
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterodinuclear zinc and magnesium catalysts for epoxide/CO2 ring opening copolymerizations†
Gemma
Trott
a,
Jennifer A.
Garden
 b and
Charlotte K.
Williams
*a
b and
Charlotte K.
Williams
*a
aChemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
bSchool of Chemistry, University of Edinburgh, EH9 3FJ, UK
First published on 26th March 2019
Abstract
The ring-opening copolymerization of carbon dioxide and epoxides is a useful means to make aliphatic polycarbonates and to add-value to CO2. Recently, the first heterodinuclear Zn(II)/Mg(II) catalyst showed greater activity than either homodinuclear analogue (J. Am. Chem. Soc.2015, 137, 15078–15081). Building from this preliminary finding, here, eight new Zn(II)/Mg(II) heterodinuclear catalysts featuring carboxylate co-ligands are prepared and characterized. The best catalysts show very high activities for copolymerization using cyclohexene oxide (TOF = 8880 h−1, 20 bar CO2, 120 °C, 0.01 mol% catalyst loading) or cyclopentene oxide. All the catalysts are highly active in the low pressure regime and specifically at 1 bar pressure CO2. The polymerization kinetics are analysed using in situ spectroscopy and aliquot techniques: the rate law is overall second order with a first order dependence in both catalyst and epoxide concentrations and a zero order in carbon dioxide pressure. The pseudo first order rate coefficient values are compared for the catalyst series and differences are primarily attributed to effects on initiation rates. The data are consistent with a chain shuttling mechanistic hypothesis with heterodinuclear complexes showing particular rate enhancements by optimizing distinct roles in the catalytic cycles. The mechanistic hypothesis should underpin future heterodinuclear catalyst design for use both in other (co)polymerization and carbon dioxide utilization reactions.
Introduction
The alternating copolymerization of carbon dioxide and epoxides yields aliphatic polycarbonates (CO2-PC) which contain 30–50 wt% CO2.1,2 It is a useful means to add-value to a common industrial waste and to reduce greenhouse gas emissions from polymer manufacture.3,4 Low molar mass CO2-PC products have properties which allow them to replace petrochemical polyols in the production of polyurethanes.5,6 High molar mass products show promising properties for use as elastomers, rigid plastics, scratch resistant coatings and even anti-microbial materials.7–9 Future product enhancements and the widespread uptake of these materials still depend upon the development of more highly active and selective catalysts.10–15 Heterogeneous catalysts can be highly active but yield much lower CO2 content in the resulting polymers.1 On the other hand, homogeneous catalysts can produce perfectly alternating copolymers but remain restricted to a relatively narrow ligand/metal scope.10–13 Amongst the most active homogeneous catalysts are Cr(III), Co(III) or Zn(II) complexes coordinated by ligands such as salens,13,16–19 phenoxy-amines,20–22 and β-diiminates.23–28Zn(II) or Mg(II) catalysts are attractive due to the relatively low cost, toxicity, lack of colour and redox activity of these metals.20,22 Most research has focussed on zinc complexes and detailed studies indicate many of the best catalysts operate by dinuclear mechanisms.15,23–31 For example, optimization of di-zinc β-diiminate catalysts over the last decade has led to very high activities and structure–activity studies indicate that rates depend upon the intermetallic separation and ligand flexibility.25–28 In a few cases, it has been shown that replacing the Zn(II) metal with Mg(II) in a dinuclear complex increases the activity whilst reducing weight which is particularly desirable for any larger-scale application.22,32 Currently the most active catalysts are all homodinuclear complexes, i.e. featuring two identical metal centres.15,23–31 In 2015, our group reported the first heterodinuclear catalyst, 1, featuring both Zn(II) and Mg(II) metals which are coordinated by a symmetrical macrocycle (Scheme 1).33,34 Complex 1 shows significantly higher activity than either the di-zinc or di-magnesium complexes or than mixtures of them.34 Recently some other hetero-catalysts have been reported including a series of zinc tris(lanthanide) complexes some of which show excellent activities at 10 bar pressure.35–37
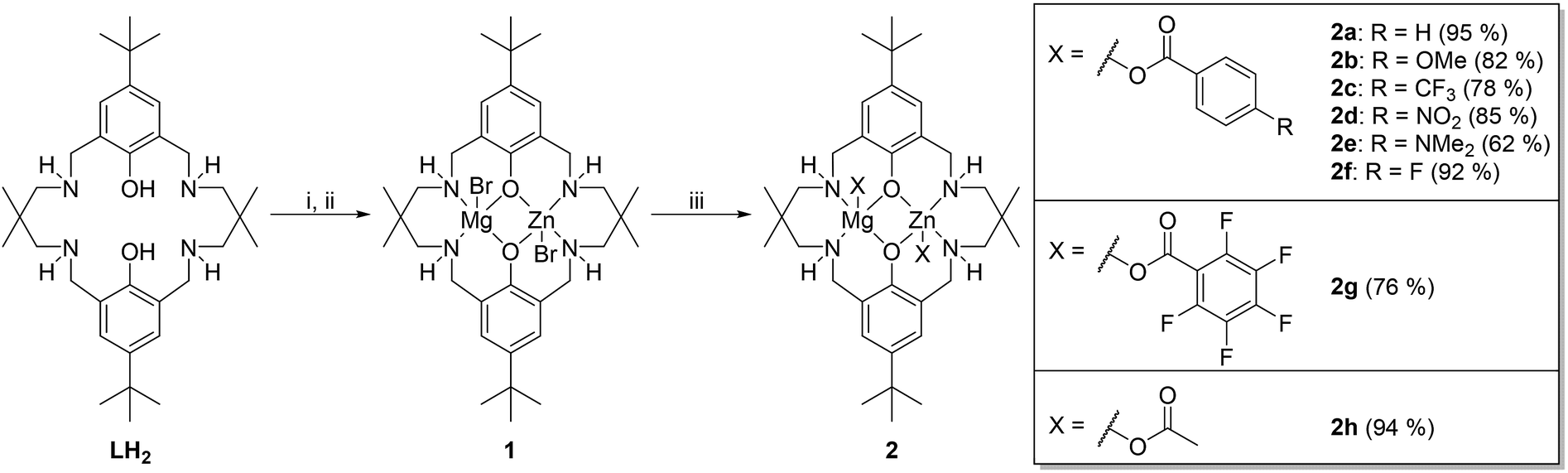 | ||
| Scheme 1 The synthesis of a series of hetero-Zn(II)/Mg(II) catalysts. Reagents: (i) ZnEt2, THF, 25 °C, 16 h; (ii) MgBr2, THF, −78 °C → RT, 1 h; (iii) KX, THF, RT, 16 h. Isolated yields in parentheses. | ||
In general, there are very few reports of the synthesis or properties of well-defined heterodinuclear Zn(II)/Mg(II) complexes in the literature, perhaps because these metals are known to be labile, similarly sized and to show equivalent coordination chemistry.38–41 In order to properly compare and understand the significance of heterodinuclear synergic relationships it is important to use symmetrical ligands but this provides a further difficulty in terms of complex synthesis. In short it requires a means to mono-metallate a symmetrical ancillary ligand.
