DOI:
10.1039/C9SC00323A
(Edge Article)
Chem. Sci., 2019,
10, 4509-4514
Secondary amines as coupling partners in direct catalytic asymmetric reductive amination†
Received
20th January 2019
, Accepted 12th March 2019
First published on 14th March 2019
Abstract
The secondary amine participating asymmetric reductive amination remains an unsolved problem in organic synthesis. Here we show for the first time that secondary amines are capable of effectively serving as N-sources in direct asymmetric reductive amination to afford corresponding tertiary chiral amines with the help of a selected additive set under mild conditions (0–25 °C). The applied chiral phosphoramidite ligands are readily prepared from BINOL and easily modified. Compared with common tertiary chiral amine synthetic methods, this procedure is much more concise and scalable, as exemplified by the facile synthesis of rivastigmine and N-methyl-1-phenylethanamine.
Introduction
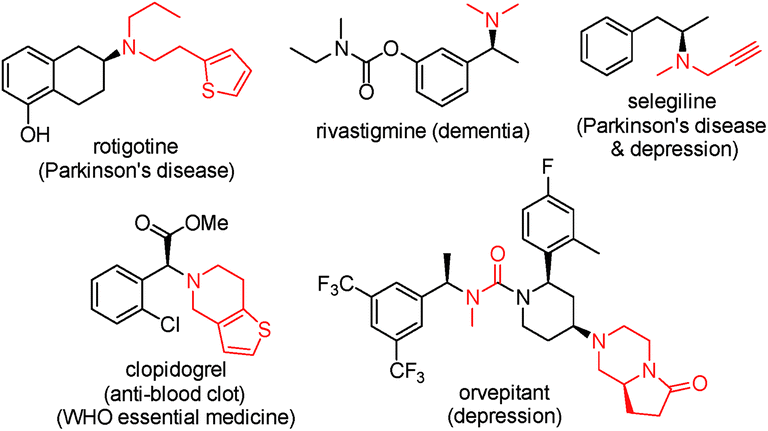
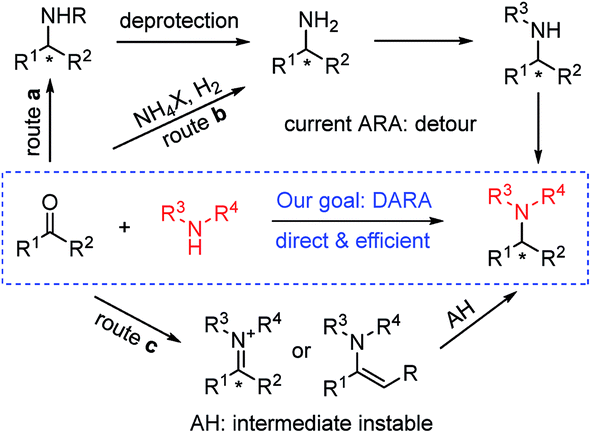
Chiral amines are one of the most important classes of compounds displaying biological activities. In fact, about 40% of new chemical entities (NCEs) among FDA approved drugs contain chiral amine moieties.1 Tertiary amines account for 60% of the total number of medicinal amines (a small selection of chiral ones is shown in Fig. 1).2 Consequently, the development of efficient synthetic methods toward chiral amines, especially chiral tertiary amines, remains atop the organic chemists' priority list and it is an intensively studied and challenging research area.3 In many cases chiral tertiary amines are prepared from the corresponding primary or secondary amines. Among those various methods, the protocols utilizing molecular hydrogen as the reductant, asymmetric hydrogenation (AH) of imine/enamine4 or iminium salts,5 and direct asymmetric reductive amination (DARA),4a,6 are preeminently attractive due to their high atom economy and the vast available chiral ligand pool. In many cases, imines are difficult to prepare by AH and their instability operationally complicates the following reduction (Scheme 1, route c). Currently DARA also takes a circuitous route through primary amines to tertiary amines (Scheme 1, route b) and/or N-deprotection (Scheme 1, route a).
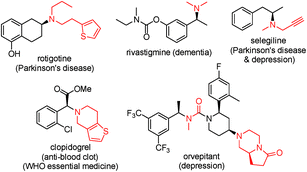 |
| | Fig. 1 Selected chiral tertiary amine drugs. | |
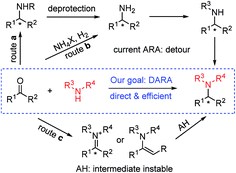 |
| | Scheme 1 Routes for the synthesis of chiral tertiary amines. | |
Reductive amination (RA) is nature's choice to synthesize essential biomonomers including amino acids inside organisms,7 and the most practical method for the artificial synthesis of pharmaceutical related and bulk fundamental amines.8 As for DARA, since its maiden voyage in the preparation of herbicide metolachlor,9 progress has been made through biocatalysis,10 organocatalysis11 and transition-metal catalysis.12 This research journey is highlighted by its successful application in the production of two pharmaceutical drugs, sitagliptin10a and suvorexant.12e Nevertheless, compared with the prevailing utilization of achiral RA in both small-scale synthesis and practical industrial production, DARA lags largely behind. It requires revolutionary improvement in aspects of substrate scope and catalyst reactivity to make DARA a general tool in chiral amine synthesis i.e., there are no reports on secondary amines as the N-source to construct tertiary chiral amines. If secondary amines are successfully applied in DARA as N-sources, chiral tertiary amines could be obtained directly without a detour (Scheme 1). The secondary amine participating RA is considered as a challenge even in an achiral manner and typically requires high reaction temperature (60 °C or above, even upto 120 °C).8c This difficulty probably stems from the steric congestion during the imine/enamine formation and the reduction thereafter. Other challenges include the formation of the alcohol side-product resulting from ketone reduction and the steric-control issue owing to the possible E/Z isomers of the imine/enamine intermediates. We envisioned that two strategies may help to address these problems: (1) the addition of appropriate additives which facilitate the imine/enamine intermediate formation,13 and (2) the application of robust and properly sized chiral ligands which could minimize the inhibitory effect of the amine starting material, imine/enamine intermediate and amine product, and at the same time sterically accommodate the bulky imine/enamine intermediates. Via the proposed route, the efficiency of the synthesis of related important tertiary chiral amine intermediates and active pharmaceutical ingredients (APIs) would be greatly enhanced.
