. Tamiaki and H. Tsukube, Chem. Commun., 2005, 4291 RSC;
(e) H. Miyake, M. Hikita, M. Itazaki, H. Nakazawa, H. Sugimoto and H. Tsukube, Chem.–Eur. J., 2008, 14, 5393 CrossRef CAS.
-
(a) S. Hiraoka, K. Hirata and M. Shionoya, Angew. Chem., Int. Ed., 2004, 43, 3814 CrossRef CAS;
(b) S. Hiraoka, T. Tanaka and M. Shionoya, J. Am. Chem. Soc., 2006, 128, 13038 CrossRef CAS;
(c) R. Kubota, S. Tashiro and M. Shionoya, Chem. Sci., 2016, 7, 2217 RSC.
-
(a) J. Yuasa, H. Ueno and T. Kawai, Chem.–Eur. J., 2014, 20, 8621 CrossRef CAS;
(b) J. Yuasa, T. Ohno, H. Tsumatori, R. Shiba, H. Kamikubo, M. Kataoka, Y. Hasegawa and T. Kawai, Chem. Commun., 2013, 49, 4604 RSC.
-
(a) M. Hutin and J. Nitschke, Chem. Commun., 2006, 1724 RSC;
(b) R. Lin, H. Zhang, S. Li, L. Chen, W. Zhang, T. Bin Wen, H. Zhang and H. Xia, Chem.–Eur. J., 2011, 17, 2420 CrossRef CAS;
(c) H. Isla, M. Srebro-Hooper, M. Jean, N. Vanthuyne, T. Roisnel, J. L. Lunkley, G. Muller, J. A. G. Williams, J. Autschbach and J. Crassous, Chem. Commun., 2016, 52, 5932 RSC;
(d) A.-C. Chamayou, G. Makhloufi, L. A. Nafie, C. Janiak and S. Lüdeke, Inorg. Chem., 2015, 54, 2193 CrossRef CAS.
-
(a) S. Zahn and J. W. Canary, Science, 2000, 288, 1404 CrossRef CAS;
(b) S. Mortezaei, N. R. Catarineu and J. W. Canary, J. Am. Chem. Soc., 2012, 134, 8054 CrossRef CAS;
(c) S. Zahn, D. Das and J. W. Canary, Inorg. Chem., 2006, 45, 6056 CrossRef CAS.
-
(a) Y. Zheng, Y.-Y. Pan, Y.-P. Ren, L.-S. Long, R.-B. Huang and L.-S. Zheng, Chem. Commun., 2014, 50, 14728 RSC;
(b) J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2007, 129, 12912 CrossRef CAS.
-
(a) W. Cullen, C. A. Hunter and M. D. Ward, Inorg. Chem., 2015, 54, 2626 CrossRef CAS;
(b) T.-Z. Xie, K. Guo, Z. Guo, W.-Y. Gao, L. Wojtas, G.-H. Ning, M. Huang, X. Lu, J.-Y. Li, S.-Y. Liao, Y.-S. Chen, C. N. Moorefield, M. J. Saunders, S. Z. D. Cheng, C. Wesdemiotis and G. R. Newkome, Angew. Chem., Int. Ed., 2015, 54, 9224 CrossRef CAS.
-
(a) A. Malviya, H. S. Jena, A. K. Mondal and S. Konar, Eur. J. Inorg. Chem., 2015, 2015, 2901 CrossRef CAS;
(b) H.-G. Jeon, H. K. Lee, S. Lee and K.-S. Jeong, Chem. Commun., 2018, 54, 5740 RSC;
(c) Y. Liu, F. C. Parks, W. Zhao and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 15477 CrossRef CAS.
