
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible perovskite electrocatalysts for oxygen reduction/oxygen evolution†
Kieren
Bradley
a,
Kyriakos
Giagloglou
a,
Brian E.
Hayden
 *ab,
Hugo
Jungius
a and
Chris
Vian
a
*ab,
Hugo
Jungius
a and
Chris
Vian
a
aIlika Technologies, Kenneth Dibben House, Enterprise Road, Southampton Science Park, Southampton, SO16 7NS, UK. E-mail: beh@soton.ac.uk
bChemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 19th March 2019
Abstract
The identification of electrocatalysts mediating both the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are prerequisite for the development of reversible fuel cells and rechargeable metal–air batteries. The question remains as to whether a bifunctional catalyst, or a single catalyst site, will exhibit potentials converging to +1.23 VRHE. Transition metal-based perovskites provide tunable catalysts where site substitution can influence both ORR and OER, however substitution in the pseudo-binary phases results in an anti-correlation in ORR and OER activities. We reveal that LaxMnyNi1−yO3−δ, compositions with lanthanum A-site sub-stoichiometry exhibit reversible activity correlating with the appearance of the Mn3+/Mn4+ redox couple. The Mn3+/Mn4+ couple is associated with Mn4+ co-existing with Mn3+ in the bulk, as La3+ is substituted by Ni2+ at the A-site to create a mixed valent system. We also show that a direct A-site substitution by the Ca2+ cation in LaxCa1−xMnyO3−δ perovskites also results in the creation of Mn4+, the appearance of the Mn3+/Mn4+ redox couple, and a concomitant reversible activity. These results highlight a general strategy of optimizing oxide electrocatalysts with reversible activity.
Introduction
Considerable effort has focused on the search for active and stable electrocatalysts for both the oxygen reduction reaction (ORR) in low temperature fuel cells (LTFC), and the oxygen evolution reaction (OER) which are more cost effective than those containing precious metals. As an additional challenge, there is an increasing interest in electrocatalysts for reversible low temperature fuel cells (RLTFC, also known as unitized regenerative fuel cells, URFC) because of their potential in distributed energy storage: a RLTFC will require catalysts with simultaneously low overpotentials for both ORR and OER. There is also a requirement for reversible ORR/OER electrocatalysts for efficient metal/air batteries.1 While the search for such “bi-functional catalysts” continues, the question also remains as to whether a single catalyst, or indeed a single site at such a catalyst, will produce potentials which converge towards the equilibrium potential of +1.23 VRHE for ORR/OER.Transition-metal oxides, and in particular perovskites, have been investigated as alternatives to the precious metal based catalysts as they can exhibit good OER or ORR activities.2 Studies of stoichiometric perovskites and A-site or B-site substituted perovskites are reported to exhibit a volcano activity dependence based on the common descriptor of the filling of the eg-orbital of the transition metal B-site which determined the enthalpy of formation of adsorbed OH intermediate.3,4 The strategy of using the eg-orbital descriptor to optimise A or B-site substituted perovskites for OER activity has been successful,4 although the approach did not necessarily predict an increase ORR activity for the same catalyst.5 The experimental investigations can be complicated, however, by the fact that there can be a synergic effect between perovskite and carbon supports, and the competition between 4e− reduction, and 2e− reduction to the peroxide intermediate. There appear to be a number of possible factors which are determining the effects of A-site and B-site substitution on OER and ORR activity, and it remains challenging to draw holistic conclusions to identify a unique strategy to optimise catalytic activity.2 The introduction of a small A-site deficiency (5%) in LaFeO3 was reported to give a large increase in OER activity together with a slight improvement in ORR activity:6 this was ascribed to the creation of surface oxygen vacancies, highlighting a potential strategy for enhancing ORR and OER activity.
In the case of manganese oxides, the oxidation state of the bulk appears to strongly influence the ORR activity of the surface, with nano-structured alpha-Mn2O3 exhibiting high ORR and OER activity.7 This bi-functional activity has been rationalized through the application of density functional calculations which also suggest that the active surface for ORR is 1/2 ML HO* on Mn3+ and for OER is O* covered Mn4+.8 Amongst various manganese oxides, carbon supported Mn2O3 exhibited the highest activity for the ORR reaction, and a direct correlation activity and the potential of the Mn4+/Mn3+ redox couple.9 The proximity of the Mn4+/Mn3+ redox couple to the ORR/OER equilibrium potential appears to be an important factor in determining the activity of Mn oxide based catalysts.8
In Mn based perovskites, the oxidation state of Mn at the B-site can be altered by substitution of cations at the A-site, partial substitution of manganese for a transition-metal in a B-site, or by altering the stoichiometry of oxygen. For example, Sr2+ and Ca2+ have been substituted for La3+ at the A-site, converting a proportion of the Mn3+ to Mn4+ on the B-site, resulting in an increase in the ORR activity.10–12 In the case of La1−xSrxMnO3, it was found that either La0.4Sr0.6MnO3 (nominal valence Mn3.6+) or La0.77Sr0.33MnO3 exhibit higher ORR activity compared to the other Sr doped perovskites.10,12 It has also been found that if the perovskite is predominantly Mn4+ this results in reduced ORR activity.3,13 In applications to the Zn–air cell, increasing Ca concentrations in graphite supported La1−xCaxMnO3 resulted in an increase in the ORR activity, with La0.6Ca0.4MnO3 appearing to be the most active catalyst.14 The ORR activity of La0.6Ca0.4MnO3 appeared improved in a number of A-site sub-stoichiometric compositions.15 However, it has also been found that increasing the electron density at the Mn site in LaCaMnO3 results in an increase in ORR activity, with LaMnO3 exhibiting the lowest overpotential for the reaction.16
To understand better the role of A and B-site substitution, and the role of A-site and B-site sub-stoichiometry, in Mn based perovskites, we have investigated both the ORR and OER activities of the LaxMnyNi1−yO3−δ and LaxCa1−xMnyO3−δ perovskites. The intrinsic activity of continuous thin film catalysts has been measured to exclude any effects of a carbon-based support, which may influence the reaction in the alkali environment. We have applied a combinatorial methodology employing metal evaporative sources combined with an oxygen plasma atom source has been shown17 to produce stoichiometric SrTi1−xFexO3−y perovskites which can be screened on chip for ORR and OER activity. The LaMnNiO3 (B-site substituted) system has been studied extensively for other purposes including the magnetic properties of the materials.18–20 Demina et al.21 synthesised a number of compositions across the entire ternary composition space and developed a structural phase diagram. ORR catalytic activity is reported in the alkali environment for the LaNi0.5Mn0.5O3−δ perovskite: LaNi0.5Mn0.5O3−δ exhibits the largest current density and lowest overpotential in the series of LaNi0.5M0.5O3−δ perovskites (M = Ni, Co, Fe, Mn, and Cr).3,22,23 A-site substituted La1−xCaxMnO3 perovskites have also been studied for their ORR electrocatalytic activity in alkaline media (above).
