
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of selective protein–protein interaction inhibitors using efficient in silico peptide-directed ligand design†
Andrew M.
Beekman
 *a,
Marco M. D.
Cominetti
a,
Samuel J.
Walpole
a,
Saurabh
Prabhu
a,
Maria A.
O'Connell
a,
Jesus
Angulo
a and
Mark
Searcey
*ab
*a,
Marco M. D.
Cominetti
a,
Samuel J.
Walpole
a,
Saurabh
Prabhu
a,
Maria A.
O'Connell
a,
Jesus
Angulo
a and
Mark
Searcey
*ab
aSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, Norfolk, NR47TJ, UK. E-mail: A.Beekman@uea.ac.uk; M.Searcey@uea.ac.uk
bSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich, Norfolk, NR47TJ, UK
First published on 22nd March 2019
Abstract
The development of protein–protein interaction (PPI) inhibitors with therapeutic value is of increasing importance as the first clinical agent has now been approved, but PPIs remain difficult targets for the development of small molecule ligands. This article describes a highly efficient approach to the development of inhibitors of the p53/hDMX or hDM2 interaction that involves the design of small molecules in silico based upon a peptide/protein structure. The process for molecule design, starting from a virtual library of just over 1200 fragments, led to the eventual synthesis of twenty compounds, of which ten bound to either hDM2, hDMX or both in in vitro binding assays. This 50% success rate is extremely efficient compared to traditional high throughput screening. The identification of two selective hDMX inhibitors from twenty compounds highlights this efficiency as, to date, only two other hDMX-selective agents exist in the literature. Preliminary biological studies show that 20% of the compounds identified have cellular activity and activate downstream pathways associated with p53 activation.
Introduction
Targeting the hDM2 (human double minute 2) p53 protein–protein interaction (PPI) has been a paradigm for PPI inhibitor development in cancer since the discovery of nutlin-3, which is currently undergoing clinical trials.1,2 Although there are many studies of p53-hDM2 interaction inhibitors ranging from natural products to designed small molecules, the scope of this promising strategy has been severely limited by the activity of hDMX.3 Despite the highly homologous nature of hDMX to hDM2 the two proteins do not perform redundant roles.4,5hDMX can prevent p53 activity by sequestering it, but hDMX also enhances the ligase activity of hDM2 towards p53 by forming hDM2/hDMX dimers.6 The overexpression of one or both of these proteins results in the loss of p53 activity in cells.7 Therefore, dual hDM2 hDMX inhibitors have become highly desirable.The development of hDMX selective molecules is also of significant importance. It has been shown that direct inhibition of hDM2 leads to p53 dependent toxicity in healthy cells.8,9 Additionally, the non-redundant roles and common mutual exclusivity in cancers suggest selective compounds are useful.10 There are currently no small molecules that modulate hDMX/p53 and exhibit both cell and animal efficacy.11 Two compounds have been highlighted which modulate hDMX/p53 selectively, but are unsuitable for clinical development.8,9,12 Therefore, there is still a demand for chemical probes or leads that are selective for hDM2, selective for hDMX or dual hDM2 hDMX inhibitors. The p53 binding domains of hDM2 and hDMX display high structural homology, but differences exist which could be exploited.7,10
Several approaches to the identification of inhibitors of the p53-hDM2 and p53-hDMX interaction have been described. Natural products such as chlorofusin can be utilised as starting points for design.13 Oligobenzamide analogues that mimic the interactions of the helical peptide have been described14,15 and helical peptides have been stabilised and shown to have cellular activity through the introduction of stapling groups.16 Both high throughput screening and computational design against the hDM2 target have had varying degrees of success with hit rates for screening of around 0.02%.
We believe these challenging PPIs are excellent targets for the exemplification of in silico peptide directed binding, a new methodology for identifying selective PPI modulators. Using copper(I) catalysed azide–alkyne cycloaddition (CuAAC) click chemistry and taking a small commercially available library of 896 alkynes and 214 azides would require the synthesis of 191![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 744 compounds to screen all possible compounds. Using these compounds as fragments for screening would need high resolution NMR with 15N-labelled protein or high throughout crystallography, relatively specialised techniques. Our approach has been to exploit the peptidic dual inhibitor Ac-Phe-Met-Aib-Pmp-6-Cl-Trp-Glu-Ac3c-Leu-NH2 (1) to develop small molecule probes that target hDM217 and hDMX.18 Briefly, the crystal structure of 1 bound to both proteins is known (hDMX: PDB ID 2GV2, hDM2: PDB ID 3FEA). The sequence of the high affinity peptide binder is used to generate two smaller peptides with reactive azide and alkyne terminals (Fig. 1).19In silico screening of the small molecule fragments then allows the identification of peptide/small molecule hybrids with restored affinity for the target site. The restoration of binding affinity implies that the small molecule fragment in some way emulates the peptide section it has replaced. The small molecule portion of the hybrid hits are then combined through in silico click chemistry and rescreened to identify potential small molecules with high affinity for the target site. Computational modelling is used to perform the entire peptide directed binding process, identifying small molecules to be prepared. This use of virtual design in peptide directed binding further improves the rapid and economic nature of this process, identifying compounds not highlighted by experimental peptide directed binding.19 Herein, we demonstrate that this approach led to the synthesis of twenty small molecules of which ten bound to hDM2 or hDMX, representing a 50% success rate. Furthermore, one compound was selective for hDMX both in vitro and in tissue culture, outperforming previously reported selective modulators.