Despite their limited synthetic precedent di-Zn(II)/Mg(II) complexes are invoked as actives sites in conjugate addition reactions,42 as highly reactive agents in Pd(II) catalyzed cross-coupling reactions,40 in carbonyl addition reactions39 and in stoichiometric fluoroarene substitution reactions.43 Several metalloenzymes also feature heterodinuclear Zn(II)/Mg(II) cores, for example DNA polymerases, aminopeptidases and alkaline phosphatases, and structural studies have been undertaken to model these active sites.44–46 These broader applications for heterodinuclear Zn(II)/Mg(II) complexes underscore the importance of both developing their synthetic chemistry and improving the understanding of reactivity.
Results and discussion
Heterodinuclear complex synthesis
The synthesis of 1 was achieved by the reaction of LH2 with, first, diethyl zinc and, second, magnesium bromide (Scheme 1). The reaction improves upon a previously published route in that both steps can now be conducted sequentially and in the same pot by using only THF as the solvent. The improved synthesis allowed the isolation of complex 1 at larger scale (multi-gram scale) and in yields of >90%. By carrying out metathesis reactions using complex 1 a series of 8 new heterodinuclear catalysts featuring benzoate co-ligands were isolated. All the potassium carboxylate metathesis reactions proceeded to high conversions and yielded only the heterodinuclear complex. In a few cases the isolated yields are slightly lower than expected on the basis of the reaction conversion (determined by aliquot analysis) due to differences in the amount of solvent needed to fully remove the potassium bromide by-product.All the complexes were analysed using NMR spectroscopy, mass spectrometry and elemental analyses (see ESI†). All the 1H NMR spectra (d4-methanol) show 8 distinct methylene resonances and this lack of symmetry is particularly diagnostic of heterodinuclear complex formation (Fig. S1†). In comparison, the C2-symmetric di-zinc or di-magnesium complexes show only four methylene resonances.20,22,34,47,48 The N–H resonances could not be identified when using d4-methanol as the NMR solvent, presumably due to rapid H/D exchange. The 1H NMR spectra were also determined in d8-THF and although all the methylene resonances were broadened, due to fluxional processes, the phenolate aromatic signals show two distinctive coupled doublets. In all cases the coupling was confirmed by COSY NMR. These two doublets are an important additional indicator of heterodinuclear complex formation since the homodinuclear analogues show singlet resonances and mixtures of homodinuclear complexes show two resonances but without any coupling (Fig. S2†).47 The metathesis reactions do not result in any significant metal re-distribution reactions. To use these complexes in polymerization catalysis it is essential to understand the extent, if any, of aggregation in solution. DOSY NMR spectra were determined for all complexes in THF solution so as to mimic the conditions for copolymerization reactions which are conducted in neat epoxide. In all cases a single diffusion coefficient was observed and, when benchmarked against known zinc complexes, the molar masses are in line with the formation of discrete dinuclear complexes (Fig. S3–S10†).
CO2/CHO polymerization catalysis
The new complexes were tested under a common set of conditions used previously to evaluate homodinuclear epoxide/CO2 catalysts: specifically at 0.1 mol% catalyst loading, in neat epoxide, 80 °C and at 1 bar pressure of CO2.22,34 The low pressure polymerizations were all conducted in Schlenk tubes with magnetic stirring. The catalytic activity was assessed by running reactions for fixed time periods and characterizing the extent of epoxide conversion. This allows determination of both the turn-over-number (TON) and turn-over-frequency (TOF). In all cases, polycarbonates were produced with a high proportion of carbonate linkages and showed carbon dioxide uptake >99% (where 100% corresponds to a completely alternating copolymer). Furthermore, in all cases the selectivity for polycarbonate was very high with no cyclic carbonate by-product detected by either 1H NMR or IR spectroscopy.The heterodinuclear catalysts are all significantly more active than homodinuclear analogues featuring the same co-ligands. For example, 2h has a TOF of 98 h−1 whereas the di-zinc complex achieves 18 h−1 and di-magnesium 30 h−1, under the same conditions.22,34 The activity also differs depending on the nature of the carboxylate co-ligand and even shows reproducible differences with only minor changes to the benzoate co-ligands (Table 1, entries 2–7). The most active catalyst, 2d (p-nitro-benzoate), is ∼3 times more active than the least active catalyst, 2g (pentafluorobenzoate).
| # | Cat. | TONb | Initiation (min)c | TOF (h−1)d | CO2e (%) | Polymerf (%) | k obs (×10−5 s−1)g | M n [Ð]h |
|---|---|---|---|---|---|---|---|---|
a Reactions were run using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 catalyst:CHO, in neat CHO ([CHO]0 = 9.88 M), 80 °C, 1 bar pressure of CO2, 6 h or where ‡ shown for 3 h, stirring rate = 600 rpm.
b TON = number of moles of cyclohexene oxide consumed/number of moles of catalyst (see ESI for more information).
c Determined using ATR-IR spectroscopy as the time taken for the on-set of polycarbonate absorptions (Fig. S11–S19).
d TOF = TON/polymerization time, i.e. corrected to account for the initiation period; the average error in TOF is ±4%.
e Determined from the 1H NMR spectrum by comparing normalised integrals for carbonate (4.65 ppm) and ether (3.45 ppm) resonances (Fig. S29).
f Determined from the 1H NMR spectrum by comparing normalised integrals for polycarbonate (4.65 ppm) and cyclic carbonate (4.00 ppm) resonances (Fig. S29).
g Determined by an initial rates method (see ESI and Fig. S20–S28).
h Determined by SEC, in THF, calibrated with narrow molar mass polystyrene standards [dispersity values in parentheses] (Fig. S30–S38). :1000 catalyst:CHO, in neat CHO ([CHO]0 = 9.88 M), 80 °C, 1 bar pressure of CO2, 6 h or where ‡ shown for 3 h, stirring rate = 600 rpm.
b TON = number of moles of cyclohexene oxide consumed/number of moles of catalyst (see ESI for more information).
c Determined using ATR-IR spectroscopy as the time taken for the on-set of polycarbonate absorptions (Fig. S11–S19).
d TOF = TON/polymerization time, i.e. corrected to account for the initiation period; the average error in TOF is ±4%.
e Determined from the 1H NMR spectrum by comparing normalised integrals for carbonate (4.65 ppm) and ether (3.45 ppm) resonances (Fig. S29).
f Determined from the 1H NMR spectrum by comparing normalised integrals for polycarbonate (4.65 ppm) and cyclic carbonate (4.00 ppm) resonances (Fig. S29).
g Determined by an initial rates method (see ESI and Fig. S20–S28).
h Determined by SEC, in THF, calibrated with narrow molar mass polystyrene standards [dispersity values in parentheses] (Fig. S30–S38).