Experimental
Compositional gradient thin film libraries were deposited using a High Throughput Physical Vapour Deposition (HT-PVD) system described in more detail elsewhere.24 This system has previously been used to synthesise a number of different metal oxide perovskites which have subsequently been screened for their ferroelectric25 paraelectric,25–27 dielectric,28 ion conductive29 and electrocatalytic17 properties. The constituent elements were deposited simultaneously using multiple off-axis Knudsen cell (k-cell) or electron beam (e-beam) sources, each source independently shadowed by a partial “wedge” shutter which controls the flux distribution across the substrate. The position of the wedge shutters controls the compositional region being deposited on the substrate. Lanthanum (Absco 99.95%) was deposited from either a high temperature k-cell or an e-beam, nickel (Testborne 99.99%) was deposited from an e-beam, calcium (Alfa Aesar 99.5%) and manganese (Alfa Aesar 99.95%) were deposited from a k-cell. Oxygen atoms were co-deposited at the substrate by means of a plasma atom source (Oxford Applied Research or Mantis). Simultaneous mixing of the elements provides a low kinetic energy pathway to solid state oxide formation enabling a low temperature route to amorphous and crystalline stoichiometric oxides. Shadow masks were used to define confine oxide growth to discrete fields on the electrochemical chip.The thin film libraries were deposited onto 35 × 35 mm Si/SiO substrates (〈100〉 silicon with 100 nm thermal silicon oxide) for composition and X-ray diffraction measurements. For electrochemical measurements, depositions were made on an electrochemical screening chip based on the design described elsewhere,30 but with indium tin oxide (ITO) tracks and pads. The ITO conductive layer was chosen for stability and electrochemical inactivity, and the connecting tracks were passivated by a layer of silicon oxide. All depositions were made with the substrate at 550 °C which was sufficient to produce the crystalline perovskite phases.
Elemental composition was measured by Energy-Dispersive X-ray Spectroscopy (EDX) (Tescan Vega 3 LMU SEM incorporating an Oxford Instruments X-Max 50, 50 mm2 large area SDD detector). The composition and thickness of the films were derived using LayerProbe software from Oxford Instruments. Crystal structures were determined by X-ray diffraction (XRD) (Bruker D8 diffractometer with GADDS detector and an Incoatec Cu Kα spot X-ray source). Compositions are reported in atomic percent (at%) with an experimental error of ±2.5 at%.
Electrochemical measurements were carried out using a glass cell, instrumentation and software specifically developed for high-throughput electrochemical screening.31 Combining the use of a single channel potentiostat and a multi-channel current follower, fast sequential monitoring of the electrode currents is achieved. The electrochemical measurements were performed in a three-compartment glass cell, especially designed to accommodate the high throughput array. A reference electrode (Hg/HgO, Sentek) was mounted in a Luggin capillary with a tip situated a few millimetres away from the electrochemical array. Prior to each set of experiments the potential of the Hg/HgO reference electrode was compared to a Gaskatel HydroFlex reference hydrogen electrode after 10 minutes equilibration in the experimental electrolyte. The counter electrode (a platinum mesh of area ∼4 cm2 and pitch 0.5 mm) was separated from the array under test by a glass sinter. All experiments were conducted in 300 mL of 0.1 M KOH (>99%, Sigma-Aldrich) solution the solution was either used saturated by oxygen or deoxygenated using argon bubbled through the cell. The ignition potentials which are extracted from the cyclic voltammetry measurement of ORR have been extracted at sufficiently low currents in this high throughput configuration32 to avoid influenced from transport limitation of reactants. As part of the study, the resistance of the thin films was measured using a four-point probe. The resistivity of the films (ESI†) was found to be sufficiently low to have a negligible effect on the electrochemical measurements. The catalysts were also subjected to an extended cycling routine of 200 cycles covering the potentials of both the OER and ORR regions in order to ensure that they were stable under the measurement conditions. There was no significant change in the voltammetry after the extended cycling.