Results and discussion
Establishment of feasibility
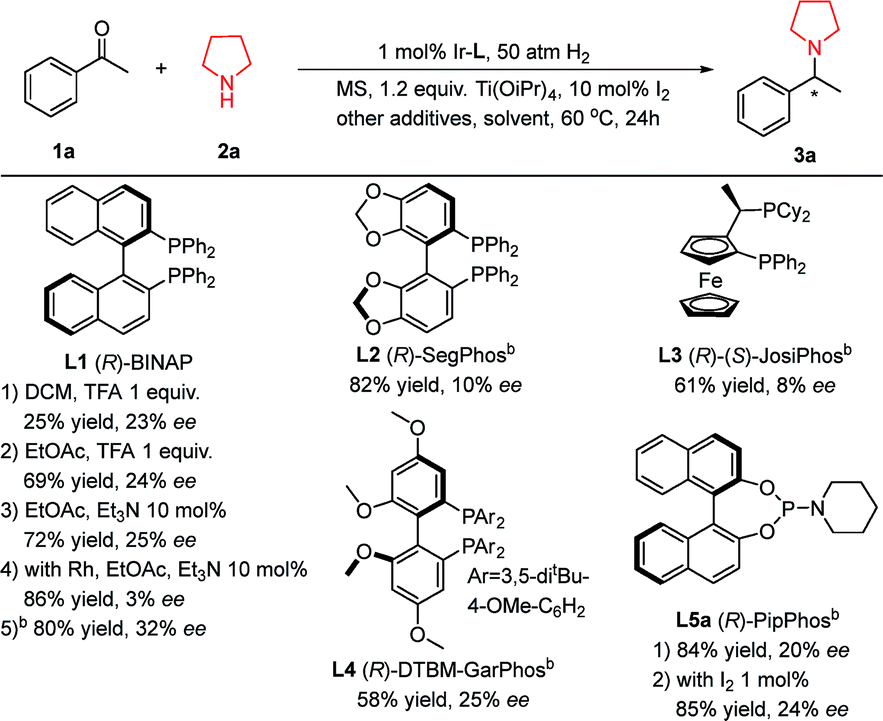
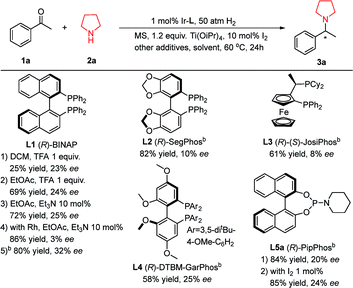
Molecular sieves, Brønsted acids12d and titanium isopropoxide12b have been proven to be efficient additives in DARA to accelerate the formation of imine intermediates. At the same time, molecular iodine is widely used in asymmetric hydrogenation of imines.3b,13 So we selected molecular sieves (MS, 4 Å), titanium isopropoxide (Ti(OiPr)4), and trifluoroacetic acid (TFA) as the additive set with iridium-(R)-BINAP (Table 1, L1) as the catalyst for the DARA of acetophenone 1a and pyrrolidine 2a. Unfortunately the reaction did not afford a satisfactory result (25% yield and 23% ee, Table 1, L1-1). Further investigation revealed that basic Et3N was a better additive than trifluoroacetic acid. With the EtOAc and CH2Cl2 solvent pair, the yield and ee were improved to 80% and 32%, respectively. Rh-(R)-BINAP furnished much lower ee compared with the corresponding iridium catalyst. From the brief screening of several common chiral ligands, none of them engendered satisfactory results. Considering that the BINOL-based phosphoramidite ligands including PipPhos (Table 1, L5a) have been widely applied in asymmetric catalysis,14 and their highly attractive features are that their two components, BINOL and the amino moiety, are easily assembled together, and each part is highly modulated, we decided to focus on this type of ligand. Exploiting the easiness of modification, we can tailor and fine-tune their steric and electronic properties for adapting our specific reaction.
Table 1 Initial DARA investigation of acetophenone and pyrrolidinea
Reaction conditions: Ir–L 1 mol%, [Ir]/L (bisphosphine) = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or [Ir]/L (monophosphine) = 1:2; 1a 0.2 mmol, 2a 0.2 mmol, solvent 2 mL, 60 °C, 20 h; MS = molecular sieves, 0.1 gram; TFA = trifluoroacetic acid; I2 10 mol%; yields and enantiomeric excesses were determined by chiral HPLC.
The solvent was EtOAc:CH2Cl2 = 1:1 with the addition of 10 mol% Et3N. :1 or [Ir]/L (monophosphine) = 1:2; 1a 0.2 mmol, 2a 0.2 mmol, solvent 2 mL, 60 °C, 20 h; MS = molecular sieves, 0.1 gram; TFA = trifluoroacetic acid; I2 10 mol%; yields and enantiomeric excesses were determined by chiral HPLC.