744 compounds to screen all possible compounds. Using these compounds as fragments for screening would need high resolution NMR with 15N-labelled protein or high throughout crystallography, relatively specialised techniques. Our approach has been to exploit the peptidic dual inhibitor Ac-Phe-Met-Aib-Pmp-6-Cl-Trp-Glu-Ac3c-Leu-NH2 (1) to develop small molecule probes that target hDM217 and hDMX.18 Briefly, the crystal structure of 1 bound to both proteins is known (hDMX: PDB ID 2GV2, hDM2: PDB ID 3FEA). The sequence of the high affinity peptide binder is used to generate two smaller peptides with reactive azide and alkyne terminals (Fig. 1).19In silico screening of the small molecule fragments then allows the identification of peptide/small molecule hybrids with restored affinity for the target site. The restoration of binding affinity implies that the small molecule fragment in some way emulates the peptide section it has replaced. The small molecule portion of the hybrid hits are then combined through in silico click chemistry and rescreened to identify potential small molecules with high affinity for the target site. Computational modelling is used to perform the entire peptide directed binding process, identifying small molecules to be prepared. This use of virtual design in peptide directed binding further improves the rapid and economic nature of this process, identifying compounds not highlighted by experimental peptide directed binding.19 Herein, we demonstrate that this approach led to the synthesis of twenty small molecules of which ten bound to hDM2 or hDMX, representing a 50% success rate. Furthermore, one compound was selective for hDMX both in vitro and in tissue culture, outperforming previously reported selective modulators.
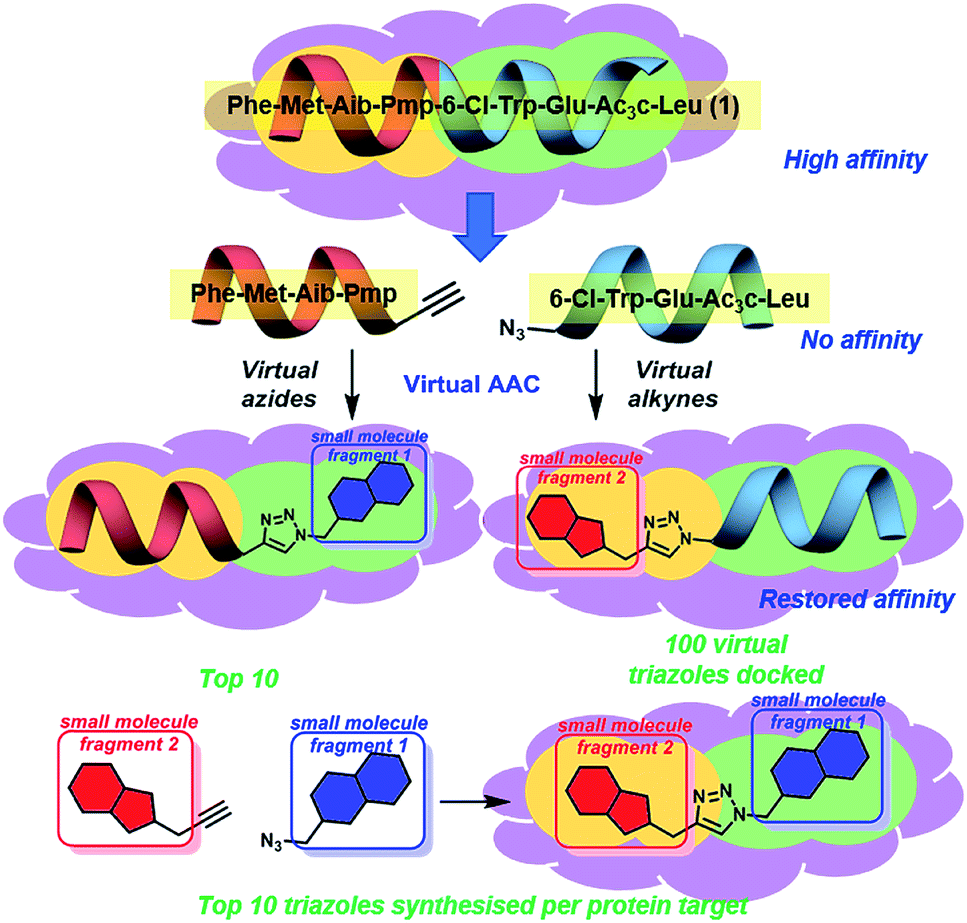 | ||
| Fig. 1 Concept of peptide directed binding utilised in this work with in silico methods to improve the rapid and economic nature of finding new PPI modulators. Peptide 1 binds to hDM2/X, and is separated into two smaller peptides with no appreciable binding despite having two key residues each. Azide–alkyne cycloaddition (AAC) identifies small molecule fragments which restore binding affinity. The identified small molecule fragments can be combined to generate a small molecule substitute for the initial peptide. | ||
Results
In silico peptide directed binding identifies candidate small molecule hDM2/X binders