-
(a) M. Fujita, O. Sasaki, T. Mitsuhashi, T. Fujita, J. Yazaki, K. Yamaguchi and K. Ogura, Chem. Commun., 1996, 20, 1535 RSC;
(b) T. Weilandt, R. W. Troff, H. Saxell, K. Rissanen and C. A. Schalley, Inorg. Chem., 2008, 47, 7588 CrossRef CAS PubMed;
(c) M. Ferrer, A. Pedrosa, L. Rodríguez, O. Rossell and M. Vilaseca, Inorg. Chem., 2010, 49, 9438 CrossRef CAS;
(d) A. Sautter, D. G. Schmid, G. Jung and F. Würthner, J. Am. Chem. Soc., 2001, 123, 5424 CrossRef CAS;
(e) T. Yamamoto, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 12309 CrossRef CAS.
-
(a) S. Bandi, A. K. Pal, G. S. Hanan and D. K. Chand, Chem.–Eur. J., 2014, 20, 13122 CrossRef CAS;
(b) S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975 CrossRef CAS;
(c) S. Neogi, G. Schnakenburg, Y. Lorenz, M. Engeser and M. Schmittel, Inorg. Chem., 2012, 51, 10832 CrossRef CAS;
(d) P. D. Frischmann, V. Kunz and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 7285 CrossRef CAS;
(e) T. Haino, T. Fujii, A. Watanabe and U. Takayanagi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10477 CrossRef CAS PubMed;
(f) K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300 CrossRef CAS;
(g) S. Hiraoka, T. Yi, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 14510 CrossRef CAS.
-
(a) J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 566 CrossRef CAS;
(b) J. Yuasa, A. Mitsui and T. Kawai, Chem. Commun., 2011, 47, 5807 RSC;
(c) T. Ogawa, J. Yuasa and T. Kawai, Angew. Chem., Int. Ed., 2010, 49, 5110 CrossRef CAS;
(d) N. Inukai, J. Yuasa and T. Kawai, Chem. Commun., 2010, 46, 3929 RSC;
(e) J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 15976 CrossRef CAS.
-
(a) N. Inukai, T. Kawai and J. Yuasa, Chem.–Eur. J., 2014, 20, 15159 CrossRef CAS;
(b) Y. Imai, T. Kawai and J. Yuasa, Chem. Commun., 2015, 51, 10103 RSC;
(c) Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, Angew. Chem., Int. Ed., 2018, 57, 8973 CrossRef CAS.
-
(a) W. Meng, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 1017 CrossRef CAS;
(b) X.-P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062 CrossRef CAS;
(c) D. Samanta and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 12483 CrossRef CAS;
(d) A. Gerus, K. Ślepokura and J. Lisowski, Inorg. Chem., 2013, 52, 12450 CrossRef CAS;
(e) L. R. Holloway, H. H. McGarraugh, M. C. Young, W. Sontising, G. J. O. Beran and R. J. Hooley, Chem. Sci., 2016, 7, 4423 RSC;
(f) G. Haberhauer, Angew. Chem., Int. Ed., 2010, 49, 9286 CrossRef CAS;
(g) Y. Kubota, S. Sakamoto, K. Yamaguchi and M. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4854 CrossRef CAS;
(h) L. Zhang, L. Lin, D. Liu, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2017, 139, 1653 CrossRef CAS;
(i) H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS;
(j) S. P. Morcillo, D. Miguel, L. Álvarez de Cienfuegos, J. Justicia, S. Abbate, E. Castiglioni, C. Bour, M. Ribagorda, D. J. Cárdenas, J. M. Paredes, L. Crovetto, D. Choquesillo-Lazarte, A. J. Mota, M. C. Carreño, G. Longhi and J. M. Cuerva, Chem. Sci., 2016, 7, 5663 RSC.
-
(a) B. Wang, Z. Zang, H. Wang, W. Dou, X. Tang, W. Liu, Y. Shao, J. Ma, Y. Li and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 3756 CrossRef CAS;
(b) D. M. Wood, W. Meng, T. K. Ronson, A. R. Stefankiewicz, J. K. M. Sanders and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 3988 CrossRef CAS PubMed.