Results and discussion
Synthesised thin film catalysts of LaxMnyNi1−yO3−δ which were screened for their electrocatalytic activity (Fig. 1–3) were fully characterised structurally by X-ray diffraction (ESI†). The perovskite is synthesised exclusively at all compositions in the region of the pseudo-binary line, and characterised by the four Bragg peaks (100), (110), (111) and (200). Bragg peaks associated with NiO and MnO2 phases are only observed at compositions with less than 30 at% La, well away from the pseudo-binary line. It is evident that the LaxMnyNi1−yO3−δ perovskites can accommodate a wide range of Mn/La sub-stoichiometry. Along the pseudo-binary composition line there is a linear dependence of the (110) 2θ position which correspond to a gradual change from the cubic LaMnO3 (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) to the rhombohedral LaNiO3 (Rc) lattice. For compositions below ca. 10% Ni, however, there is a large scatter in the data towards higher 2θ values. There is also a range of 2θ values obtained for the cubic LaMnO3 (Pmm) which corresponds to a smaller lattice dimension that would be expected.33 We suggest that this is a result of La3+ substitution by Mn2+ at the A-site. It is interesting to note that in the ternary compositions, there is also a region of La sub-stoichiometric compositions in the Ni poor composition region (up to y = 0.2 in LaxMnyNi1−yO3−δ) where the (110) 2θ angle remains constant near to the value for LaMnO3. A likely explanation for this is that either Ni2+ or Mn2+ ions are also being co-ordinated in the La A-site, leading to a compensation in the lattice size as the B-site is accommodating Ni3+. Moreover, Mn2+ and Ni2+ exhibit same ionic radius (70 pm)34 and could be either Mn2+ or Ni2+ on the A-site. Mn2+ has been substituted on the A-site in the LaMnO3,35,36 in the Ca1−xMnxTiO3
m) to the rhombohedral LaNiO3 (Rc) lattice. For compositions below ca. 10% Ni, however, there is a large scatter in the data towards higher 2θ values. There is also a range of 2θ values obtained for the cubic LaMnO3 (Pmm) which corresponds to a smaller lattice dimension that would be expected.33 We suggest that this is a result of La3+ substitution by Mn2+ at the A-site. It is interesting to note that in the ternary compositions, there is also a region of La sub-stoichiometric compositions in the Ni poor composition region (up to y = 0.2 in LaxMnyNi1−yO3−δ) where the (110) 2θ angle remains constant near to the value for LaMnO3. A likely explanation for this is that either Ni2+ or Mn2+ ions are also being co-ordinated in the La A-site, leading to a compensation in the lattice size as the B-site is accommodating Ni3+. Moreover, Mn2+ and Ni2+ exhibit same ionic radius (70 pm)34 and could be either Mn2+ or Ni2+ on the A-site. Mn2+ has been substituted on the A-site in the LaMnO3,35,36 in the Ca1−xMnxTiO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 and in the Sr1−xMnxTiO3 system.38
37 and in the Sr1−xMnxTiO3 system.38
 | ||
| Fig. 1 The compositional dependence on LaxMnyNi1−yO3−δ electrocatalysts of the charge associated with (A) the Ni2+/Ni3+ couple (integrating the cathodic peak over the potential range 1.2 < VRHE < 1.45). (B) The Ni0/Ni2+ couple (integrating the cathodic peak over the potential range 0.0 < VRHE < 0.6). (C) The Mn3+/Mn4+ couple (integrating the anodic peak over the potential range 0.9 < VRHE < 1.15). The data has been extracted from cyclic voltammetry (0.1 M KOH electrolyte, 5 mV s−1 scan rate, room temperature) recorded in deoxygenated 0.1 M KOH, on a series of LaxMnyNi1−yO3−δ electrocatalyst thin film libraries. | ||
 | ||
| Fig. 2 The compositional dependence of the overpotential measured on LaxMnyNi1−yO3−δ thin film electrocatalysts using cyclic voltammetry for: (A) the Oxygen Reduction Reaction (ORR) calculated from the ignition potential at −20 μA cm−2 in the cathodic scan and (B) the Oxygen Evolution Reaction OER calculated from the ignition potential at 40 μA cm−2. Measurements have been made at a scan speed of 5 mV s−1 in oxygen saturated 0.1 M KOH at 25 °C. The experimental error in the overpotentials is ±10 mV. | ||
 | ||
| Fig. 3 (A) The compositional dependence of the reversibility of the ORR/OER on LaxMnyNi1−yO3−δ thin film electrocatalysts expressed as the difference between the ORR and OER overpotentials (Fig. 2) with an experimental error of ±15 mV. (B) Cyclic voltammograms of the ORR and OER showing the reactions at for selected compositions. Measurements have been made at a scan speed of 5 mV s−1 in oxygen saturated 0.1 M KOH at 25 °C. | ||
We will show that it is this compositional region in which both the Mn3+/Mn4+ redox is evident and the perovskites exhibit the most reversible ORR/OER activity. In the absence of dissolved oxygen, cyclic voltammetry (0.1 M KOH electrolyte, 5 mV s−1 scan rate and room temperature) was used to identify the main redox couples associated with the LaxMnyNi1−yO3−δ perovskites. Some examples of these are shown in ESI† obtained at several compositions. Three of the redox couples can be ascribed to the Ni2+/Ni3+ couple,39 Ni0/Ni2+ couple40 and the Mn3+/Mn4+ couple8 centred at 1.45 VRHE, 0.4 VRHE and 1.0 VRHE respectively. The two pairs of Ni redox peaks were observed to have the largest integrated charge at high nickel concentrations in LaxMnyNi1−yO3−δ, and in the LaNiO3 binary. The compositional dependence of the charge associated with the Ni2+/Ni3+ couple centred at 1.45 VRHE (manifested as sharp anodic and cathodic peaks separated by 100 mV) was assessed by integrating the cathodic peak over the potential range 1.2 < VRHE < 1.45 and is shown in Fig. 1A. The charge for this couple increases with nickel content along the pseudo-binary composition line with the greatest charge identified in the LaNiO3 binary. There is also a region of Ni2+/Ni3+ redox activity, albeit with a much lower charge, in perovskites with lower Ni compositions, at La A-site sub-stoichiometric compositions. This corresponds to the same regions of composition which exhibited a smaller than expected lattice parameter in the diffraction (ESI†) which we associated with Mn2+ or Ni2+ at the A-site: the appearance of the Ni2+/Ni3+ redox in this composition region suggests that there is Ni2+ in the A-site. The compositional dependence of the charge associated with the Ni0/Ni2+ couple centred at 0.4 VRHE (manifested as broad anodic and cathodic peaks separated by ca. 400 mV) was assessed by integrating the cathodic peak over the potential range 0.0 < VRHE < 0.6 and is shown in Fig. 1B. The highest integrated charge is observed along the pseudo-binary composition line, increasing with the Ni content of the perovskite, but is reduced again for LaNiO3.