|
||||||||
| 1 | 1 | 247 | 160 | 78 | >99 | >99 | 3.0 | 3000 [1.18] |
| 2 | 2a | 407 | 50 | 87 | >99 | >99 | 2.8 | 13300 [1.03] |
| 6400 [1.06] | ||||||||
| 3 | 2b ‡ | 252 | 40 | 108 | >99 | >99 | 3.5 | 8700 [1.04] |
| 3500 [1.18] | ||||||||
| 4 | 2c | 383 | 30 | 89 | >99 | >99 | 3.5 | 10500 [1.03] |
| 4700 [1.13] | ||||||||
| 5 | 2d ‡ | 307 | 20 | 124 | >99 | >99 | 3.8 | 10200 [1.02] |
| 4750 [1.08] | ||||||||
| 6 | 2e ‡ | 258 | 27 | 101 | >99 | >99 | 3.3 | 9980 [1.04] |
| 4200 [1.14] | ||||||||
| 7 | 2f | 437 | 45 | 83 | >99 | >99 | 3.4 | 19000 [1.03] |
| 8000 [1.18] | ||||||||
| 8 | 2g | 199 | 80 | 43 | >99 | >99 | 1.4 | 2800 [1.23] |
| 9 | 2h | 438 | 50 | 98 | >99 | >99 | 3.1 | 12700 [1.04] |
| 5100 [1.16] | ||||||||
One of the more active catalysts, 2b, was also tested for the copolymerization of various other epoxides, including cyclopentene oxide (CPO) at 1 bar pressure of CO2 (Tables S1 and S2†). For CO2/CPO ROCOP showed a slightly lower TOF of 30 h−1 compared to a value using CHO of 108 h−1. Other catalysts have also been reported to polymerize CPO more slowly than CHO perhaps due to differences in ring strain between the two epoxides.19,49–52 The copolymerization was very selective both for polymer (vs. cyclic carbonate) and carbonate linkage formation (>99%). Under optimized conditions (0.01 mol% 2b, 20 bar CO2, 80 °C), a TOF value of 76 h−1 was achieved and poly(cyclopentene carbonate) with molar mass values as high as 42000 g mol−1 was produced. These activities and molar masses are both at the leading end of values reported for CPO polymerizations.19,49–522b was also an efficient catalyst for ROCOP using vinyl-cyclohexene oxide (TOF = 71 h−1 and carbonate selectivity >99%), cyclohexadiene oxide (TOF = 1 h−1, carbonate selectivity >99%) and 3,4-epoxytetrahydrofuran (TOF = 14 h−1, carbonate selectivity >99%) (Table S2†). However, it showed only low activity and selectivity in the ROCOP of PO/CO2 (TOF = 8 h−1, carbonate selectivity = 98%, polymer selectivity = 26%) (Table S2†).
Polymerization kinetics
In order to understand the relative activity of the series of complexes detailed analysis of the polymerization kinetics was undertaken using in situ IR spectroscopy. This analysis is recommended so as to allow determination of rate coefficients which should allow for more accurate comparison of catalyst performances than point kinetic values (TOF).The polymerization rate law was determined using 2e which is one of the most active catalysts of the series. Starting from an idealized rate law dependent upon all reagent concentrations:
| Rate = kp[2e]x[CHO]ypCO2z |
Various experiments were conducted to determine the values for x, y and z. Firstly, the order in catalyst concentration (x) was determined by conducting experiments under pseudo first order conditions and applying catalyst concentrations from 1.5–7 mM. In every case in situ ATR-IR spectroscopy was used to monitor the reactions and the initial rate coefficient, kobs, determined as the gradient of linear fits to the data over the conversion range 0–20% (see Fig. S43–S47† for each data set and associated fit). The plot of kobsvs. [2e] shows a linear fit to the data which is consistent with a first order dependence on catalyst concentration (Fig. 1a). Alternatively, plots of ln(kobs) vs. ln[catalyst] show a gradient of 0.95 which is also consistent with a first order dependence on catalyst concentration.
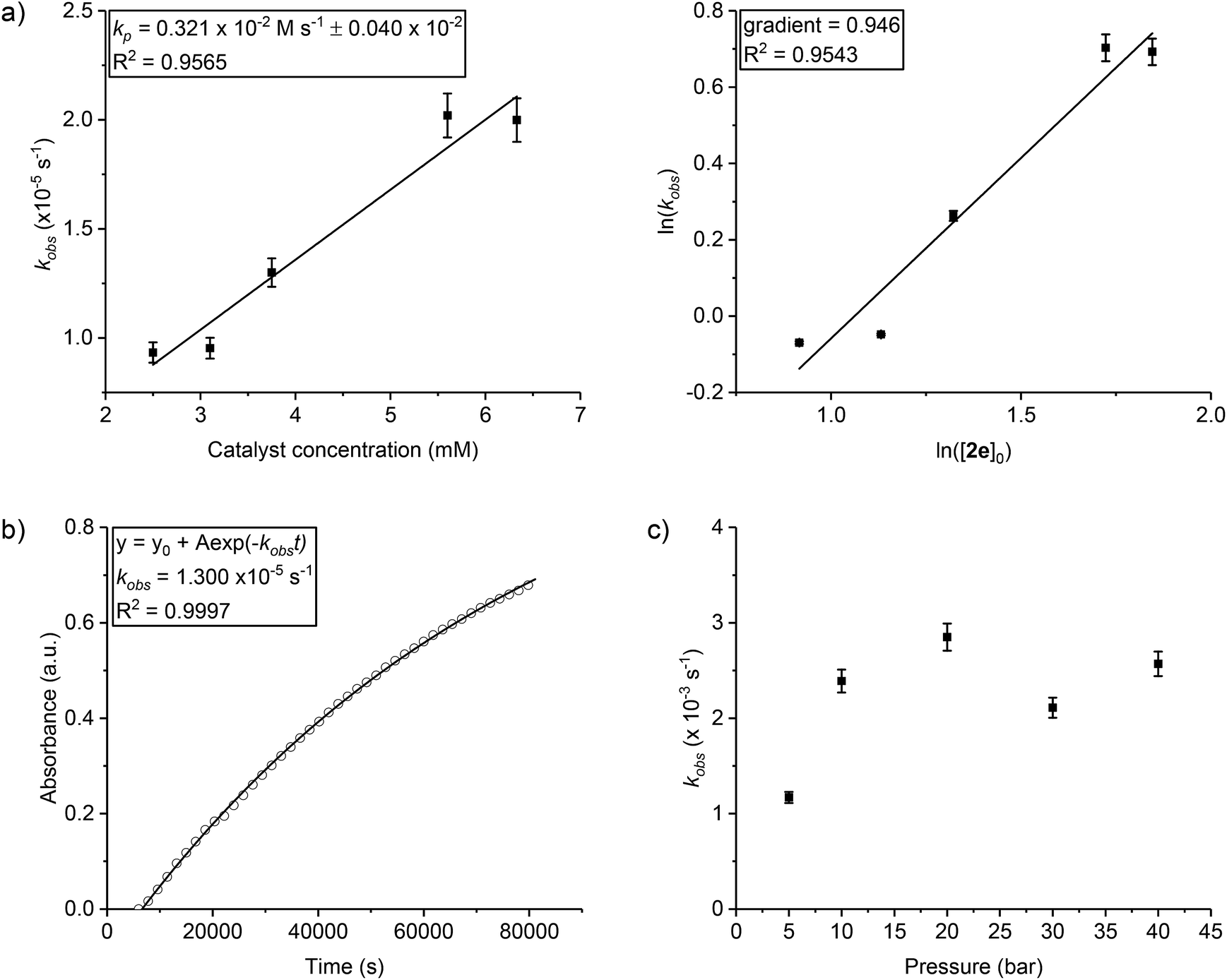 | ||
| Fig. 1 Reaction kinetic analysis to determine order in (a) catalyst (first order); (b) epoxide, CHO (first order); (c) CO2 pressure (zero order) for catalyst 2e. Reaction conditions: [CHO]0 = 5 M in diethyl carbonate, [2e]0 = 5 mM (for plots b and c), temperature = 80 °C (for plots a and b) or 120 °C (for plot c), CO2 pressure = 1 bar (for plots a and b). For clarity only every 30th data point is shown for plot b. 5% error bars shown and further details on data collection methods and analysis are available in the ESI.† | ||
The order in epoxide concentration was determined by an integrated rate law method and involved analysis of the polymerization from 0–65% conversion (after which point viscosity is sufficiently high as to become a limiting factor). Analysis of the absorbance vs. time data shows an exponential growth in the absorption intensity assigned to the polycarbonate. Applying an exponential fit to the data demonstrates that the reaction is first order in epoxide concentration and shows a kobs value of 1.3 × 10−5 s−1. It is notable that the rate coefficients obtained using either the initial rates method or the integrated rate law treatment show very good agreement which gives confidence in using the initial rates method to compare catalyst activities (e.g. for catalyst 2e the integrated rate law, kobs = 1.30 × 10−5 s−1vs. initial rates method, kobs = 1.44 × 10−5 s−1). In order to determine the order in carbon dioxide pressure a series of runs were conducted in a high pressure autoclave equipped with an ATR-IR in situ probe. The experiments were conducted at 120 °C and, therefore, show overall faster rates than equivalent studies at 80 °C. Plots of rate coefficient, kobs, vs. CO2 pressure show a weak correlation between rate and pressure only up to 10 bar and thereafter values are broadly similar (Fig. 1c and S48–S52†). The apparent increment in activity from 5–10 bar is mostly likely due to an experimental limitation: it was observed from the reactor pressure gauge that very rapid carbon dioxide consumption occurred particularly in the early stages of polymerization but the reactor has no automatic refill mechanism to top-up the consumed CO2 and maintain constant pressure. The relative change in CO2 concentration as the reaction progresses is therefore much more likely to be significant at 5 bar than at higher pressures. The results suggest that there is a zero order dependence in carbon dioxide pressure over the range 10–40 bar and indicate a zero-order dependence at lower pressures if CO2 concentration were maintained. The rate law for catalyst 1 was also determined, since it carries a slightly different halide co-ligand: it showed the same second order rate law (Fig. S53–S55†).
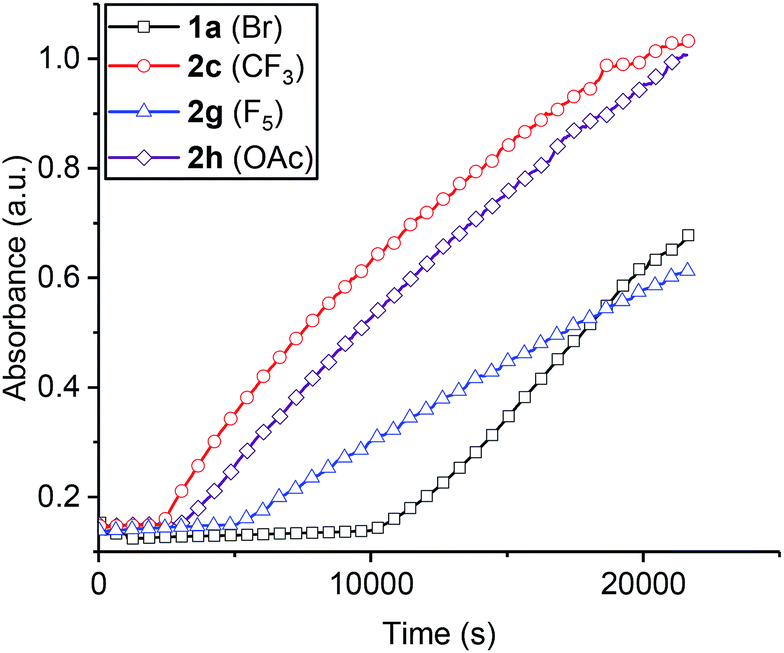 | ||
| Fig. 2 Raw data plots for selected catalysts (co-ligands) to illustrate the significance of initiation time and propagation rates (the data for all catalysts are shown in the ESI and Fig. S11–S19†). For clarity every 10th data point is shown. | ||
Overall, the kinetic data indicate a second order rate law and show a first order dependence on both catalyst and epoxide concentration but a zero order dependence on carbon dioxide (10–40 bar). It is proposed that the rate determining step is metal carbonate attack on coordinated epoxide. A feasible mechanistic interpretation for the reaction kinetics is that the polymerization follows the chain shuttling mechanism which is discussed later (Scheme 2).29,30,33
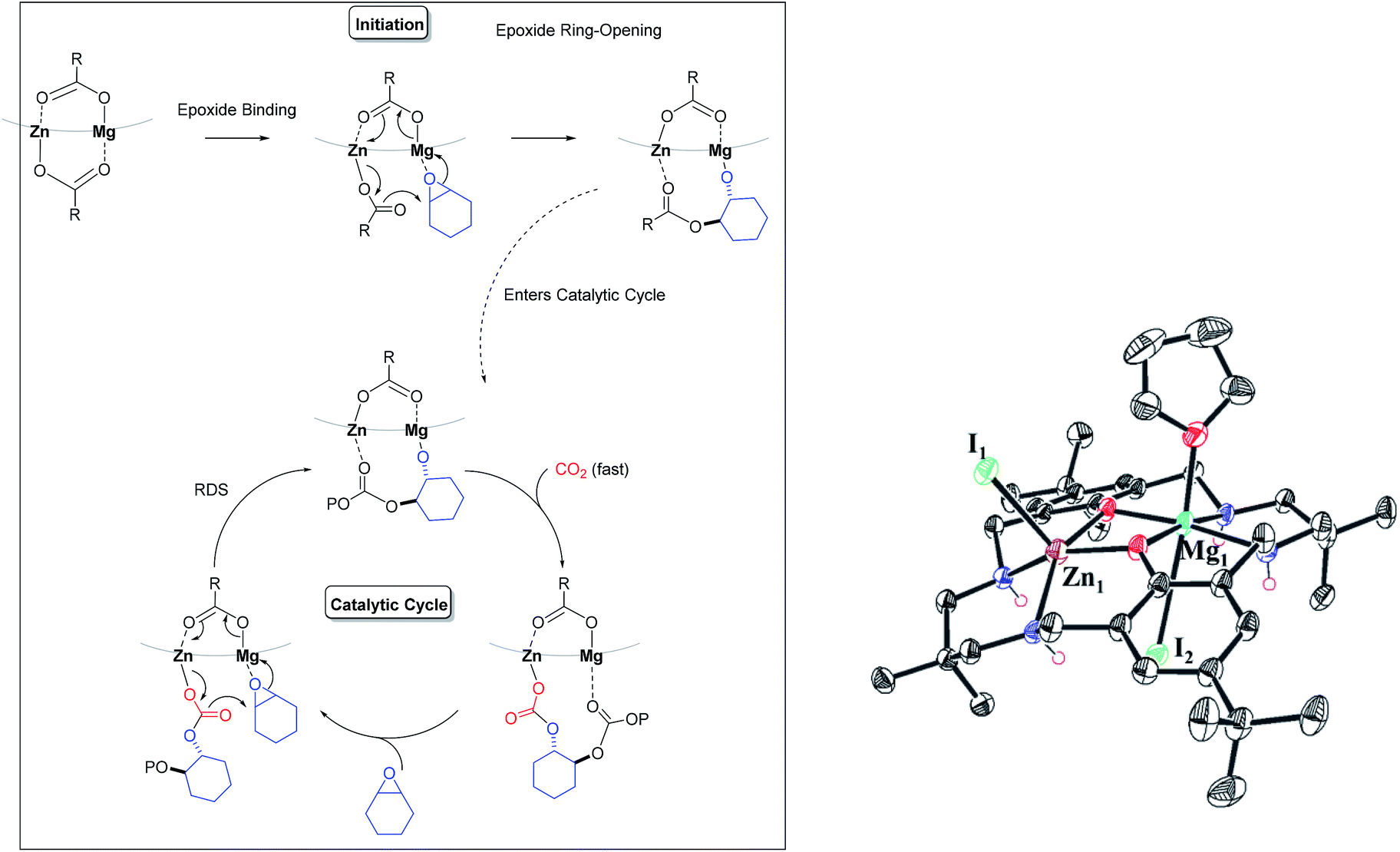 | ||
| Scheme 2 LHS: chain shuttling mechanism proposed for the heterodinuclear catalysts. The ligand framework is shown as a light grey curved line both for clarity and to reflect the finding that the ligand adopts this conformation in solid state structures (with N–H substituents being on the same, convex face of the molecule).29R is acetate- or (substituted)benzoate-co-ligand and P stands for growing polymer chain. RHS shows the recently reported structure of a related Zn(II)/Mg(II) heterodinuclear complex with THF coordinated at the Mg(II) centre.47 | ||
To compare the polymerization behaviour of the series of new complexes, in situ reaction analyses and kinetic fits were applied to all 8 catalysts. The initial rates method was used to fit the data and to differentiate between initiation periods and propagation rates for the catalysts (Fig. S20–S28†). All the catalysts showed initiation periods, during which <5% conversion was observed, and the length of these initiation times was variable (Fig. 2, Table 1). This induction period could be attributed to the dissociation of the bridging carboxylate ligand, providing a coordinative vacancy for epoxide binding, although other processes cannot be excluded such as rates of CO2 dissolution48 and/or epoxide hydrolysis to form diol species.54 Following the initiation period, the polymerization conversion vs. time data over the range 0–20% conversion was linearly fit, enabling determination of the initial rate coefficient (Table 1). Although the series showed different initiation times, nearly all the values fall within the range 2.8–3.8 × 10−5 s−1. There is no obvious correlation between electron donating/withdrawing properties of the benzoate co-ligand and propagation rate. The exception is catalyst 2g which features a pentafluorobenzoate co-ligand and shows a substantially slower polymerization rate. In terms of propagation rates, the largest difference corresponds to the three fold difference between 2g (pentafluorobenzoate) and 2d (nitro-benzoate). Generally, the higher the propagation rate, the shorter the initiation period although there are cases where this correlation breaks down (Table 1, entries 6 and 7).
The similarity between the propagation rate coefficients appears, at first sight, be contrary to the chain shuttling mechanistic hypothesis which suggests that one benzoate ligand remains coordinated during propagation (Scheme 2). There are two alternative interpretations of the activity data: either the chain shuttling mechanism needs fine-tuning to reflect that initiation occurs from both sites (i.e. the co-ligand only influences initiation) or most of these benzoate co-ligands show marginal electronic differences and thus there is no significant moderation of propagation rate with these co-ligands. In support of the second rationale is the observation that the NMR resonances for nearly all the benzoate complexes show very similar chemical shifts, i.e. the para-substituent on the benzoate ligand does not strongly influence the catalyst electronics. Complex 2g (pentafluorobenzoate) is, once again, a clear exception as its carbonyl resonance is significantly shifted upfield which indicates a different electronic environment compared to the other benzoates. The most consistent interpretation of the data for complex 2g is to invoke the chain shuttling mechanism since this allows for the co-ligand to influence both rates of initiation and propagation (Scheme 2). Future research could be directed towards investigating other co-ligands with strongly differing electronic properties. It should be noted, however, that in terms of combining the best catalyst rates with simple process conditions the selection of carboxylate ligands is desirable as they are highly active and air-stable complexes.