-
(a) T. Y. Kim, L. Digal, M. G. Gardiner, N. T. Lucas and J. D. Crowley, Chem.–Eur. J., 2017, 23, 15089 CrossRef CAS;
(b) Q.-N. Zheng, X.-H. Liu, T. Chen, H.-J. Yan, T. Cook, D. Wang, P. J. Stang and L.-J. Wan, J. Am. Chem. Soc., 2015, 137, 6128 CrossRef CAS PubMed;
(c) Y. Zheng, Z. Zhao, M. Wang, K. Ghosh, J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 16873 CrossRef CAS PubMed;
(d) G.-J. Zhao, F. Yu, M.-X. Zhang, B. H. Northrop, H. Yang, K.-L. Han and P. J. Stang, J. Phys. Chem. A, 2011, 115, 6390 CrossRef CAS PubMed;
(e) S. Leininger, J. Fan, M. Schmitz and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1380 CrossRef CAS PubMed;
(f) C. J. Kuehl, T. Yamamoto, S. R. Seidel and P. J. Stang, Org. Lett., 2002, 4, 913 CrossRef CAS;
(g) J. D. Crowley, A. J. Goshe and B. Bosnich, Chem. Commun., 2003, 2824 RSC;
(h) K. Ghosh, J. Hu, H. S. White and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 6695 CrossRef CAS PubMed.
- For a rare example
of C3-symmetric ligands that are potentially undergoes alternation of the direction of metal–ligand interactions relying on ligand conformation, see: B. Brusilowskij, S. Neubacher and C. A. Schalley, Chem. Commun., 2009, 785 RSC.
-
(a) R. Katoono, K. Fujiwara and T. Suzuki, Chem. Commun., 2014, 50, 5438 RSC;
(b) R. Katoono, Y. Obara, K. Fujiwara and T. Suzuki, Chem. Sci., 2018, 9, 2222 RSC;
(c) R. Katoono, S. Kawai and T. Suzuki, Chem. Sci., 2016, 7, 3240 RSC.
- For coordination-driven stacked assemblies, see
(a) C. Lescop, Acc. Chem. Res., 2017, 50, 885 CrossRef CAS PubMed;
(b) B. Nohra, R. Reau and C. Lescop, Eur. J. Inorg. Chem., 2014, 1788 CrossRef CAS;
(c) B. Nohra, S. Graule, C. Lescop and R. Réau, J. Am. Chem. Soc., 2006, 128, 3520 CrossRef CAS PubMed.
-
(a) H. Y. Lee, J. Park, M. S. Lah and J.-I. Hong, Chem. Commun., 2007, 5013 RSC;
(b) H.-J. Kim, D. Moon, M. S. Lah and J.-I. Hong, Angew. Chem., Int. Ed., 2002, 41, 3174 CrossRef CAS;
(c) L. Yan, Z. Wang, M.-T. Chen, N. Wu, J. Lan, X. Gao, J. You, H.-M. Gau and C.-T. Chen, Chem.–Eur. J., 2008, 14, 11601 CrossRef CAS PubMed;
(d) O. Mamula, M. Lama, S. G. Telfer, A. Nakamura, R. Kuroda, H. Stoeckli-Evans and R. Scopelitti, Angew. Chem., Int. Ed., 2005, 44, 2527 CrossRef CAS PubMed.
- The 4:1 complex is CD silent (Fig. 1a red lines), which probably results from diastereomerization of the tetrahedral complex, or less excitonic interactions between the ligands in the complex.
- The isosbestic points did not clearly switch at [Zn2+]/[Im(RorS)3Bz]0 = 0.25 (Fig. 1b), indicating that there is a possibility that the formation of the (Im(RorS)3Bz)4(Zn2+)3 would start before the completion of the formation of (Im(RorS)3Bz)4Zn2+.
- A. Pastor and E. Martínez-Viviente, Coord. Chem. Rev., 2008, 252, 2314 CrossRef CAS.