The solvent was EtOAc:CH2Cl2 = 1:1 with the addition of 10 mol% Et3N.
|
|
Evolution of chiral monodentate phosphoramidite ligands
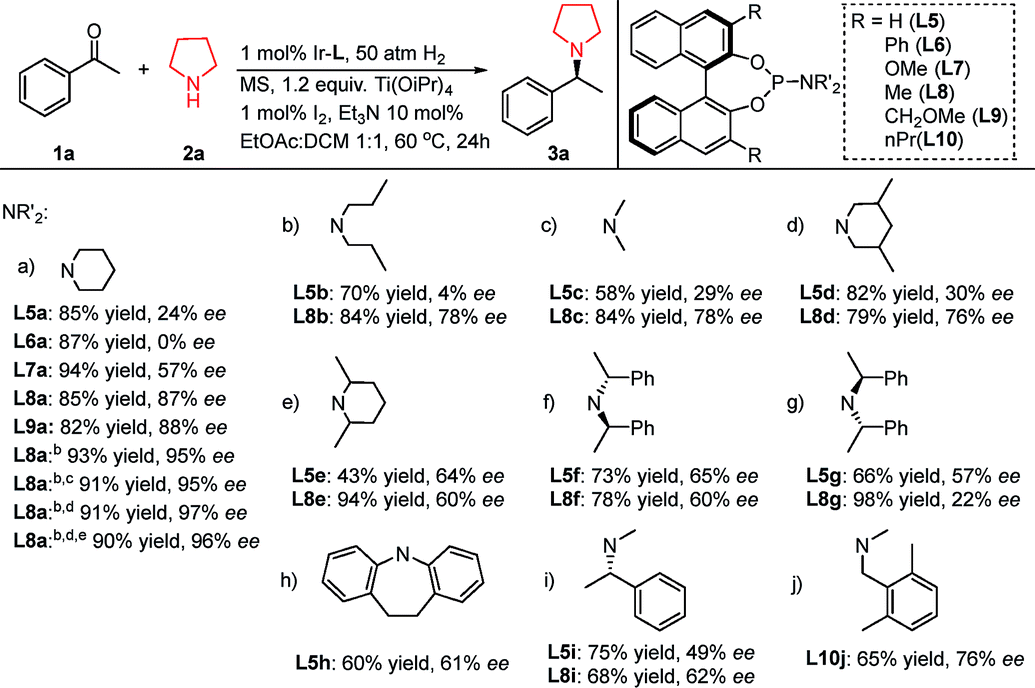
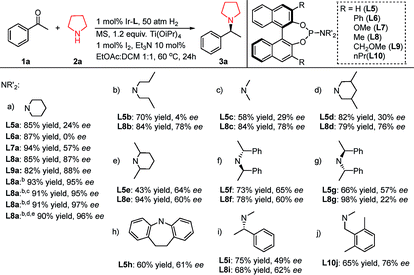
Initially several chiral phosphoramidite ligands (Table 2, L5a–h) with different structural features were synthesized and applied in the reductive coupling of 1a with 2a. It appeared that the bulky amino moiety on L5 positively impacted the enantioselectivity: from L5a to L5e, the ee value was improved from 24% to 60%, then this trend reached a limit (Table 2, L5f, L5h) at around 64% ee. So another strategy was required to further improve the stereoselectivity. Taking full advantage of its easy modification feature, the 3,3′-positions of the BINOL back-bone were embedded with several common substituents (–Ph, –Me, –OMe and –CH2OMe) to synthesize ligands L6a–L9a. Fortunately this approach gave rise to better results. We observed that the enantioselectivity was boosted to 87%, in which L8a afforded the best result (85% yield and 87% ee). The improvement specifically functioned better for the piperidine moiety on the chiral ligands than for other groups (Table 2, L8aversusL8b–L8i). We postulated that it stems from the rigidity of the piperidine ring structure. While the amino moiety possesses chirality, the steric configuration exerted significant influence on the DARA enantioselectivity. Particularly when the 3,3′-positions of the BINOL back-bone were embedded with substituents (Table 2, L8fversusL8g), the ee difference was as high as 38%. Further reaction condition optimization enhanced the ee to 95% by replacing 10 mol% Et3N with the same amount of 1,4-diazabicyclo[2.2.2]octane (DABCO), changing the solvent to THF/DCM/DCE (1:1:1.5), and decreasing the reaction temperature to room temperature. We found that molecular sieves actually exerted no obvious influence on the reaction. With 0.1 mol% of Ir–L8a at lower temperature (0 °C), the reaction still proceeded smoothly to further improve the enantioselectivity to 97%. When the H2 pressure was decreased to 10 atm, the desired product was obtained without noticeable loss of yield or stereoselectivity.
Table 2 Examination of chiral monodentate phosphoramidite ligands in DARA of acetophenone and pyrrolidinea
|
Reaction conditions: [Ir]/L/1a/2a = 1:2:100:100; 1a 0.1 mmol, solvent 2 mL, 60 °C, 20 h; MS = molecular sieves, 0.1 gram; yields were isolated yields; enantiomeric excesses were determined by chiral HPLC.
10 mol% 1,4-diazabicyclo[2.2.2]octane (DABCO) was added instead of Et3N; reaction solvent was DCM/THF/DCE (1:1:1.5); reaction temperature was room temperature.
The H2 pressure was 10 atm.
The reaction temperature was 0 °C.