The compositional dependence of the charge associated with the Mn3+/Mn4+ couple centred at 1.0 VRHE (manifested as broad anodic and cathodic peaks separated by 200 mV) was assessed by integrating the anodic peak over the potential range 0.9 < VRHE < 1.15 and is shown in Fig. 1C. It is observed with the highest charge for perovskites with a lower Ni content, where y > 0.25 in LaxMnyNi1−yO3−δ. The compositional region where it is observed, however, is exclusively for compositions with La A-site sub-stoichiometry (Fig. 1C): the maximum in the Mn3+/Mn4+ charge density for the anodic sweep is observed for La0.85Mn0.7Ni0.45O3−δ. The apparent substitution of La3+ at the A-site by what we believe is likely to be mainly Ni2+ is apparently directly correlated with the appearance of the Mn3+/Mn4+ redox couple. We suggest that it is the multivalent Mn cation environment created by a divalent ion substitution (Ni2+ at the A-site creating a Mn4+ environment at the B-site).
The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) were measured on the catalyst libraries using cyclic voltammetry. Examples of ORR activities in cathodic sweeps of a series of catalysts with pseudo-binary compositions measured in oxygen saturated electrolyte are shown in ESI.† The ORR activity of LaNiO3 is seen to be very poor, and substitution with Mn increases activity, with the ignition potential (−20 μA cm−2) moving to higher values, and the reduction currents increase more steeply. The best performing ORR catalyst are observed for LaMn0.45Ni0.05O3−δ, and ORR activity drops at lower Ni content. The use of planar electrodes without hydrodynamic flow results in the current being mass transfer limited above a small overpotential. Consequently, ignition potential is used to define the capability of the materials as catalysts:17 additionally, to aid interpretation of the results, the ignition potentials are reported as an over potential with respect to the reversible oxygen potential of 1.23 V in Fig. 2A over the compositions investigated. This clearly shows that the compositions of highest ORR activity are for LaMnO3 and increase further for a range of A-site La sub-stoichiometric compositions of LaMn0.45Ni0.05O3−δ.
Examples of OER activities in anodic sweeps of a series of catalysts with pseudo-binary compositions are shown in ESI.† The OER activity is greatest for Ni rich compositions in LaxMnyNi1−yO3−δ, with the lowest activity for compositions at low nickel substitution, it should be noted that the addition of manganese generally has a negative effect on the OER activity. However, as in the case of the ORR reaction, the compositional dependence of the OER reaction is complex. The overpotential for the OER reaction based on the ignition potential (40 μA cm−2) is plotted as a function of composition for the catalysts investigated in Fig. 2B. The OER activity along the pseudo-binary compositions of LaxMnyNi1−yO3−δ decrease with decreasing Ni content, although increase again slightly for LaMnO3.
The compositional dependence of the ORR and OER activities along the pseudo-binary compositions LaxMnyNi1−yO3−δ highlight that there is, in general, an anti-correlation in the activities of the catalysts: overpotentials for OER are lowest for Ni rich compositions, and overpotentials for ORR are lowest for Mn rich compositions. The overpotentials we observe for the ORR active LaMnO3 catalyst, and the OER active LaNiO3 are in good agreement to those reported elsewhere.3,4 It is also clear, by comparison with the main redox activities of Ni and Mn, shown in Fig. 1, that there is no direct correlation of ORR or OER activity with the redox processes identified.
There is, however, a region of compositional space that evidently does not show this anti-correlation in the ORR and OER activities (Fig. 2). Catalysts which exhibit both ORR and OER activities are found for a region of A-site deficient compositions of LaxMnyNi1−yO3−δ. These are for compositions with Ni contents below 50 at%. To identify compositions that exhibit both good OER and ORR activities, i.e. the catalysts exhibiting the most reversible OER/ORR behaviour, Fig. 3A shows the compositional dependence of the difference in the ORR and OER overpotentials for the catalysts investigated. The most reversible catalysts are clearly seen in the region which is expected to be A-site sub-stoichiometric of the perovskite. The most active reversible electrocatalyst corresponds to compositions around La0.85Mn0.7Ni0.45O3−δ. Fig. 3B provide examples of the convergence of the ORR and OER overpotentials towards equilibrium. The strongest performing catalyst (orange) has the lowest ORR over potential, yet unlike the other voltammetry taken from the pseudo-binary tie-line, there is not a correlating poor OER over potential, resulting in a reversible catalyst. La0.85Mn0.7Ni0.45O3−δ exhibits an ORR overpotential even lower than LaMnO3, (considered an excellent ORR electrocatalyst) but now also an overpotential for OER similar to that found for one of the most active OER electrocatalysts Ba0.5Sr0.5Co0.8Fe0.2O3+δ.3,4
Striking is the correlation between catalyst compositions which exhibit the most reversible OER/ORR activities (Fig. 3A), and those which exhibit the highest charge associated with the Mn3+/Mn4+ redox activities (Fig. 1C). There is evidently a role that the Mn3+/Mn4+ couple plays in catalysing both the ORR and OER reactions simultaneously. The reaction sites that support either the ORR or OER reaction along the pseudo-binary compositions of LaxMnyNi1−yO3−δ (but not both reactions simultaneously) are evidently not the same site associated with the Mn3+/Mn4+ which is not observed for these stoichiometries (Fig. 1C).
The association of the Mn3+/Mn4+ redox couple with ORR activity is consistent with the observation that the oxidation state on manganese oxides which can result in this couple, and increase ORR activity.9 The uniqueness of the ability of the Mn3+/Mn4+ to also sustain reversible ORR/OER is also consistent with the arguments that it is OH adsorbed at the Mn3+ site which is active in ORR, and O adsorbed at Mn4+ for the OER activity.8 It will therefore be the proximity of the Mn3+/Mn4+ to the ORR/OER equilibrium potential together with the reversibility of the surface redox which will determine the ultimate reversibility of this reaction at such a bifunctional site.