Peptide 1 binds with high affinity to both hDM2 and hDMX, which allows its exploitation to develop small molecule dual inhibitors as both chemical probes and potential therapeutic lead molecules. In order to identify small molecules to be synthesised and evaluated against hDM2 and hDMX the crystal structures for the peptide 8-mer 1 bound to hDM2 (PDB ID: 2GV2)17 and hDMX (PDB ID: 3FEA)18 were edited to identify small molecule fragments that emulate a section of the 8 mer. The peptide was split, Phe-Met-Aib-Pmp-propargyl glycine and azidoacetamide-6-Cl-Trp-Glu-Ac3c-Leu (Fig. 1), allowing two key binding residues to be present in both smaller peptides (Phe and Pmp, 6-Cl-Trp and Leu). The site left vacant by the removed peptide fragment represents a more tractable binding site for a small molecule. Covalent docking was then performed simulating the Huisgen 1,3-dipolar cycloaddition (see ESI,† pg 6), with the commercially available library of alkynes or azides (ESI,† pg 12). The semi-peptide was held in place with flexibility in the reactive terminal and the final residue, allowing for the possibility that the added small molecule fragment is held in the correct orientation for binding. The results were scored and ranked20 and the top ten azides and top ten alkynes that were synthetically viable were used to generate one hundred virtual triazoles. The generated triazoles were then induced-fit docked to the protein, scored and ranked to identify the top ten small molecules that bound in silico with high affinity to each protein and that would be synthesised (Fig. 1).21 The identified triazole small molecules are likely to bind with the same binding elements as identified by the hybrid, but may sit in a slightly altered orientation to allow for tighter binding. This process was carried out for both hDM2 and hDMX and as such twenty small molecule triazoles were synthesised following published procedures with CuSO4 and sodium ascorbate in a tBuOH/H2O mix, followed by purification by reverse phase HPLC (see ESI† pg. 9 for all synthesised compounds).13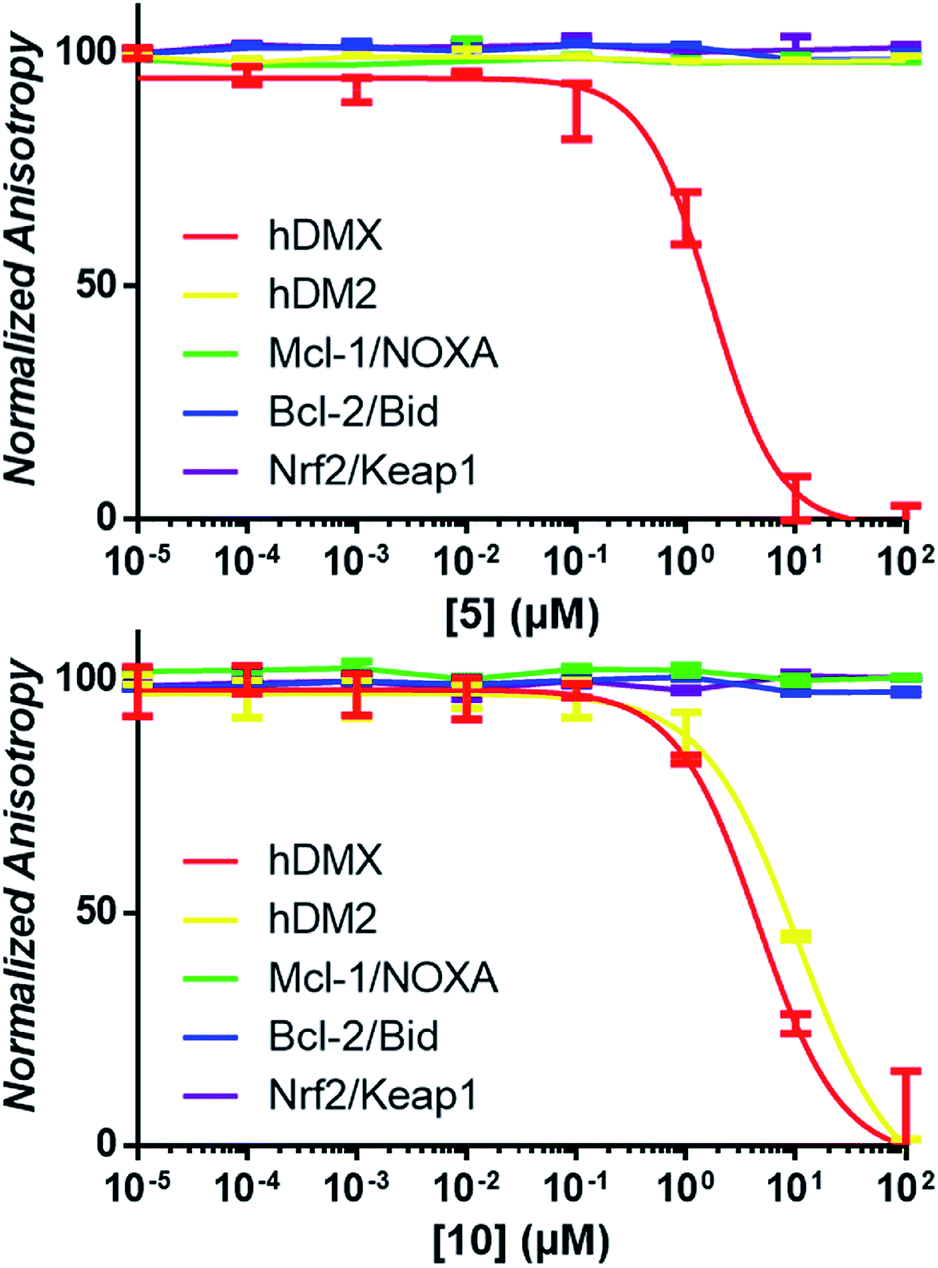 | ||
| Fig. 2 Examples of competitive-displacement assays of small molecules towards a selection of PPIs. Compounds 5 and 10 used to exemplify the titration of compounds towards the hDMX/p53, hDM2/p53, Mcl-1/NOXA, Bcl-2/Bid and Nrf2/Keap1 interactions. Dose response curves can be seen for 5 towards hDMX, and for 10 towards hDMX and hDM2, with no competitive binding towards any other PPIs observed. | ||