Catalytic conditions
Next, the influences of the reaction conditions on activity and selectivity were explored. These studies were conducted using three of the best performing catalysts (2b, c, e) at 1 bar (Table 2). Increasing the reaction temperature from 80 to 120 °C resulted in a significant increase in the catalytic activity and TOF values reached ∼400 h−1 for all three catalysts. Even at this elevated reaction temperature there was no evidence for any by-product formation and high proportions of carbonate linkages were observed. Using high temperature conditions and an autoclave to ensure sufficient carbon dioxide concentration (20 bar) allowed the loading of catalyst 2c to be reduced significantly and resulted in a very high TOF value of 8800 h−1. The catalyst even retained acceptable activity at a loading as low as 1:20000 (2c:CHO).
| Cat. | Catalyst (mol%) | Temp. (°C) | Pressure (bar) | PCHCb (%) | TONb | TOF (h−1)b | M n [Ð]b |
|---|---|---|---|---|---|---|---|
| a Reactions were carried out in a Parr high pressure vessel with an impeller at 20 bar. b See Table 1 and ESI for all data (Fig. S56–S60). | |||||||
| 2b | 0.1 | 120 | 1 | >99 | 435 | 377 | 12280 [1.04] |
| 5340 [1.13] | |||||||
| 2c | 0.1 | 120 | 1 | >99 | 466 | 419 | 14490 [1.06] |
| 5930 [1.15] | |||||||
| 2e | 0.1 | 120 | 1 | >99 | 645 | 430 | 21760 [1.04] |
| 9090 [1.15] | |||||||
| 2c | 0.01 | 120 | 20 | >99 | 4415 | 8830 | 44400 [1.04] |
| 21200 [1.05] |
|||||||
| 2c | 0.005 | 120 | 20 | >99 | 5435 | 1359 | 54380 [1.04] |
| 26550 [1.04] |
|||||||
Polymerization control
The catalysts also showed good polymerization control, as assessed by linear increases in polymer molar mass vs. conversion (Fig. 3b). The polycarbonates generally showed bimodal molar mass distributions, albeit with narrow dispersity in each distribution. Such bimodality has frequently been observed previously in the literature and has been extensively studied.4,22,53,54 Bimodality commonly results because of a side-reaction between residual water and cyclohexene oxide to generate 1,2-cyclohexanediol (CHD).55 Since all chains propagate at the same rate those initiated from mono-functional groups (i.e. catalyst carboxylates) show approximately half the molar mass of chains initiated from di-functional groups (i.e. the diol).21,54–56 MALDI-ToF analysis of the chain end groups substantiates this hypothesis since two series of chains are observed due to carboxylate and hydroxyl telechelic chains, respectively. When reactions were conducted with the addition of 10 equivalents of CHD or water, monomodal molar mass distributions were observed with narrow dispersity. The MALDI-ToF spectra, under these conditions, showed only chains end-capped with hydroxyl groups (Fig. 3a). The excess chain transfer agent reduces slightly the activity and productivity of the catalyst, in particular when using excess water, possibly due to accelerated catalyst decomposition pathways (Table 3).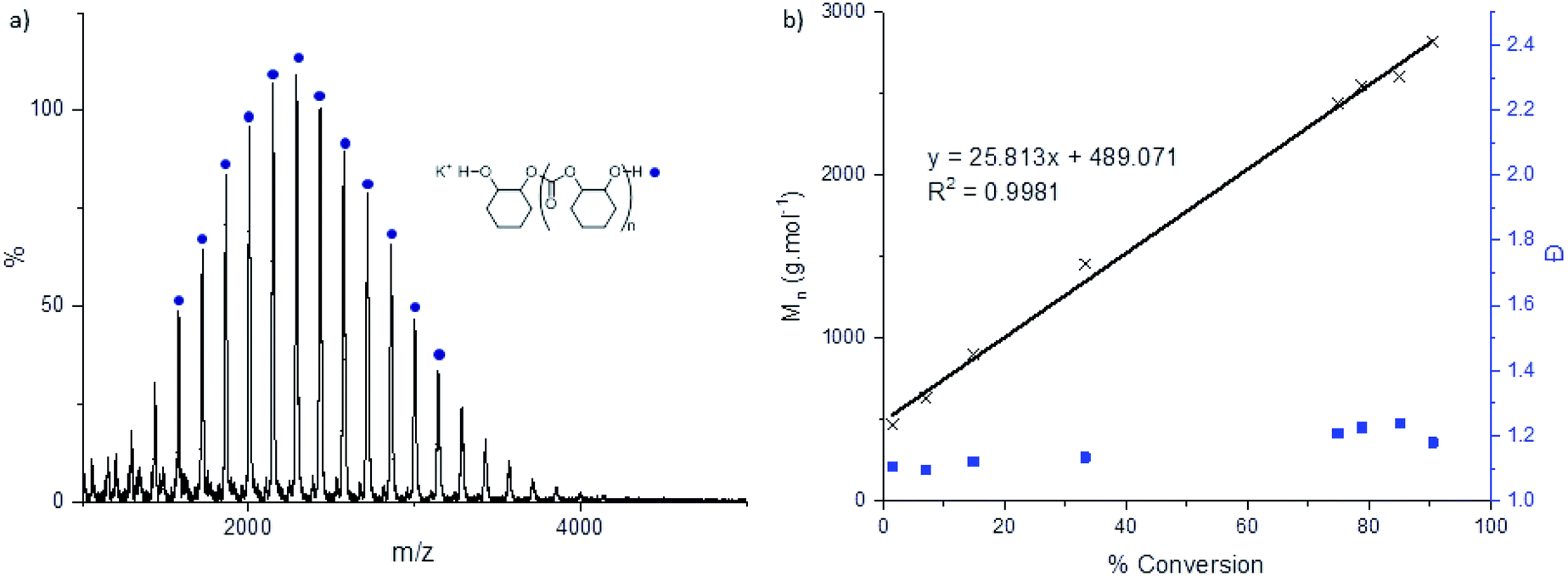 | ||
| Fig. 3 (a) MALDI-ToF mass spectrum of poly(cyclohexene carbonate) formed from catalyst 2c (0.1 mol%) with 10 equivalents of added CHD. (b) Plot of the polycarbonate Mn (black crosses) and Ð (blue squares) versus % CHO conversion. Reaction conditions: 2b/CHO/CHD = 1/1000/10, 80 °C, 5 M in toluene (Fig. S65–S72†). | ||
| Entry | Catalyst | CTA (eq.) | CO2b (%) | Polymerb (%) | TONb | TOF (h−1)b | M n [Ð]b |
|---|---|---|---|---|---|---|---|
|
a Reactions were run using a catalyst:CHO molar ratio of 1:1000 at 80 °C under 1 bar pressure of CO2 for 6 hours.
b See Table 1 and ESI for all data (Fig. S61–S64).
c CHD = 1,2-cyclohexanediol.