The explanation for the appearance of the Mn3+/Mn4+ redox couple and the concomitant ORR/OER reversible activity at an A-site sub-stoichiometry compositions in LaxMnyNi1−yO3−δ can be explained for this B-site substituted perovskite if one assumes that Ni3+ can not only be incorporated at the B-site, but can be incorporated as Ni2+ at the A-site.41,42 This is equivalent to the classical divalent substitution of the La A-site which is known to achieve charge balance by inducing similar concentrations of Mn4+.43 It is apparently the creation of Mn4+ in addition to Mn3+ in a mixed valance system that is important in introducing the Mn3+/Mn4+ redox couple. This model is also consistent with the observation that the optimum catalyst has a composition around La0.85Mn0.7Ni0.45O3−δ: the limit of lattice stability with A-site deficiency has been shown to be around an A-site sub-stoichiometry of about 10 at%.42,44,45
In order to test the thesis that A-site sub-stoichiometry of Mn based perovskites can lead to the appearance of the Mn3+/Mn4+ redox couple, and a concomitant increase in the ORR/OER reversible activity, we have also investigated the LaxCa1−xMnyO3−δ perovskites. The inclusion of Ca should result in a direct substitution of the La A-site by the divalent cation.15,46,47 This type of substitution of the A-site by a divalent ion is well known for the formation of a mixture of Mn3+ and Mn4+ cations in the lattice.48–52 A series of the LaxCa1−xMnyO3−δ perovskite libraries were synthesised and characterised for their electrochemical behaviour (Fig. 4–6), and characterised by X-ray diffraction (ESI†). The perovskite (Pnma) was identified over the region of compositions investigated around the pseudo-binary composition line, although the region where the Ca:La ratio was greater than ca. 3:1 catalysts were poorly crystallised. Along the perovskite pseudo-binary composition line LaxCa1−xMnyO3−δ, a continuous A-site substitution in a cubic (Pmm) to orthorhombic (Pnma) perovskite solid solution is exhibited.
 | ||
| Fig. 4 The compositional dependence of the charge associated with Mn3+/Mn4+ redox couple (integrating the anodic peak over the potential range 0.9 < VRHE < 1.15) on LaxCa1−xMnyO3−δ electrocatalysts. The data has been extracted from cyclic voltammetry (0.1 M KOH electrolyte, 5 mV s−1 scan rate, room temperature) recorded in deoxygenated 0.1 M KOH, on a series of LaxCa1−xMnyO3−δ electrocatalyst thin film libraries. | ||
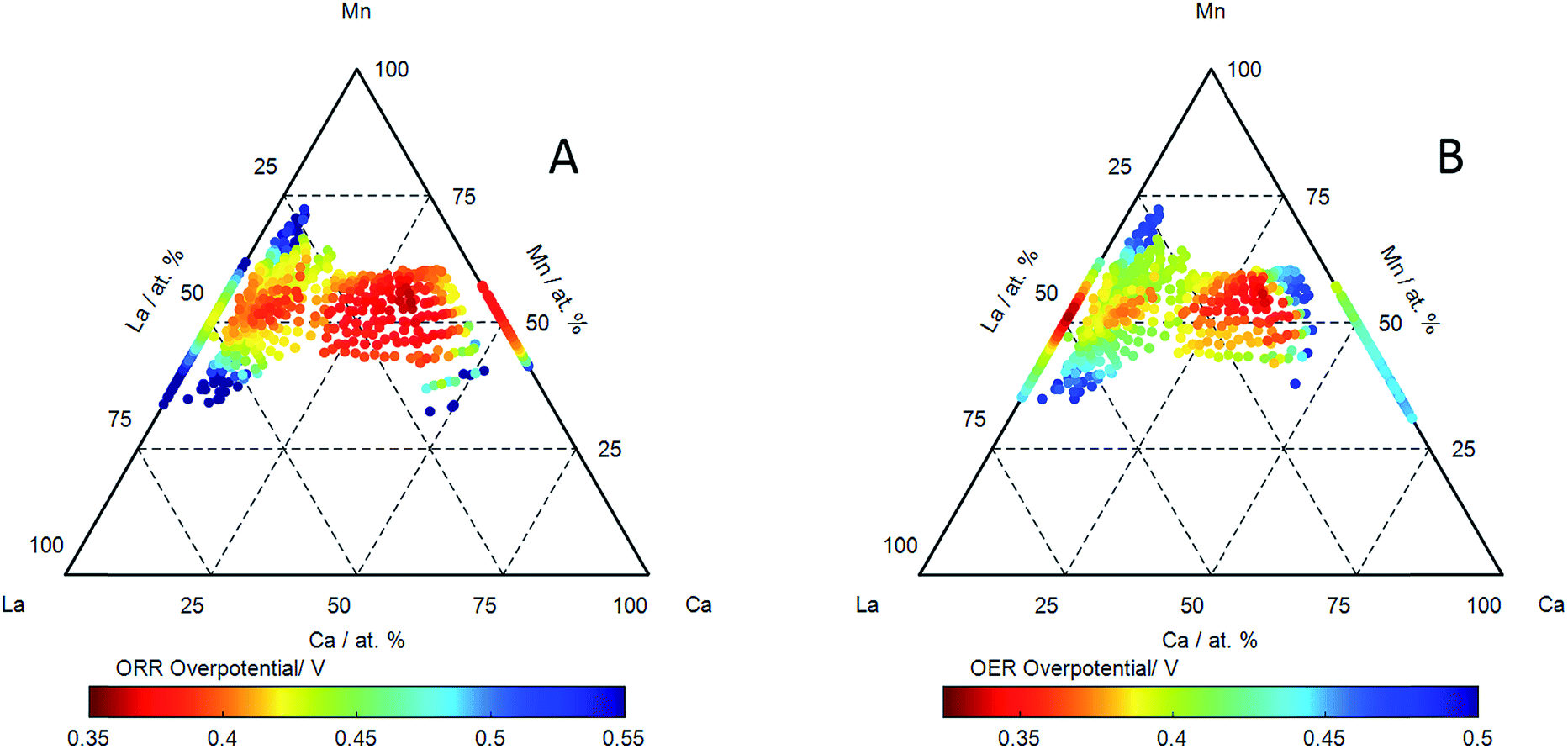 | ||
| Fig. 5 The compositional dependence of the overpotential measured on LaxCa1−xMnyO3−δ thin film electrocatalysts using cyclic voltammetry for: (A) the Oxygen Reduction Reaction (ORR) calculated from the ignition potential at −70 μA cm−2 in the cathodic scan and (B) the Oxygen Evolution Reaction OER calculated from the ignition potential at 40 μA cm−2. Measurements have been made at a scan speed of 5 mV s−1 in oxygen saturated 0.1 M KOH at 25 °C. The experimental error in the overpotentials is ±10 mV. | ||
 | ||
| Fig. 6 (A) The compositional dependence of the reversibility of the ORR/OER on LaxCa1−xMnyO3−δ thin film electrocatalysts expressed as the difference between the ORR and OER overpotentials (Fig. 5) with an experimental error of ±15 mV. (B) Cyclic voltammograms of the ORR and OER showing the reactions at for selected compositions. Measurements have been made at a scan speed of 5 mV s−1 in oxygen saturated 0.1 M KOH at 25 °C. | ||
In the absence of dissolved oxygen, three manganese redox couples can be identified in the cyclic voltammetry (ESI†). The Mn2+/Mn3+, Mn3+/Mn4+ and Mn2+/Mn4+ couples8 centred at 0.6 VRHE, 0.9 < VRHE < 1.0 and 1.1 VRHE respectively, are identified over the compositional range of LaxCa1−xMnyO3−δ investigated. The Mn2+/Mn3+ couple is identified at potentials which shift with composition in the region 0.4 to 0.8 VRHE53 and is present across the LaxCa1−xMnyO3−δ perovskites compositional range. Substitution of Ca in the lattice results in a reduction in the charge associated with the Mn2+/Mn3+ couple. The Mn3+/Mn4+ couple observed at potentials 0.9–1.2 VRHE,8 is in the same potential range where the couple is observed in LaxMnyNi1−yO3−δ (ESI†). With the increasing calcium content, the peak is seen to shift to higher potentials. But most importantly, the integrated charge density of this couple increases with Ca substitution in LaxCa1−xMnyO3−δ. The integrated charge of the cathodic peak of this couple is plotted as a function of composition in Fig. 4. Increasing the Ca content in LaxCa1−xMnyO3−δ along the pseudo-binary composition line results in an increase in the charge associated with the Mn3+/Mn4+ couple with a maximum at La0.3Ca0.7MnO3−δ. A slightly higher maximum in the charge is observed for La0.3Ca0.6Mn1.1O3−δ. At higher concentrations of Ca, this couple decreases in charge, and the Mn2+/Mn4+ couple predominates in CaMnO3, a charge is still observed for the high Ca samples despite a lack of the Mn3+/Mn4+ couple; the Mn2+/Mn4+ peaks are large and broad, thus making complete dissociation difficult. This observation is consistent with the model in which the introduction of a divalent ion such as Ca in the A-site causes a charge deficiency in the structure resulting in the generation of Mn4+ at the B-site.43 As was observed in the case of LaxMnyNi1−yO3−δ (Fig. 3C), the generation of the Mn3+/Mn4+ couple is associated with the formation of Mn4+ in the lattice.
The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) were measured on the catalyst libraries using cyclic voltammetry. Examples of ORR activities (measured in cyclic voltammetry) in cathodic sweeps of a series of catalysts with pseudo-binary compositions measured in oxygen saturated electrolyte are shown in ESI.† An ignition potential for the ORR reaction has been extracted (at a current of −70 μA cm−2) and reported an effective overpotential with respect to the reversible oxygen potential of 1.23 V, and this is shown as a function of composition of LaxCa1−xMnyO3−δ in Fig. 5A. The ORR activity is seen to be good at all compositions along the pseudo-binary compositions of LaxCa1−xMnyO3−δ, however the addition of calcium to LaMnO3 (which shows the highest overpotential) results in a continuous reduction in the ORR overpotential. The lowest overpotential for the ORR reaction was observed for a calcium A-site substituted lattice composition of La0.3Ca0.6Mn1.1O3−δ of 0.362 V, along the pseudo-binary line, the highest overpotential was 0.368 V for La0.3Ca0.7MnO3−δ.
Examples of OER activities (measured in cyclic voltammetry) in anodic sweeps of a series of catalysts with pseudo-binary compositions measured in oxygen free electrolyte are shown in ESI.† The effective overpotential for the OER reaction (extracted from the ignition potential at 40 μA cm−2) as a function of composition in LaxCa1−xMnyO3−δ is shown in Fig. 5B. The overpotential for OER is relatively low on LaMnO3. LaMnO3 also exhibits the highest overpotential for ORR. The introduction of Ca in LaMnO3 initially results in an increase in the OER overpotential in LaxCa1−xMnyO3−δ up to ca. 20% Ca. The ORR reaction exhibits a reduction in overpotential with increase in Ca concentration for these compositions. These compositions (for Ca < 20%) of LaxCa1−xMnyO3−δ are therefore characterised by an anti-correlation of OER and ORR activities. However, a decrease in the overpotential for the OER is observed in LaxCa1−xMnyO3−δ for concentrations above ca. 20 at% Ca: a continuing reduction in ORR overpotential is also observed. This results in a region of LaxCa1−xMnyO3−δ compositions which exhibit a high activity for both OER and ORR reactions. This effect is again best exemplified in a plot of the difference between the OER and ORR potential as a function of composition: this reflects the effectiveness of the catalyst in the reversible reaction. This is shown in Fig. 6A, with some examples of the corresponding OER and ORR voltammograms shown in Fig. 6B. The region of highest reversible activity along the pseudo-binary composition line is centred at La0.3Ca0.7MnO3−δ with the most reversible activity observed for La0.3Ca0.6Mn1.1O3−δ. Comparison of Fig. 6A with Fig. 4 also reveals that the region of composition exhibiting the highest reversible activity also corresponds to the region of compositions exhibiting the most prominent Mn3+/Mn4+ redox activity.