Identified small molecules selectively modulate the p53-hDM2/X PPIs
It was expected that some of these molecules would display selectivity for the target used in their design, but inevitably dual modulators would be identified due to the homogeneity of the hDM2/X binding sites and the use of the same peptide 1. The synthesised compounds were first analysed with competitive fluorescence anisotropy (FA) assays. A FAM tagged p4 peptide22,23 was used as the fluorescently tagged peptide for both hDM2, reported previously,13 and hDMX. The wild-type p53 peptide (residues 15–27) was employed as a positive control and an indication that the FA assays were performing adequately. Of the twenty compounds synthesised, 50% demonstrated binding in the FA assays to either hDM2 or hDMX, with IC50 values less than 100 μM (Table 1). Strikingly, one compound displayed significant selectivity for hDMX over hDM2 (5), while overall 40% of compounds designed for hDMX modulated the hDMX interaction (2–5). Two of these compounds, 2 and 3, had an IC50 in the tens of nanomolar range. Of the compounds designed for hDM2, 50% demonstrated the ability to modulate the hDM2 PPI (6–10), with three showing selectivity for hDM2 over hDMX (7–9). Interestingly, one of the compounds designed for hDM2 (11) had affinity for hDMX but no appreciable binding to hDM2. To confirm selectivity, these compounds were analysed in other PPI FA assays routinely utilised in our lab. Pleasingly, compounds 2–11 demonstrated no appreciable binding at 100 μM towards the PPIs Mcl-1/Noxa, Bcl-2/Bid19 and Nrf2/Keap1,24 despite similarities to compounds reported to bind to these PPIs (Fig. 2).| a IC50 values determined by non-linear regression of at least three independent experiments (see ESI,† pg 10). Errors are 95% confidence intervals (CI). Fmoc, 9-fluorenylmethylcarbonyl. |
|---|

|
The compounds that exhibited binding in the FA assay were validated with differential scanning fluorimetry (DSF).25 Compounds displayed the ability to increase the melting temperature of proteins compared to a vehicle control (see ESI,† pg. 41).26 Thermal stability was demonstrated using the hydrophobic dye Sypro orange, mimicking the observed FA assay results (see ESI,† pg. 40).