The catalyst loading was 0.1 mol%.
|
|
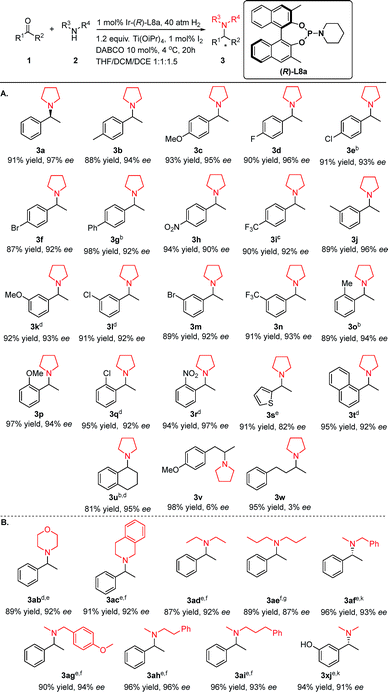
Examination of substrate scope
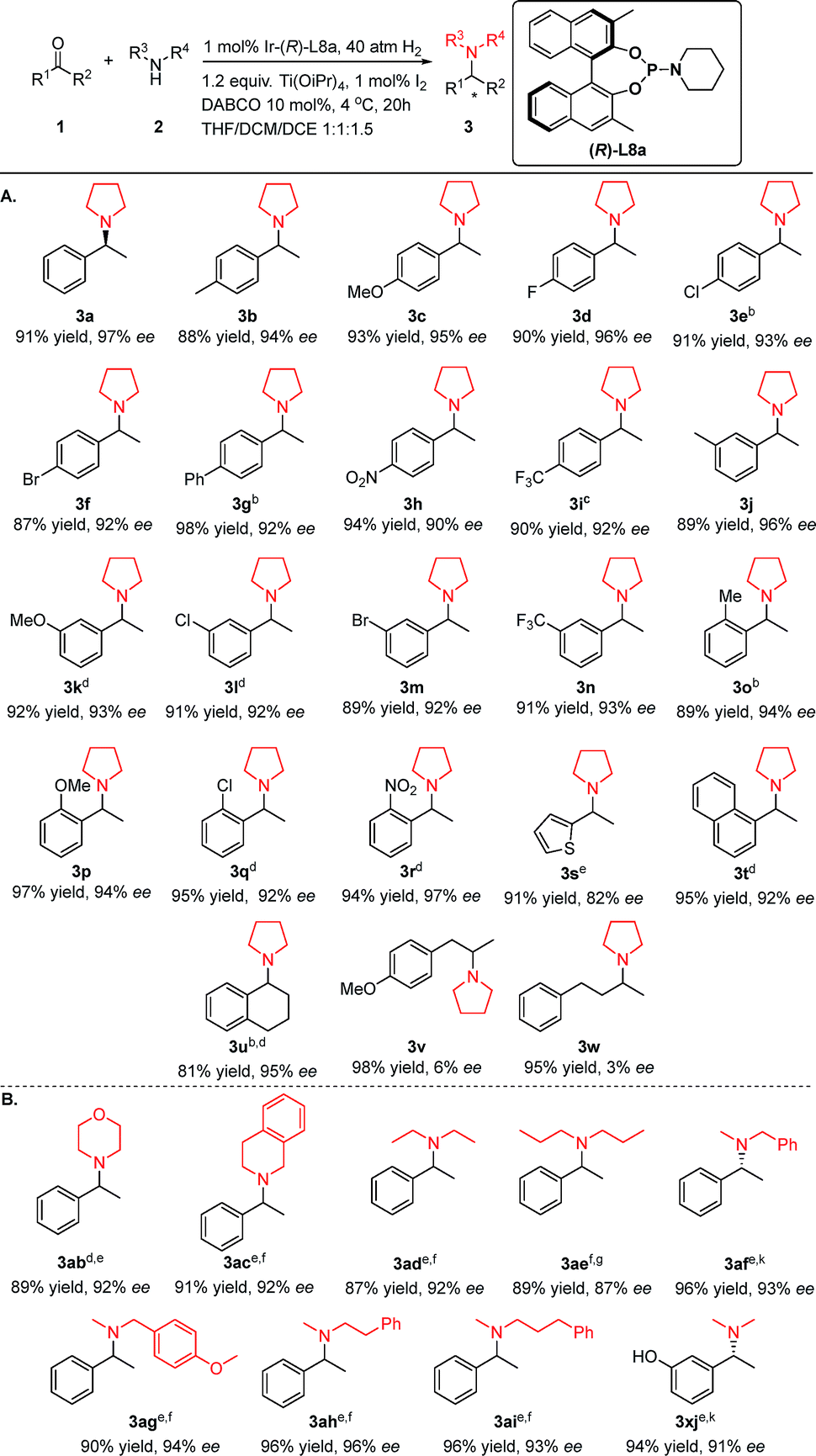
Under the established optimized reaction conditions, the applicable substrate scope with respect to both various ketones and amino coupling partners was investigated (Table 3). The additive set and catalytic system worked well for a series of aromatic ketones (Table 3a). In general, the electronic properties or positions of the substituent on the aromatic ring appeared to have limited effects on the experimental results. Regardless of whether the ketones have para-, meta-, or ortho-aryl substituents that are electron-donating (1c, 1k, and 1p), electron-withdrawing (1d, 1e, 1h, 1i, 1n, and 1r) or relatively sizeable (1g), the corresponding products were generated in excellent yields of 87–98% and between 90% and 97% ee. Notably, sterically hindered ketones (1o–1r, 1t), which were problematic substrates for previously reported DARA reactions,11a,12m could be smoothly transformed into the desired products without noticeable differences in yields and stereoselectivity from less hindered substrates (3pversus3c and 3rversus3h). In addition, the polar and reducible group –NO2 (3h & 3r) was well-tolerated in the reactions. Moreover, the methodology worked fairly well for heteroaromatic ketone 1s (91% yield and 82% ee). The aryl–alkyl hybrid ketones 1v and 1w were not suitable substrates, as only 6% and 3% ee were achieved for 3v and 3w, respectively.
Table 3 Exploration of the substrate scopea
|
Reaction conditions: [Ir]/L8a/1a/2a = 1:2:100:110; 1a 0.3 mmol, total solvent volume 3 mL, 4 °C, 20 h; DABCO = 1,4-diazabicyclo[2.2.2]octane; yields were isolated yields; enantiomeric excesses were determined by chiral HPLC or 1H NMR with a chemical shift reagent.