d Catalyst and CHO were stirred in air for 1 h at 25 °C before purging the system with CO2 and heating to 80 °C.
|
|||||||
| 1 | 2c | None | >99 | >99 | 634 | 106 | 7000 [1.04] |
| 3400 [1.07] | |||||||
| 2 | 2c | H2O (10) | >99 | >99 | 274 | 46 | 600 [1.14] |
| 3 | 2c | CHD (10)c | >99 | >99 | 440 | 73 | 1700 [1.10] |
| 4 | 2b | None | >99 | >99 | 559 | 93 | 2870 [1.13] |
A significant benefit of this series of compounds is their air-stability and to demonstrate how this could be exploited a reaction was conducted using 2b, dissolved in CHO, which was stirred in air for 1 h prior to addition of CO2. The polymerization was successful and resulted in good activity and selectivity values (Table 3). The resulting polymer showed a monomodal molar mass distribution without requiring any addition of chain transfer agent.
Discussion
Overall this series of catalysts shows high activities and selectivity and performs successfully in the low CO2 pressure regime. In order to provide context to the activity and selectivity values these new heterodinuclear catalysts are compared against some of the most successful literature catalysts (Fig. 4). The best Zn(II)/Mg(II) catalyst (2d, TOF = 124 h−1) is significantly more active than either the di-Zn(II) or di-Mg(II) analogues (18 h−1 and 30 h−1, respectively, under equivalent conditions).33 The activity is also higher than previously reported di-Zn(II) catalysts coordinated by a non-macrocyclic ligand (TOF < 3 h−1).57,58 The activity matches that of proline ligated di-Zn(II) catalysts (TOF ∼149 h−1)59 but is lower than optimized Co(III)-salen catalysts (TOF = 263 h−1).60 One advantage of these Zn(II)/Mg(II) heterodinuclear catalysts is that very high CO2 and polymer selectivity is maintained even at elevated temperatures which contrasts with simple Co(III) salen catalyst systems that are well-known to be thermally unstable with respect to cobalt reduction and catalyst death. For the Zn(II)/Mg(II) complexes, the best activity value reaches 654 h−1 which is at the upper end of values for the low pressure regime.10,14,15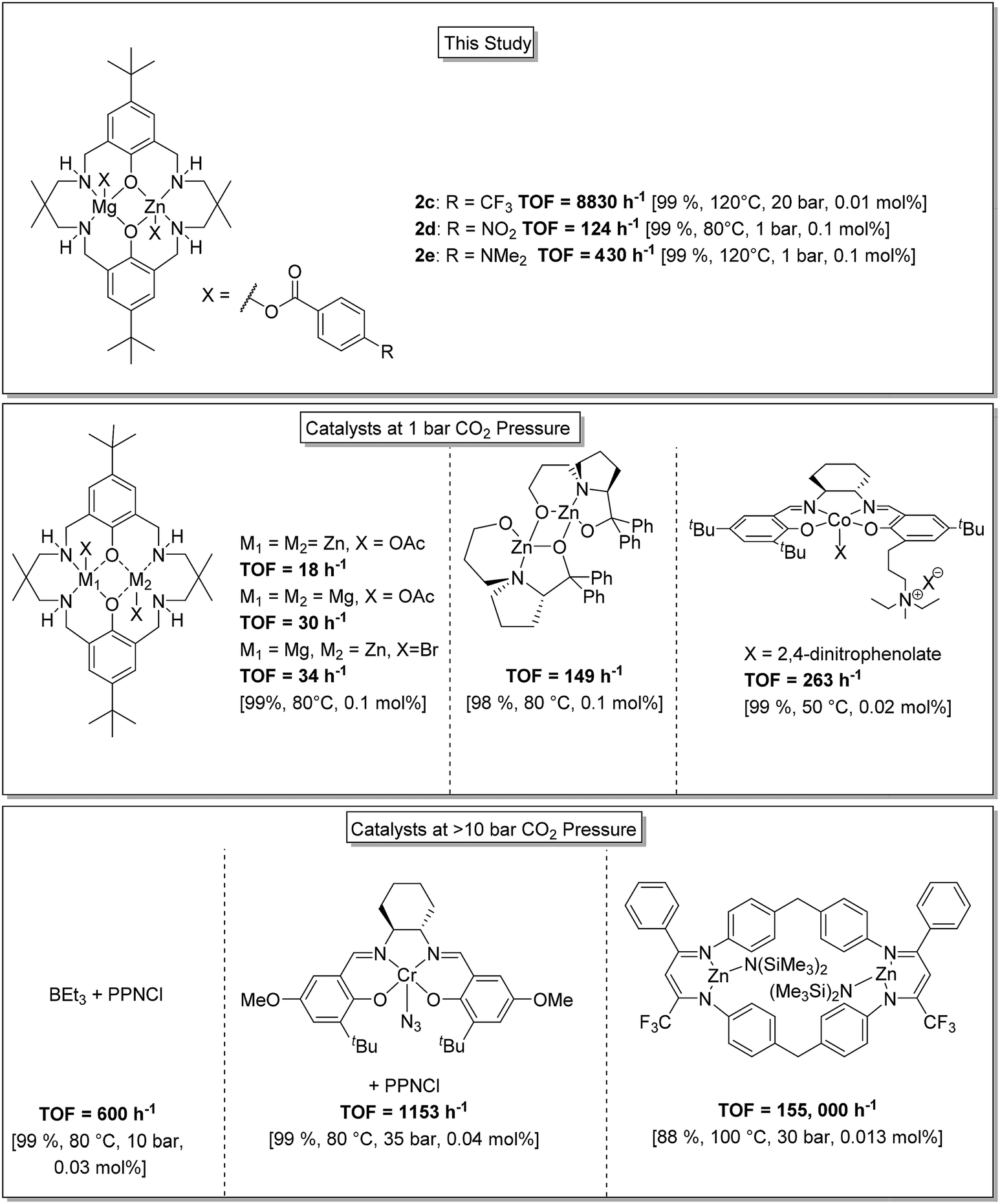 | ||
| Fig. 4 Illustration of the structures, activity and selectivity for some of the highest performing catalysts reported for CO2/CHO ROCOP.22,25,33,34,59,61 | ||
At higher CO2 pressures (20 bar), catalyst 2c showed an activity of 8800 h−1. This value significantly exceeds the performance of tetranuclear Zn(II)/Ln(III) catalysts (TOF = 200–300 h−1, 10 bar).36 It is also higher than a series of new organocatalysts, formed in situ by reaction between boranes and ionic salts (TOF = 600 h−1, 10 bar),61 and higher than for optimized Cr(III)-salen catalyst systems (1153 h−1).62 Indeed, the activity is only surpassed by a tethered di-zinc(II) β-diiminate catalyst (TOF = 155000 h−1), but its selectivity for CO2 is significantly higher (>99% for 2cvs. 80% for di-zinc catalyst).25 The activity values obtained using cyclopentene oxide/CO2 are also high compared to other catalysts.19,49–52
Polymerization mechanism
The polymerization rate law is second order and shows a first order dependence on catalyst and epoxide concentration but appears largely independent of CO2 pressure. The polymerization kinetics are interpreted using a chain shuttling mechanism (Scheme 2).29,33 According to the mechanism, with each monomer insertion step the growing polymer chain moves metal. The rate law indicates the rate determining step is metal carbonate attack on an epoxide molecule coordinated at the second metal. DFT studies indicate that once the epoxide is ring-opened the polymer chain shuttles to the metal originally coordinating the epoxide.29 To balance charge, the carboxylate co-ligand also changes its anionic coordination site to the opposite metal. The fast step in the catalytic cycle is proposed to be carbon dioxide insertion and DFT studies have indicated this step also involves migration of the metal-alkoxide growing polymer chain end from one metal to the other.29 Once again, the carboxylate co-ligand changes its site of anionic coordination to the opposite metal. The proposed mechanism requires that the polymer chain ‘shuttles’ between the two metal