Use of STD NMR indicates that compounds bind in the p53 binding site of hDM2/X
To provide structural details of the binding of these compounds in the p53 binding site, we employed an STD NMR initial growth-rates approach to identify the binding mode and the interactions with the binding grooves of hDM2 and hDMX.27–29 Compound 4 was chosen for analysis by STD NMR because of its solubility in aqueous buffer and its relatively weak binding to both hDM2 and hDMX, allowing the binding kinetics to fall within the fast-exchange conditions necessary for STD NMR. This criteria precludes tighter binders (2 & 3) from being analysed with this technique. Compound 4 displayed STD signals in the presence of either hDM2 or hDMX, indicating binding to both proteins.Additionally, the ligand binding epitope of compound 4 was mapped using the STD-NMR initial growth rates method (ESI,† pg 15). In order to gain some structural information docking calculations were used to identify a binding pose. We were able to validate the binding pose within the hDM2 binding groove using CORCEMA-ST,30 which calculates theoretical STD intensities based on a given 3D model complex. The NOE R-factor between the experimental and theoretical STD intensifies was 0.17, indicating good agreement. Interestingly, preliminary CORCEMA-ST trials of 4 binding to hDMX suggested the formation of oligomers. Dimerization induced by structurally similar compounds has been observed previously,8 and suggests a possible selective binding mode which could be exploited for hDMX modulation. Fig. 3 displays the binding pose of 4 to hDM2 demonstrating tight binding of the fluorinated aryl ring in the pocket vacated by chlorotryptophan of 1. This STD/CORCEMA-ST validated binding pose, along with the binding affinities determined by the competitive fluorescence anisotropy assay indicate these compounds are binding in the p53 binding groove. The identified binding epitope places the fluorine atom in the same pocket as that occupied by the chlorine atom of the Cl-Trp in 1. This same pocket also exists in the hDMX binding site and may explain the improved binding of the fluoroaryl group (2, 10 & 11 compared to S8, S2 & S9).
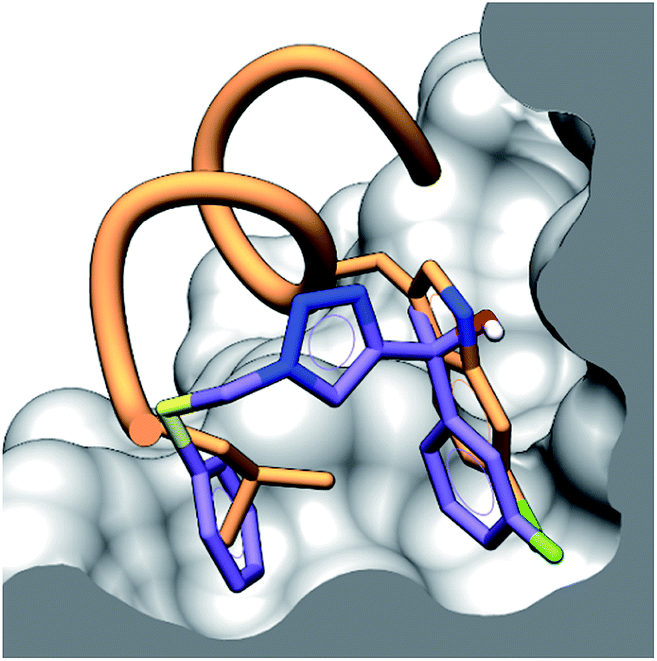 | ||
| Fig. 3 Binding pose of 4 (purple) to hDM2 in the p53 binding site determined by CORCEMA-ST, with an overlay of peptide 1 (orange) showing the binding residues of Leu and Cl-Trp. The surface of hDM2 is cut along the plane of the centroid of the aromatic rings of compound 4. | ||
Compounds inhibit cancer cell growth in a selective fashion
Compounds that had activity in the FA assay were examined for their ability to inhibit cell growth and affect metabolism in an array of cancer cell lines that demonstrate varying expressions of the p53 and hDM2/X proteins. The human cancer cell lines A549,31 HCT1163,31 and MCF-731 express both hDM2 and hDMX, SJSA-1 is hDM2 dependent11 and JEG3 is hDMX dependent.3 All cell lines express wild type p53. Gratifyingly, 10% of the prepared compounds demonstrated the ability to inhibit cell growth. Compound 10, which was shown to bind to both hDM2 and hDMX, had activity towards cell lines that are dependent on hDMX, dependent on hDM2 or dependent on both, as expected (Table 2). Compound 5, which showed selectivity for hDMX over hDM2, demonstrated activity towards the hDMX dependent JEG3, but not towards the hDM2 dependent SJSA-1, a further suggestion of selectivity of 5 for hDMX. The remaining compounds did not demonstrate activity in the MTS assay, perhaps due to issues with cell penetration. Alterations to the in silico screening process are currently underway to increase the output of compounds with desirable characteristics, such as cell permeability.