The applied ligand was (R)-L9a.
The reaction solvent was DMF/CH3CN/THF (1:1:1).
The applied solvent was acetonitrile/2-methyltetrahydrofuran 1:1.
The applied ligand was (Sa,Rc)-L9i.
The additive set was: 10 mol% Et3N, 5 mol% I2; reaction solvent was DMF/CH3CN/THF 1:1:1; reaction temperature was room temperature.
The applied ligand was (R)-L10j.
2 equiv. Ti(OiPr)4 was used; 10 mol% Et3N was used instead of DABCO; reaction solvent was CH3CN/THF 4:1; reaction temperature was 12 °C; H2 pressure was 50 atm.
|
|
Other N-sources were also successfully applied in this method (Table 3B). Cyclic amines, morpholine 2b and tetrahydroisoquinoline 2c, coupled with 1a efficiently to afford the desired products 3ab and 3ac with good yields and enantiopurity. Acyclic amines 2d, 2e and 2f also led to satisfactory results. Among the various alkylated amines, N-methylamines are of special interest because of their role in regulating biological functions.8c,15 We examined the DARA of 1a with N-methylbenzylamine 2f. This particular secondary amine was selected as the N-source because benzyl is one of the most used N-protecting groups and could be easily removed with various readily available reagents. The corresponding N-methyl-N-benzyl-1-phenylethylamine product 3af was obtained in excellent yield and enantioselectivity. Several other N-methyl amines 2g–2j were also explored as the N-sources and good to excellent results were achieved. It is noteworthy that the protic polar and acidic –OH group (1x) was well tolerated in this methodology. To better fit substrates with different electronic and steric properties, reaction solvents and chiral ligands have been adjusted accordingly. Acetonitrile, a polar solvent that has been rarely utilized in asymmetric hydrogenation, along with 2-methyltetrahydrofuran gave better results for some meta- and ortho-substituted, bulkier and more polar substrates 1k, 1l, 1q, 1r, 1t, 1ab and 1af than the DCM:THF:DCE solvent trio. Dimethyl formamide, another polar solvent, also worked better for amine sources other than pyrrolidine 2a (Table 3B). A chiral ligand with a longer side-arm L9a was applied for 1e, 1g, and 1o, as L9i was used for various secondary amines 2b–2d and 2f–2j. The above results have demonstrated the versatility and power of the readily fine-tuned phosphoramidite ligand series.
Practical applications
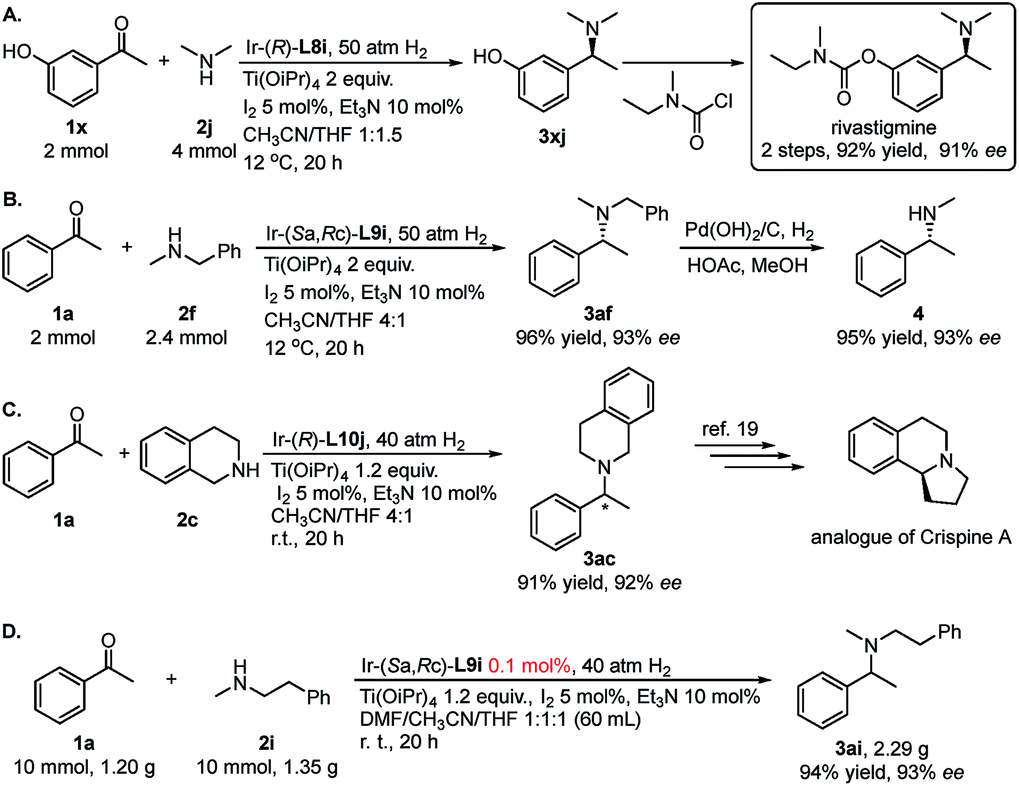
To further showcase the utility of this DARA strategy, we next made efforts on the synthesis of rivastigmine (API for Exelon, for the treatment of Alzheimer's type and Parkinson's type diseases and Lewy bodies, Fig. 1). Currently its industrial production depends on racemate resolution.16 Several efficient catalytic asymmetric synthetic routes have been developed, including AH of the corresponding ketone,17 AH of N-methyl imines11b and asymmetric hydroamination of alkynes,18 as the key transformation. In comparison, our method is much more efficient in terms of step-economy and operational simplicity. Starting from readily available and cheap meta-hydroxyacetophenone 1x, (S)-rivastigmine was synthesized in 2 steps with 92% overall yield and 91% ee (Scheme 2A). Moreover, with the aforementioned N-methyl benzylamine 2c as the N-source, after N-benzyl deprotection of the resulting product 3af from the DARA with 1a, N-methyl amine 4 could be obtained without losing enantiopurity (Scheme 2B). This extended the application of this protocol to the synthesis of chiral secondary amines. The product 3ac, from 1a and tetrahydroisoquinoline 2c, could be further elaborated to synthesize the analogue of the natural product and anti-tumor alkaloid (+)-crispine A (Scheme 2C).19 This reaction can easily be conducted on a large scale. Catalyzed with 0.1 mmol% of Ir–L9i, the reductive amination of 1.20 g (10 mmol) of ketone 1a and amine 2i gave 2.29 g of chiral amine 3ai (94% yield and 93% ee).