centres twice per complete cycle of insertions and that one of the benzoate co-ligands remains coordinated throughout the polymerization and serves to counter-balance charge and polymer chain movement on the opposite face of the catalyst. The results of the structure-kinetic study presented here show only a weak correlation between propagation rate and benzoate co-ligands but this appears most likely due to the limited electronic differences between the benzoates. In the case of the pentafluorobenzoate co-ligand there is NMR spectroscopic evidence for a stronger electronic influence and concomitant evidence of a significant difference in propagation rate. This data can be best interpreted according to the chain shuttling process since the benzoate remains coordinated during propagation and can influence the propagation rate.The chain shuttling mechanism allows for the key roles in catalysis to be differentiated and may also rationalize the improved performances of heterodinuclear catalysts vs. homodinuclear counterparts. Since the rate limiting step is likely metal epoxide attack by the second metal carbonate group, an obvious question is: which metal coordinates the epoxide and which the carbonate? A detailed DFT study was conducted comparing the rate limiting steps for di-zinc, di-magnesium and a Zn/Mg complex (see ESI† for detailed discussion and results). For the heterodinuclear complex the propagation cycles were examined for either zinc-epoxide or magnesium-epoxide coordination pathways. The DFT results substantiated the mechanistic hypothesis by showing a substantially lower barrier for epoxide molecule coordination at Mg(II) compared to Zn(II) [with the epoxide coordinated intermediates showing relative stabilities of −7.7 kcal mol−1 (Mg(II) coordination) vs. −1.4 kcal mol−1, (Zn(II) coordination), respectively]. All the catalysts showed transition state barriers, for the rate limiting step, that were not significantly different, within the error limits for DFT. Although this finding does not allow DFT to provide any further mechanistic insight, it is consistent with the observed differences in rates. Very recently a solid state structure was obtained for another heterodinuclear complex (analogue of complex 1) obtained by growing crystals from a THF solution (Scheme 2b).47 The structure shows the two different metals and has a molecule of THF coordinated at the Mg(II) centre.
Accordingly the chain shuttling mechanistic hypothesis can be fine-tuned for Zn(II)/Mg(II) complexes. The polymer chain is alternately coordinated by the two metal centres. In the rate limiting step the zinc centre provides the carbonate group which attacks the magnesium coordinated epoxide molecule. This results in the formation of a magnesium coordinated alkoxide group which undergoes fast CO2 insertion and forms a zinc carbonate intermediate. As the Zn(II)/Mg(II) complexes show significantly greater rates than di-zinc or di-magnesium analogues it is also clear that the proximity of the magnesium centre labilizes the zinc carbonate group. This could occur through electronic communication through the phenolate oxygen atom which bridges the two metal centres and provides a means to control electron density and hence reactivity at the metals.63,64 The mechanistic findings from these hetero-Zn(II)/Mg(II) catalysts are expected to be more broadly applicable to other dinuclear CO2 copolymerization catalysts. A clear recommendation is to prepare Zn(II)/Mg(II) complexes of dinucleating bis(β-diiminate) ligands which would be expected to show even higher activity than the best dizinc(II) complexes. The chain shuttling pathway is also expected to apply to other polymerization processes and can be exploited to enhance rates of epoxide/anhydride alternating polymerizations and epoxide/heterocumulene processes. Generally there is intense interest and motivation to activate carbon dioxide in chemistry and the concepts illustrated in this work of reagent shuttling and activation between two dissimilar metal sites as a means to increase catalytic performances warrant investigation for other CO2 utilization processes.