| SJSA-1 IC50 (μM) | A549 IC50 (μM) | HCT116 IC50 (μM) | MCF-7 IC50 (μM) | JEG3 IC50 (μM) | |
|---|---|---|---|---|---|
| a IC50 values determined by non-linear regression of at least three experiments (see ESI,† pg 13). Errors are the transformed greater extreme of the standard error. | |||||
| 5 | >100 | 16.2 ± 0.06 | 20.5 ± 0.07 | 29.8 ± 0.13 | 35.5 ± 0.10 |
| 10 | 30.0 ± 2.29 | 65.7 ± 0.22 | >100 | >100 | 38.1 ± 0.03 |
Compounds induce events downstream of p53 activation
To ascertain whether compounds 5 and 10 were inducing p53 related cell death we examined the induction of apoptosis with a caspase 3/7 activation assay. A549 and JEG-3 cells were treated with vehicle (DMSO), positive control (staurosporine), 5 and 10. Cleavage of the DEVD sequence was monitored to observe caspase 3/7 activity. Activation of caspase 3/7 was observed for both 5 and 10 in line with the observed IC50 values (Fig. 4A). The transcriptional activation of targets downstream of p53 was examined with quantitative PCR (qPCR).3 JEG-3 cells were treated with vehicle control, 5 and 10 for 6 h at 37 °C, followed by RNA isolation, reverse transcription, and qPCR analysis of the obtained cDNA using the primers of downstream p53 targets p53, hDM2, p21 and MIC-1. Both compounds 5 and 10 significantly increased the expression of each of the target genes when compared to the vehicle control (Fig. 4B). | ||
| Fig. 4 (A) A549 and JEG-3 cells were treated with vehicle (DMSO), positive control (staurosporine, STS), 5 and 10 at 100 μM and 50 μM for 6 h, 4 h and 2 h at 37 °C. Treatment with Caspase-Glo 3/7 reagent (Promega) allowed for the monitoring of the cleavage of pro-luminescent substrate by caspase 3/7. Data are mean ± SEM for at least n = 3. RLU – relative luminescent units. STS – staurosporine. (B) JEG-3 cells were treated with vehicle (DMSO), 5 and 10 at 100 μM for 6 h at 37 °C, and transcriptional upregulation of p53, hDM2, p21 and MIC-1 was evaluated by qPCR analysis. 18S was used as a control. Y-axis is the mRNA-fold induction vs. the vehicle control (DMSO). All p values were less than 0.05 when compared to the vehicle. | ||
Virtual peptide directed binding was employed to identify new modulators of p53/hDM2 and p53/hDMX. This use of computational peptide directed binding represents a complementary method to identify PPI modulators in a rapid and economic manner. Twenty compounds were prepared, ten designed for hDM2 and ten designed for hDMX, and 50% of the compounds were found to modulate the PPI. This hit rate is a significant improvement compared to recent studies which utilise a chemical array (0.04%)32 and high-throughput screen (0.07%).33 Applying peptide directed binding to the p53-hDM2/X PPIs has identified new scaffolds for modulating these PPIs, which have excellent lead like features.34 Importantly, this process has identified selective inhibitors for both hDM2 and hDMX, despite the high structural homology of the binding sites of these proteins. Only two small molecules have been previously reported which selectively modulate the p53-hDMX PPI, highlighting the power of this technique. Virtual peptide directed binding has identified compounds that interact with the p53 binding site of hDM2 and hDMX, supported by activity in competitive fluorescence anisotropy assays and, more significantly, quantitative STD NMR analysis. A subset of the identified modulators was shown to have activity towards cancer cell lines that are known to have dependence on hDM2, hDMX or both, and these compounds were shown to induce p53 activation downstream events.