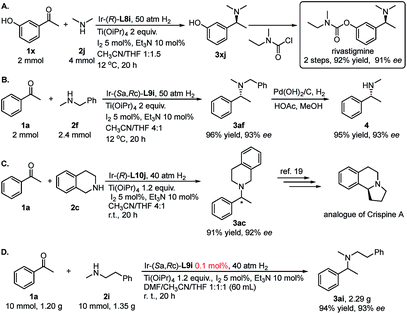 |
| | Scheme 2 Practical application of DARA of secondary amines to prepare tertiary amines. | |
The above syntheses exemplify the versatility and potential utilities of the DARA method for the expedited and straightforward construction of important and/or medicinally relevant enantioenriched molecules.
Mechanism study
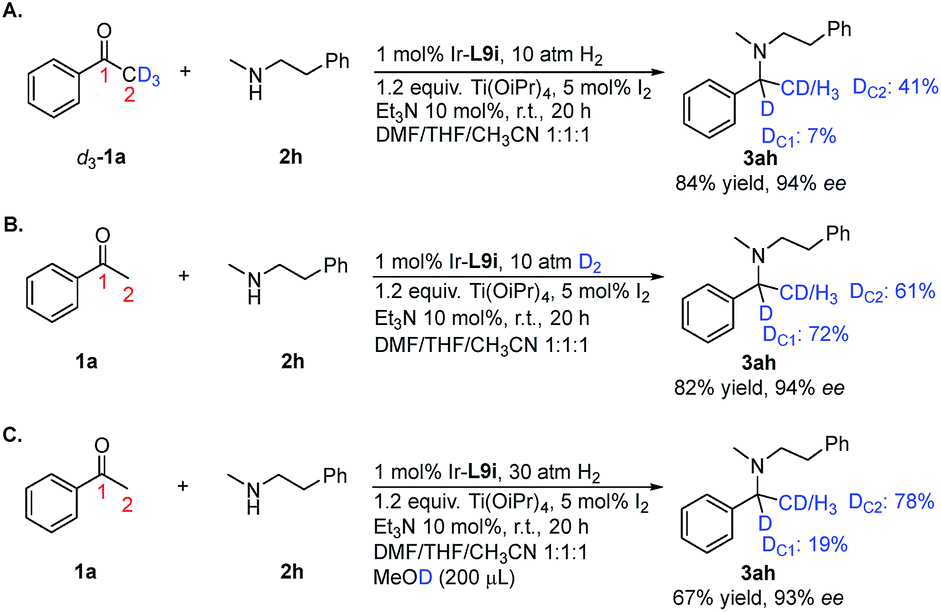
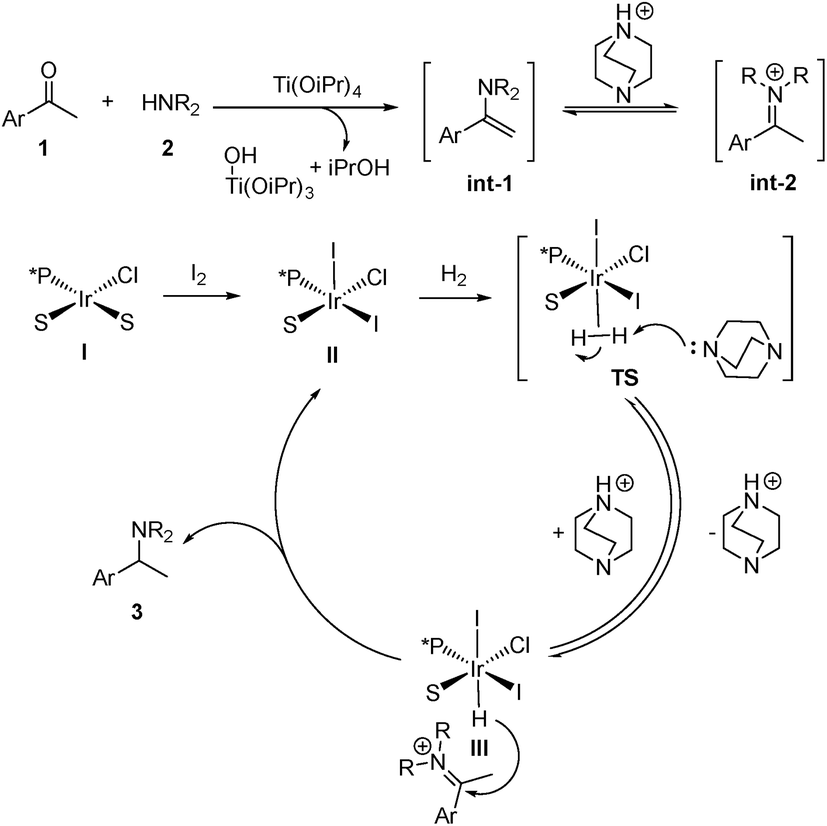
To gain insight into the reaction pathway, we conducted isotopic labeling experiments of 1a with 2h. From the 1H NMR integration, using methyl deuterated substrate 1a, the deuterium abundance at the chiral center C1 of 3ah was 7% and that on C2 was 41% (Scheme 3A). When applying deuterium gas as the reductant, deuterium incorporation at the C1 and C2 positions was 72% and 61%, respectively (Scheme 3B). The above results indicated that 1a and 2h formed the enamine intermediate, which underwent rapid tautomerization with the iminium form under the applied reaction conditions; also, the hydride addition to the iridium center step in the catalytic cycle was reversible. The results from the addition of MeOD further confirmed the involvement of tautomerization and the reversibility of the hydride addition in the reaction pathway (Scheme 3C). For non-aromatic ketones 1t and 1u, two possible enamine intermediates may be formed, along with the E/Z isomers, resulting in inferior stereoselectivity for the corresponding products. Although in our experiment the Ir/ligand ratio was 1:2, the 31P NMR spectra revealed only one P coordinated to Ir (see ESI†). After stirring the Ir–L9i complex in CDCl3 with 10 equiv. of Et3N for 24 h, the 31P NMR spectrum displayed no obvious change, which excluded the possibility of cyclometalated iridium complex formation.20 Based on the above results, we proposed the possible reaction pathway (Scheme 4). With the help of Ti(OiPr)4, ketone 1 reacts with amine 2 to form the enamine intermediate int-1, which accepts a proton and transforms into iminium int-2. int-1 and int-2 undergo fast interconversion. The iridium(I) complex I is oxidized to iridium(III) by molecular iodine.21 Then DABCO or Et3N facilitates the heterolytic cleavage of the hydrogen molecule by iridium to form III. This step is reversible, which results in the hydrogen incorporation at C1 of 3ah (Scheme 3).
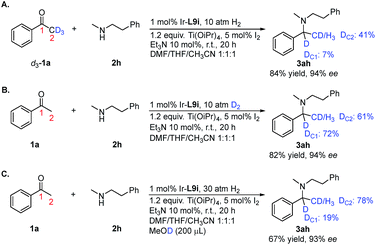 |
| | Scheme 3 Deuterium incorporation studies. | |
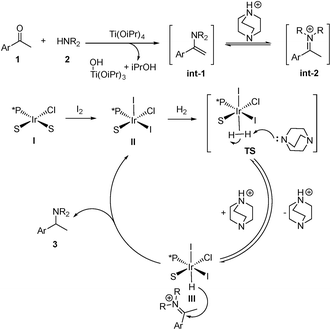 |
| | Scheme 4 Proposed reaction pathways. | |
Conclusions