Conclusion
A thin film combinatorial synthetic methodology has been used successfully to synthesise libraries of hundreds of thin film LaxMnyNi1−yO3−δ and LaxCa1−xMnyO3−δ perovskite electrocatalysts. These two ternary perovskites are examples of B-site and A-site substitution along the pseudo-binary composition lines, respectively. X-ray diffraction has been used to characterise the perovskite structures. In both cases, we show there is a large region of compositions away from these lines where a perovskite structure is observed. In the case of LaxMnyNi1−yO3−δ there appears to be a continual distortion of the LaMnO3 cubic structure to a rhombohedral LaNiO3 structure along the tie-line. There is, however, evidence of some A-site substitution of Mn2+ in the LaMnO3, and more extensive A-site substitution of Ni2+ in La sub-stoichiometric compositions of LaxMnyNi1−yO3−δ. In the case of the LaxCa1−xMnyO3−δ perovskites, there appears to be a continual distortion of the LaMnO3 cubic structure (Pmm) to an orthorhombic structure (Pnma) in LaxCa1−xMnyO3−δ, and with a change back to a cubic (Pmm) structure in CaMnO3.
High throughput electrochemical screening has been undertaken a systematic study of the surface redox behaviour, and the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) on the synthesised perovskites. In the case of LaxMnyNi1−yO3−δ compositions along the tie-line, there is an anti-correlation in the ORR and OER activities. We identify, however, a region of catalyst compositions with lanthanum A-site sub-stoichiometry where both high ORR and OER activity is observed, and this also correlates with the appearance of the Mn3+/Mn4+ redox couple. The optimal reversible ORR/OER activity is observed for La0.85Mn0.7Ni0.45O3−δ. This compositional region of reversible ORR/OER elecrocatalytic activity and Mn3+/Mn4+ redox activity also corresponds to compositions where A-site substitution of La3+ by Ni2+ was suggested based on the X-ray diffraction results. We suggest that the appearance of the Mn3+/Mn4+ couple is associated with the creation of Mn4+ in the bulk as La3+ is substituted by Ni2+ at the A-site to create a mixed valent system.
To support this interpretation, we also show that a direct A-site substitution of La3+ by the divalent Ca2+ cation in LaxCa1−xMnyO3−δ perovskites, which is expected to result in the mixed valent Mn2+ and Mn4+ perovskite, also results in the appearance of the Mn3+/Mn4+ redox couple in cyclic voltammetry centred near to the tie-line, with the maximum charge observed for La0.3Ca0.6Mn1.1O3−δ. We also show that the compositional distribution of catalysts which exhibit the most reversible ORR/OER electrocatalysts also correlates with the presence of the Mn3+/Mn4+ redox couple.
In these two La–Mn based perovskite systems investigated, therefore, La A-site substitution by a divalent cation (either Ni2+ or Ca2+) which results in the creation of a mixed valent Mn3+ and Mn4+ bulk lattice also results in the appearance of the Mn3+/Mn4+ redox couple. The appearance of the Mn3+/Mn4+ redox couple is also shown to be responsible for the creation of the most active reversible ORR/OER electrocatalysts. These observations are consistent with ab initio calculations on Mn oxides which predict the active site for ORR being HO* on Mn3+ and for OER is O* on Mn4+.8 It is this single bifunctional site mediating the Mn3+/Mn4+ redox couple in the perovskites resulting in the most reversible electrocatalysis. Mixed valent Mn3+ and Mn4+ in the bulk lattice of the perovskite appears to be sufficient to generate this active site. It is the proximity of the potential of the Mn3+/Mn4+ redox couple to the reversible ORR/OER potential in the alkali environment which results in the reversible activity. We suggest that a similar strategy of A-site substitution could be more widely used in producing mixed cation occupation at the B-site of perovskites generally, and the resulting couple tuned to support single (bifunctional) site reversible ORR/OER activity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the Toyota Research Institute for funding this work.References
- R. Gao, L. Liu, Z. B. Hu, P. Zhang, X. Z. Cao, B. Y. Wang and X. F. Liu, J. Mater. Chem. A, 2015, 3, 17598–17605 RSC.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- E. Fabbri, R. Mohamed, P. Levecque, O. Conrad, R. Kotz and T. J. Schmidt, ChemElectroChem, 2014, 1, 338–342 CrossRef.
- Y. L. Zhu, W. Zhou, J. Yu, Y. B. Chen, M. L. Liu and Z. P. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- N. Ramaswamy, R. J. Allen and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 12650–12664 CrossRef CAS.
- H. Y. Su, Y. Gorlin, I. C. Man, F. Calle-Vallejo, J. K. Norskov, T. F. Jaramillo and J. Rossmeisl, Phys. Chem. Chem. Phys., 2012, 14, 14010–14022 RSC.
- A. S. Ryabova, F. S. Napolskiy, T. Poux, S. Y. Istomin, A. Bonnefont, D. M. Antipin, A. Y. Baranchikov, E. E. Levin, A. M. Abakumov, G. Kéranguéven, E. V. Antipov, G. A. Tsirlina and E. R. Savinova, Electrochim. Acta, 2016, 187, 161–172 CrossRef CAS.
- K. A. Stoerzinger, W. Lu, C. Li, Ariando, T. Venkatesan and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 1435–1440 CrossRef CAS PubMed.
- M. Yuasa, N. Yamazoe and K. Shimanoe, J. Electrochem. Soc., 2011, 158, A411–A416 CrossRef CAS.
- J. Tulloch and S. W. Donne, J. Power Sources, 2009, 188, 359–366 CrossRef CAS.
- K. A. Stoerzinger, M. Risch, J. Suntivich, W. M. Lü, J. Zhou, M. D. Biegalski, H. M. Christen, Ariando, T. Venkatesan and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 1582–1588 RSC.
- J. Hu, L. Wang, L. Shi and H. Huang, J. Power Sources, 2014, 269, 144–151 CrossRef CAS.
- X. Z. Yuan, X. X. Li, W. Qu, D. G. Ivey and H. J. Wang, ECS Trans., 2011, 35, 11–20 CAS.
- V. Celorrio, L. Calvillo, E. Dann, G. Granozzi, A. Aguadero, D. Kramer, A. E. Russell and D. J. Fermín, Catal. Sci. Technol., 2016, 6, 7231–7238 RSC.
- B. E. Hayden and F. K. Rogers, J. Electroanal. Chem., 2018, 819, 275–282 CrossRef CAS.
- M. C. Sánchez, J. García, J. Blasco, G. Subías and J. Perez-Cacho, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 144409 CrossRef.