Discussion
The deployment of in silico peptide directed binding has demonstrated an extraordinarily high hit rate for new PPI small molecule modulators of the p53 hDM2/X interaction. Perhaps more interestingly, this process has identified new binding motifs for these PPIs, as well as highlighting selective scaffolds for hDMX, a notoriously challenging task.35,36 The nature of peptide directed binding provides structure activity relationship information without producing specific modifications. The fluoroaryl group shows enhanced binding to hDMX, and the 2,4-difluoroaryl compound may offer selectivity for hDMX (i.e.5 and 11). The Fmoc azoalanine drives binding to hDM2, a phenomenon we have shown previously,13 but does not guarantee binding (S1 – see ESI,† pg. 9). The presence of the sugar moiety imparts potent binding, but again does not guarantee it (S8). Despite the FP and DSF results supporting tight binding, the hydrophilic sugar moiety raises concerns about compounds 2 and 3 as leads, as the p53 binding site is known to be hydrophobic. Docking suggested the polar sugar occupying the Phe binding pocket of 1 (see ESI,† pg 14), but docking results should be interpreted with caution. Modifying the sugar with nonpolar protecting groups reduces potency, but affinity is still observed (i.e.10 and 11), suggesting key binding moieties may still be available. Investigation of 2 and 3 as leads is ongoing. The in silico calculations highlighted the zidovudine structure (azide section of 9) as a likely potent binder, but experimentally resulted in a poor hit rate, perhaps highlighting modifications to be made in compound selection process or docking calculations (see ESI,† pg 9).The compounds identified which selectively modulate the p53-hDMX interaction outperform the two selective molecules reported in the literature. The first known selective binder of hDMX – SJ-172550 – was demonstrated to bind with less affinity to hDMX than p53. Both 5 and 11 demonstrate greater binding affinity to hDMX than p53 (ESI,† pg 10).35 Additionally, the cellular activity of these compounds is reported in the millimolar range, with our compounds demonstrating at least two orders of magnitude improvement. SJ-172550 is also known to not be a competitive binder, but rather induces a conformational change through covalent bond formation, which does not exist in a reducing environment.37 Unfortunately, this limits the value of SJ-172550 as a lead candidate.
CTX1, the second small molecule shown to have selectivity for hDMX, has not been validated in in vitro protein assays, with this compound being identified through a cellular screen.9 CTX1 demonstrated IC50 values in the tens to hundreds of micromolar range. Compound 5 possesses slightly improved potency, but has other important advantages. CTX1 is an acridine based molecule, known to bind to DNA, and act as a PAIN molecule. This characteristic makes CTX1 unusable in many light based assays, such as fluorescence, due to interference. It is also unknown whether CTX1 is a competitive inhibitor of p53 or acts through some other mechanism (some suggestion the CTX1 mode of action overlaps with the action of 9-aminoacridine),9 making its use as a chemical tool limited.
SJ-172550 was found as one of three hits in a library of 295848 compounds.35 CTX1 came from a screen of over 20000 compounds.9 In this instance we have prepared twenty molecules and identified two selective p53-hDMX modulators. The true value of the significant advance in the improvement of success rate we have demonstrated here is the power of peptide directed binding to allow researchers in academia and the pharmaceutical industry, chemistry and biology to quickly and cheaply develop modulators for the protein–protein interaction they wish to target. The need for enormous libraries of molecules and expensive high throughput screening facilities is not necessary for the identification of small molecule modulators, as has been the case for so many challenging targets.
Notably, the compounds prepared are unoptimised but still display strong activity in the fluorescence anisotropy assay and low micromolar cellular activity, highlighting the rapid ability of peptide directed binding to identify selective chemical probes.
Conclusions
This work has exemplified the power of peptide directed binding to rapidly identify small molecule PPI modulators, at a high hit rate, that show selectivity for challenging targets. This method complements experimental peptide directed binding and is expected to be readily applied to other PPIs such as 14-3-3/tau, bromodomains or Nrf2/Keap1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SJW is funded by the Biotechnology and Biological Sciences Research Council (BBSRC) Doctoral Training Partnership PhD studentship. SP is funded by BBSRC grant no BB/N008448/1. We acknowledge the EPSRC UK National Mass Spectrometry Facility at Swansea University. We thank Professor Aart Jochemsen (Leiden) for the kind gift of hDMX.Notes and references
- L. T. Vassilev, J. Med. Chem., 2005, 48, 4491–4499 CrossRef CAS PubMed.
- V. Tisato, R. Voltan, A. Gonelli, P. Secchiero and G. Zauli, J. Hematol. Oncol., 2017, 10, 133 CrossRef PubMed.
- F. Bernal, M. Wade, M. Godes, T. N. Davis, D. G. Whitehead, A. L. Kung, G. M. Wahl and L. D. Walensky, Cancer Cell, 2010, 18, 411–422 CrossRef CAS PubMed.