- We considered that the dramatic NMR resolution enhancement (Fig. 3) was caused by the prevention of NMR line broadening, not by dramatic formation of (Im3Bz)4(Zn2+)3 at this molar ratio. We have also measured the 1H NMR at low temperature (243 K), however the NMR line broadening (due to the faster ligand exchange around Zn2+) was not prevented even at the low temperature (Fig. S4†).
- We have measured the CD spectra of Im(RorS)3Bz in the presence of 1 equiv. of Zn2+ with the changing of the ratio of ImR3Bz and ImS3Bz (the total concentration is fixed) to investigate the chiral self-sorting effect in the present system (Fig. S7†). The observed CD intensity (Δε) increases linearly with increasing the enantiomeric excess (ee) of Im(RorS)3Bz, indicating that no appreciable chiral self-sorting takes place in this system.
-
N. Berova, K. Nakanishi and R. W. Woody, Circular Dichroism: Principles and Applications, New York, Wiley-VCH, 2nd edn, 2004 Search PubMed.
-
(a) J. Kumar, T. Nakashima, H. Tsumatori and T. Kawai, J. Phys. Chem. Lett., 2014, 5, 316 CrossRef CAS PubMed;
(b) T. Ikai, Y. Wada, S. Awata, C. Yun, K. Maeda, M. Mizuno and T. M. Swager, Org. Biomol. Chem., 2017, 15, 8440 RSC;
(c) M. Inouye, K. Hayashi, Y. Yonenaga, T. Itou, K. Fujimoto, T. Uchida, M. Iwamura and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 14392 CrossRef CAS PubMed.
-
(a) J. Yuasa, T. Ohno, K. Miyata, H. Tsumatori, Y. Hasegawa and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9892 CrossRef CAS PubMed;
(b) T. Y. Bing, T. Kawai and J. Yuasa, J. Am. Chem. Soc., 2018, 140, 3683 CrossRef CAS PubMed;
(c) J. Kumar, B. Marydasan, T. Nakashima, T. Kawai and J. Yuasa, Chem. Commun., 2016, 52, 9885 RSC;
(d) Y. Okayasu and J. Yuasa, Mol. Syst. Des. Eng., 2018, 3, 66 RSC.
- Hence, there is a possibility that a mass peak at m/z = 1901.3 in Fig. 6 due to [(ImH−)2(ImS3Bz)2(Zn2+)3(OSO2CF3)3]+ is the fragment mass of [(ImH2)m(ImS3Bz)2(Zn2+)3] (m = 4–6).
- At a molar ratio of [ImH2]/[(Im3Bz)4(Zn2+)3]0 = 12.0, dissociation of the self-assembly (Scheme 3E) caused the signals to become identical to those of free Im3Bz (Fig. 7c).
- Sharp signals “m” and “l” correspond to the imidazole (aromatic) protons of ImH2, which slightly shifts during the titration (Fig. 7d).
- The strong coordination ability of ImH2 to Zn2+ is a primary requirement for the conversion of (ImH2)3(Im3Bz)4(Zn2+)3 complex into (ImH2)m(Im3Bz)2(Zn2+)3. We have confirmed that complex formation of ImH2 and Zn2+ seems to be also almost stoichiometric similarly to the case of complex formation between he C3-symmetric ligand and Zn2+, which was confirmed by UV/vis and 1H NMR titration experiments (Fig. S15 and S16†). The direct comparison of the coordination ability of ImH2 and the C3-symmetric ligand to Zn2+ could be difficult, because the complex formation is almost stoichiometric in both cases (the binding constants are too large to be determined accurately). However, the coordination ability of ImH2 should be potentially stronger than the C3-symmetric ligand, because the (ImH2)m(Im3Bz)2(Zn2+)3 complex was finally dissociated in the presence of 12 equivalents of ImH2 (Scheme 3E, Fig. 4, 5, and 7).
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and additional data. See DOI: 10.1039/c9sc00399a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*