In summary, we have unprecedentedly applied secondary amines as N-sources in direct catalytic asymmetric reductive amination, thus providing a solution to an important and persisting problem in this research area. Accelerated by the additive set and iridium–phosphoramidite ligand catalysts, they coupled smoothly with various ketones to afford chiral tertiary amines in excellent yields and high levels of stereocontrol. Benefitting from the highly modulated feature of the BINOL-based phosphoramidite ligands, a variety of this series of ligands with divergent electronic and spatial properties could be rapidly developed to accommodate substrates with different structures for achieving excellent stereoselectivity. This methodology is operationally simple and cost-efficient with minimal steps by utilizing readily available bulk chemicals. With our protocol, related chiral tertiary amines can be synthesized in a more convenient and effective manner.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21772155, 21402155 and 21602172) is gratefully acknowledged.
Notes and references
- L. M. Jarvis, Chem. Eng. News, 2016, 94, 12–17 Search PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
-
(a)
Chiral Amine Synthesis: Methods, Developments and Applications, ed. T. C. Nugent, Wiley-VCH, Weinheim, 2010 Search PubMed;
(b)
Stereoselective Formation of Amines, Top. Curr. Chem., ed. W. Li and X. Zhang, 2014, p. 343 Search PubMed;
(c) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
-
(a) T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS;
(b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed;
(c) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Soc. Rev., 2012, 41, 4126–4139 RSC;
(d) D. J. Ager, A. H. M. de Vries and J. G. de Vries, Chem. Soc. Rev., 2012, 41, 3340–3380 RSC.
-
(a) M. P. Magee and J. R. Norton, J. Am. Chem. Soc., 2001, 123, 1778–1779 CrossRef CAS PubMed;
(b) Y. Ji, G.-S. Feng, M.-W. Chen, L. Shi, H. Du and Y.-G. Zhou, Org. Chem. Front., 2017, 4, 1125–1129 RSC.
- C. Wang and J. Xiao, Top. Curr. Chem., 2014, 343, 261–282 CrossRef CAS PubMed.
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef CAS.
-
(a) H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef CAS;
(b) I. B. Seiple, Z. Zhang, P. Jakubec, A. Langlois-Mercier, P. M. Wright, D. T. Hog, K. Yabu, S. R. Allu, T. Fukuzaki, P. N. Carlsen, Y. Kitamura, X. Zhou, M. L. Condakes, F. T. Szczypiński, W. D. Green and A. G. Myers, Nature, 2016, 533, 338–345 CrossRef CAS PubMed;
(c) R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed;
(d) K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS.
- H.-U. Blaser, H.-P. Buser, H.-P. Jalett, B. Pugin and F. Spindler, Synlett, 1999, 867–868 CrossRef CAS.
- Selected examples:
(a) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed;
(b) R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS;
(c) I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 8, 1076–1082 CrossRef CAS PubMed;
(d) G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- Selected examples:
(a) R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed;
(b) V. N. Wakchaure, J. Zhou, S. Hoffmann and B. List, Angew. Chem., Int. Ed., 2010, 49, 4612–4614 CrossRef CAS PubMed;
(c) X. Xiao, Y. Xie, C. Su, M. Liu and Y. Shi, J. Am. Chem. Soc., 2011, 133, 12914–12917 CrossRef CAS PubMed.