- M. N. Iliev, H. Guo and A. Gupta, Appl. Phys. Lett., 2007, 90, 151914 CrossRef.
- J. S. Kang, G. Kim, H. J. Lee, S. Kolesnik, B. Dabrowski, H. Lee, J. Y. Kim, J. Lee, B. Kim and B. I. Min, J. Appl. Phys., 2009, 105, 07d721 CrossRef.
- A. N. Demina, V. A. Cherepanov, A. N. Petrov and M. V. Klokova, Inorg. Mater., 2005, 41, 736–742 CrossRef CAS.
- J. Sunarso, A. A. J. Torriero, W. Zhou, P. C. Howlett and M. Forsyth, J. Phys. Chem. C, 2012, 116, 5827–5834 CrossRef CAS.
- X. Liu, X. Liu, J. Meng, C. Yao, X. Zhang, J. Wang and J. Meng, Int. J. Hydrogen Energy, 2016, 41, 22361–22372 CrossRef CAS.
- S. Guerin and B. E. Hayden, J. Comb. Chem., 2006, 8, 66–73 CrossRef CAS PubMed.
- B. E. Hayden and S. Yakovlev, Thin Solid Films, 2016, 603, 108–114 CrossRef CAS.
- A. David, S. Guérin, B. E. Hayden, R. Noble, J.-P. Soulié, C. Vian, I. P. Koutsaroff, S. i. Higai, N. Tanaka, T. Konoike, A. Ando, H. Takagi, T. Yamamoto, T. Fukura and H. Ieki, Cryst. Growth Des., 2014, 14, 523–532 CrossRef CAS.
- M. S. B. Darby, S. Guerin, B. E. Hayden, H.-J. Schreiner and S. Yakovlev, J. Appl. Phys., 2013, 113, 014104 CrossRef.
- C. Yada, C. E. Lee, D. Laughman, L. Hannah, H. Iba and B. E. Hayden, J. Electrochem. Soc., 2015, 162, A722–A726 CrossRef CAS.
- M. S. Beal, B. E. Hayden, T. Le Gall, C. E. Lee, X. J. Lu, M. Mirsaneh, C. Mormiche, D. Pasero, D. C. A. Smith, A. Weld, C. Yada and S. Yokoishi, ACS Comb. Sci., 2011, 13, 375–381 CrossRef CAS PubMed.
- S. Guerin, B. E. Hayden, C. E. Lee, C. Mormiche and A. E. Russell, J. Phys. Chem. B, 2006, 110, 14355–14362 CrossRef CAS PubMed.
- S. Guerin, B. E. Hayden, C. E. Lee, C. Mormiche, J. R. Owen, A. E. Russell, B. Theobald and D. Thompsett, J. Comb. Chem., 2004, 6, 149–158 CrossRef CAS PubMed.
- B. E. Hayden, S. Guerin, D. Pletcher, M. E. Rendall and J. P. Suchsland, J. Comb. Chem., 2006, 8, 679–686 CrossRef PubMed.
- M. J. Sayagués, J. M. Córdoba and F. J. Gotor, J. Solid State Chem., 2012, 188, 11–16 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Horyń, A. Sikora and E. Bukowska, J. Alloys Compd., 2003, 353, 153–169 CrossRef.
- M. Wołcyrz, R. Horyń, F. Bourée and E. Bukowska, J. Alloys Compd., 2003, 353, 170–174 CrossRef.
- W. Pon-On, S. Meejoo, A. Mehtar and I. M. Tang, Ceram. Int., 2011, 37, 2075–2079 CrossRef CAS.
- A. Tkach, P. M. Vilarinho and A. L. Kholkin, Acta Mater., 2005, 53, 5061–5069 CrossRef CAS.
- R. N. Singh, L. Bahadur, J. P. Pandey, S. P. Singh, P. Chartier and G. Poillerat, J. Appl. Electrochem., 1994, 24, 149–156 CAS.
- D. S. Hall, C. Bock and B. R. MacDougall, J. Electrochem. Soc., 2013, 160, F235–F243 CrossRef CAS.
- S. Krishnan, V. Sharma, P. Singh and R. Ramprasad, J. Phys. Chem. C, 2016, 120, 22126–22133 CrossRef CAS.
- C. Singh and M. Rakesh, Indian J. Eng. Mater. Sci., 2009, 16, 288–290 CAS.
- C. Zener, Phys. Rev., 1951, 82, 3 CrossRef.
- C. Doroftei, P. D. Popa and F. Lacomi, Sens. Actuators, A, 2012, 173, 24–29 CrossRef CAS.
- Y. B. Zuo, J. H. Li, J. X. Yi, Z. B. Wang and C. S. Chen, J. Solid State Chem., 2008, 181, 700–704 CrossRef CAS.
- K. Suresh, T. S. Panchapagesan and K. C. Patil, Solid State Ionics, 1999, 126, 299–305 CrossRef CAS.
- S. Ponce, M. A. Pena and J. L. G. Fierro, Appl. Catal., B, 2000, 24, 193–205 CrossRef CAS.
- W. Zhong, C. T. Au and Y. W. Du, Chin. Phys. B, 2013, 22, 11 Search PubMed.
- R. von Helmolt, J. Wecker, B. Holzapfel, L. Schultz and K. Samwer, Phys. Rev. Lett., 1993, 71, 2331–2333 CrossRef CAS PubMed.
- S. Jin, T. H. Tiefel, M. McCormack, R. A. Fastnacht, R. Ramesh and L. H. Chen, Science, 1994, 264, 413–415 CrossRef CAS PubMed.
- Z. Arnold, K. Kamenev, M. R. Ibarra, P. A. Algarabel, C. Marquina, J. Blasco and J. García, Appl. Phys. Lett., 1995, 67, 2875–2877 CrossRef CAS.
- Z. Wei, A. Chak-Tong and D. You-Wei, Chin. Phys. B, 2013, 22, 11 Search PubMed.
- V. Celorrio, E. Dann, L. Calvillo, D. J. Morgan, S. R. Hall and D. J. Fermin, ChemElectroChem, 2016, 3, 283–291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00412b |
| This journal is © The Royal Society of Chemistry 2019 |