Conclusions
The successful isolation of 8 new heterodinuclear Zn(II)/Mg(II) complexes results in highly active catalysts for CO2/epoxide alternating copolymerization. All the new complexes show very good performances both in terms of activity (TOF = 8800 h−1) and selectivity (>99% CO2 uptake over the entire temperature range). The catalysts are amongst the most active reported both at low CO2 pressure (1 bar) and in the higher pressure regime (>10 bar). The polymerization kinetics, analysed using in situ IR spectroscopy, showed an overall second order rate law. The rates were first order dependent on catalyst and epoxide concentrations but independent of carbon dioxide pressure (1–40 bar). Polymerization control was high in all cases and the reaction conditions were controlled to allow the preparation of polycarbonate polyols which are relevant to polyurethane manufacture. Overall, these heterodinuclear catalysts warrant further investigation both as catalysts for polymerizations and for a range of organic transformations, such as conjugate additions, fluorinations and even hydrolysis reactions. In terms of CO2/epoxide alternating copolymerization catalysis there is still significant scope to moderate both the metals, ligands and co-ligands so as to optimize performances. Heterodinuclear complexes of Zn(II)/Mg(II) using other dinucleating ancillary ligands should be explored as a means to increase activity and maintain high overall selectivity and control.Conflicts of interest
Charlotte K. Williams is a director of Econic Technologies.Acknowledgements
The EPSRC (EP/L017393/1; EP/K014668/1) and Climate KIC (Project EnCO2re) are acknowledged for research funding.Notes and references
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed
.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
- N. v. d. Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC
- S. H. Lee, A. Cyriac, J. Y. Jeon and B. Y. Lee, Polym. Chem., 2012, 3, 1215–1220 RSC
- T. Stoesser, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC
- M. A. Subhani, B. Kohler, C. Gurtler, W. Leitner and T. E. Muller, Angew. Chem., Int. Ed., 2016, 55, 5591–5596 CrossRef CAS PubMed
- Y. Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS
- A. W. Kleij, M. North and A. Urakawa, ChemSusChem, 2017, 10, 1036–1038 CrossRef CAS PubMed
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 19 CrossRef PubMed
- J. A. Garden, Green Mater., 2017, 5, 103–108 Search PubMed
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC
- S. J. Na, S. Sujith, A. Cyriac, B. E. Kim, J. Yoo, Y. K. Kang, S. J. Han, C. Lee and B. Y. Lee, Inorg. Chem., 2009, 48, 10455–10465 CrossRef CAS PubMed
- Y. Liu, W.-M. Ren, J. Liu and X.-B. Lu, Angew. Chem., Int. Ed., 2013, 52, 11594–11598 CrossRef CAS PubMed
- Y. Liu, W.-M. Ren, K.-K. He and X.-B. Lu, Nat. Commun., 2014, 5, 5687–5694 CrossRef PubMed
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS PubMed
- M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed
- M. Reiter, S. Vagin, A. Kronast, C. Jandl and B. Rieger, Chem. Sci., 2017, 8, 1876–1882 RSC
- S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Chem.–Eur. J., 2015, 21, 8148–8157 CrossRef CAS PubMed
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredrickson and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed
-
C. Romain, A. Thevenon, P. K. Saini and C. K. Williams, in Carbon Dioxide and Organometallics, ed. X. B. Lu, 2016, vol. 53, pp. 101–141 Search PubMed
- S. Ghosh, D. Pahovnik, U. Kragl and E. Mejia, Macromolecules, 2018, 51, 846–852 CrossRef CAS
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed
- J. Qin, B. Xu, Y. Zhang, D. Yuan and Y. Yao, Green Chem., 2016, 18, 4270–4275 RSC
- H. Nagae, R. Aoki, S.-n. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS PubMed
- J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC
-
R. E. Mulvey and S. D. Robertson, in Organo-Di-Metallic Compounds, ed. Z. Xi, Springer-Verlag Berlin, Berlin, 2014, vol. 47, pp. 129–158 Search PubMed
- D. R. Armstrong, W. Clegg, P. Garcia-Alvarez, A. R. Kennedy, M. D. McCall, L. Russo and E. Hevia, Chem.–Eur. J., 2011, 17, 8333–8341 CrossRef CAS PubMed
- T. D. Bluemke, W. Clegg, P. Garcia-Alvarez, A. R. Kennedy, K. Koszinowski, M. D. McCall, L. Russo and E. Hevia, Chem. Sci., 2014, 5, 3552–3562 RSC
- M. Westerhausen, Dalton Trans., 2006, 4755–4768 RSC
- B. M. Trost and M. J. Bartlett, Acc. Chem. Res., 2015, 48, 688–701 CrossRef CAS PubMed
- C. Bakewell, B. J. Ward, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 2348–2356 RSC
- P. S. Freemont, J. M. Friedman, L. S. Beese, M. R. Sanderson and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 8924–8928 CrossRef CAS
- S. K. Burley, P. R. David, A. Taylor and W. N. Lipscomb, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 6878–6882 CrossRef CAS
- T. Tanase, J. W. Yun and S. J. Lippard, Inorg. Chem., 1996, 35, 3585–3594 CrossRef CAS
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57(24), 15575–15583 CrossRef CAS PubMed
- C. Romain, J. A. Garden, G. Trott, A. Buchard, A. J. P. White and C. K. Williams, Chem.–Eur. J., 2017, 23, 7367–7376 CrossRef CAS PubMed
- Y. Z. Hua, X. C. Yang, M. M. Liu, X. X. Song, M. C. Wang and J. B. Chang, Macromolecules, 2015, 48, 1651–1657 CrossRef CAS
- D. J. Darensbourg, W.-C. Chung and S. J. Wilson, ACS Catal., 2013, 3, 3050–3057 CrossRef CAS
- D. J. Darensbourg, S.-H. Wei, A. D. Yeung and W. C. Ellis, Macromolecules, 2013, 46, 5850–5855 CrossRef CAS
- L. S. Huang, C. Y. Tsai, H. J. Chuang and B. T. Ko, Inorg. Chem., 2017, 56, 6141–6151 CrossRef CAS PubMed
- M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS
- N. Yi, J. Unruangsri, J. Shaw and C. K. Williams, Faraday Discuss., 2015, 183, 67–82 RSC
- W. J. van Meerendonk, R. Duchateau, C. E. Koning and G.-J. M. Gruter, Macromolecules, 2005, 38, 7306–7313 CrossRef CAS
- Y. Xiao, Z. Wang and K. Ding, Chem.–Eur. J., 2005, 11, 3668–3678 CrossRef CAS PubMed
- Y. Xiao, Z. Wang and K. Ding, Macromolecules, 2006, 39, 128–137 CrossRef CAS
- M. Schutze, S. Dechert and F. Meyer, Chem.–Eur. J., 2017, 23, 16472–16475 CrossRef PubMed
- W.-M. Ren, X. Zhang, Y. Liu, J.-F. Li, H. Wang and X.-B. Lu, Macromolecules, 2010, 43, 1396 CrossRef CAS
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed
- D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144 CrossRef CAS
- L. Mandal and S. Mohanta, Dalton Trans., 2014, 43, 15737–15751 RSC
- S. R. Bayly, E. R. Humphrey, H. de Chair, C. G. Paredes, Z. R. Bell, J. C. Jeffery, J. A. McCleverty, M. D. Ward, F. Totti, D. Gatteschi, S. Courric, R. S. C. Barry, B. R. Steele and C. G. Serettas, J. Chem. Soc., Dalton Trans., 2001, 1401–1414 RSC
Footnote |
| † Electronic supplementary information (ESI) available: All experimental protocols, data for all table entries, spectroscopy and kinetic data. See DOI: 10.1039/c9sc00385a |
| This journal is © The Royal Society of Chemistry 2019 |