-
(a) R. Kadyrov and T. H. Riermeier, Angew. Chem., Int. Ed., 2003, 42, 5472–5474 CrossRef CAS PubMed;
(b) Y. Chi, Y. Zhou and X. Zhang, J. Org. Chem., 2003, 68, 4120–4122 CrossRef CAS PubMed;
(c) D. Steinhuebel, Y. Sun, K. Matsumura, N. Sayo and T. Saito, J. Am. Chem. Soc., 2009, 131, 11316–11317 CrossRef CAS PubMed;
(d) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967–6969 CrossRef CAS PubMed;
(e) N. A. Strotman, C. A. Baxter, K. M. J. Brands, E. Cleator, S. W. Krska, R. A. Reamer, D. J. Wallace and T. J. Wright, J. Am. Chem. Soc., 2011, 133, 8362–8371 CrossRef CAS PubMed;
(f) Z.-P. Chen, S.-B. Hu, J. Zhou and Y.-G. Zhou, ACS Catal., 2015, 5, 6086–6089 CrossRef CAS;
(g) H. Huang, X. Liu, L. Zhou, M. Chang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5309–5312 CrossRef CAS PubMed;
(h) P. Yang, L. H. Lim, P. Chuanprasit, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12083–12087 CrossRef CAS PubMed;
(i) H. Zhou, Y. Liu, S. Yang, L. Zhou and M. Chang, Angew. Chem., Int. Ed., 2017, 56, 2725–2729 CrossRef CAS PubMed;
(j) B. Song, C.-B. Yu, Y. Ji, M.-W. Chen and Y.-G. Zhou, Chem. Commun., 2017, 53, 1704–1707 RSC;
(k) H. Huang, Y. Zhao, Y. Yang, L. Zhou and M. Chang, Org. Lett., 2017, 19, 1942–1945 CrossRef CAS PubMed;
(l) Y. Zhang, Q. Yan, G. Zi and G. Hou, Org. Lett., 2017, 19, 4215–4218 CrossRef CAS PubMed;
(m) H. Huang, Z. Wu, G. Gao, L. Zhou and M. Chang, Org. Chem. Front., 2017, 4, 1976–1980 RSC;
(n) J. Gallardo-Donaire, M. Hermsen, J. Wysocki, M. Ernst, F. Rominger, O. Trapp, A. S. K. Hashmi, A. Schäfer, P. Comba and T. Schaub, J. Am. Chem. Soc., 2018, 140, 355–361 CrossRef CAS PubMed;
(o) X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 2024–2027 CrossRef CAS PubMed;
(p) Y. Lou, Y. Hu, J. Lu, F. Guan, G. Gong, Q. Yin and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 14193–14197 CrossRef CAS PubMed;
(q) B. Song, M.-W. Chen and Y.-G. Zhou, Org. Chem. Front., 2018, 5, 1113–1117 RSC;
(r) T. Yang, Q. Yin, G. Gua and X. Zhang, Chem. Commun., 2018, 54, 7247–7250 RSC;
(s) G. Gao, S. Du, Y. Yang, X. Lei, H. Huang and M. Chang, Molecules, 2018, 23, 2207–2218 CrossRef PubMed.
- L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006–4123 CrossRef CAS PubMed.
-
(a) A. J. Minnaard, B. L. Feringa, L. Lefort and J. G. de Vries, Acc. Chem. Res., 2007, 40, 1267–1277 CrossRef CAS PubMed;
(b) W. Fu and W. Tang, ACS Catal., 2016, 6, 4814–4858 CrossRef CAS.
- J. Chatterjee, F. Rechenmacher and H. Kessler, Angew. Chem., Int. Ed., 2013, 52, 254–269 CrossRef CAS PubMed.
-
A. Gaitonde, M. Mangle and S. Pawar, International patent WO 2005/061446, 2005.
- P.-C. Yan, G.-L. Zhu, J.-H. Xie, X.-D. Zhang, Q.-L. Zhou, Y.-Q. Li, W.-H. Shen and D.-Q. Che, Org. Process Res. Dev., 2013, 17, 307–312 CrossRef CAS.
- S.-L. Shi and S. L. Buchwald, Nat. Chem., 2015, 7, 38–44 CrossRef CAS PubMed.
- L. Benmekhbi, F. Louafi, T. Roisnel and J.-P. Hurvois, J. Org. Chem., 2016, 81, 6721–6739 CrossRef CAS PubMed.
-
(a) C. A. Kiener, C. Shu, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14272–14273 CrossRef CAS PubMed;
(b) B. Tutkowski, S. Kerdphon, E. Limé, P. Helquist, P. G. Andersson, O. Wiest and P.-O. Norrby, ACS Catal., 2018, 8, 615–623 CrossRef CAS.
-
(a) D. Xiao and X. Zhang, Angew. Chem., Int. Ed., 2001, 40, 3425–3428 CrossRef CAS;
(b) B. Qu, H. P. R. Mangunuru, S. Tcyrulnikov, D. Rivalti, O. V. Zatolochnaya, D. Kurouski, S. Radomkit, S. Biswas, S. Karyakarte, K. R. Fandrick, J. D. Sieber, S. Rodriguez, J.-N. Desrosiers, N. Haddad, K. McKellop, S. Pennino, H. Lee, N. K. Yee, J. J. Song, M. C. Kozlowski and C. H. Senanayake, Org. Lett., 2018, 20, 1333–1337 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and spectra. See DOI: 10.1039/c9sc00323a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a