- V. Böttger, A. Böttger, C. Garcia Echeverria, Y. F. M. Ramos, A. J. van der Eb, A. G. Jochemsen and D. P. Lane, Oncogene, 1999, 18, 189 CrossRef PubMed.
- F. Toledo and G. M. Wahl, Int. J. Biochem. Cell Biol., 2007, 39, 1476–1482 CrossRef CAS PubMed.
- A. Shvarts, W. T. Steegenga, N. Riteco, T. Laar, P. Dekker, M. Bazuine, R. C. Ham, W. H. v. Oordt, G. Hateboer, A. J. Eb and A. G. Jochemsen, EMBO J., 1996, 15, 5349–5357 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci., 2013, 110, E3445–E3454 CrossRef CAS PubMed.
- B. Graves, T. Thompson, M. Xia, C. Janson, C. Lukacs, D. Deo, P. Di Lello, D. Fry, C. Garvie, K. S. Huang, L. Gao, C. Tovar, A. Lovey, J. Wanner and L. T. Vassilev, Proc. Natl. Acad. Sci., 2012, 109, 11788–11793 CrossRef CAS PubMed.
- G. Karan, H. Wang, A. Chakrabarti, S. Karan, Z. Liu, Z. Xia, M. Gundluru, S. Moreton, Y. Saunthararajah, M. W. Jackson, M. K. Agarwal and D. N. Wald, Mol. Cancer Ther., 2016, 15, 574–582 CrossRef CAS PubMed.
- S. Datta, M. E. Bucks, D. Koley, P. X. Lim and S. N. Savinov, Bioorg. Med. Chem., 2010, 18, 6099–6108 CrossRef CAS PubMed.
- F. Wachter, A. M. Morgan, M. Godes, R. Mourtada, G. H. Bird and L. D. Walensky, Oncogene, 2017, 36, 2184 CrossRef CAS PubMed.
- G. M. Popowicz, A. Dömling and T. A. Holak, Angew. Chem., Int. Ed., 2011, 50, 2680–2688 CrossRef CAS PubMed.
- M. M. D. Cominetti, S. A. Goffin, E. Raffel, K. D. Turner, J. C. Ramoutar, M. A. O'Connell, L. A. Howell and M. Searcey, Bioorg. Med. Chem. Lett., 2015, 25, 4878–4880 CrossRef CAS PubMed.
- J. P. Plante, T. Burnley, B. Malkova, M. E. Webb, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Commun., 2009, 5091–5093, 10.1039/B908207G.
- A. Shaginian, L. R. Whitby, S. Hong, I. Hwang, B. Farooqi, M. Searcey, J. Chen, P. K. Vogt and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 5564–5572 CrossRef CAS PubMed.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- K. Sakurai, C. Schubert and D. Kahne, J. Am. Chem. Soc., 2006, 128, 11000–11001 CrossRef CAS PubMed.
- J. Kallen, A. Goepfert, A. Blechschmidt, A. Izaac, M. Geiser, G. Tavares, P. Ramage, P. Furet, K. Masuya and J. Lisztwan, J. Biol. Chem., 2009, 284, 8812–8821 CrossRef CAS PubMed.
- A. M. Beekman, M. A. O'Connell and L. A. Howell, Angew. Chem., Int. Ed., 2017, 56, 10446–10450 CrossRef CAS PubMed.
- K. Zhu, K. W. Borrelli, J. R. Greenwood, T. Day, R. Abel, R. S. Farid and E. Harder, J. Chem. Inf. Model., 2014, 54, 1932–1940 CrossRef CAS PubMed.
- W. Sherman, H. S. Beard and R. Farid, Chem. Biol. Drug Des., 2006, 67, 83–84 CrossRef CAS PubMed.
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810–8817 CrossRef CAS PubMed.
- A. Czarna, G. M. Popowicz, A. Pecak, S. Wolf, G. Dubin and T. A. Holak, Cell Cycle, 2009, 8, 1176–1184 CrossRef CAS PubMed.
- R. J. Steel, M. A. O'Connell and M. Searcey, Bioorg. Med. Chem. Lett., 2018, 28, 2728–2731 CrossRef CAS PubMed.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212 CrossRef CAS PubMed.
- A. Madhumalar, L. H. Jun, C. J. Brown, D. P. Lane and C. S. Verma, Cell Cycle, 2009, 8, 2828–2836 CrossRef PubMed.
- J. Angulo, S. A. Goffin, D. Gandhi, M. Searcey and L. A. Howell, Chem.–Eur. J., 2016, 22, 5858–5862 CrossRef CAS PubMed.
- B. Bertrand, J. Fernandez Cestau, J. Angulo, M. M. D. Cominetti, Z. A. E. Waller, M. Searcey, M. A. O'Connell and M. Bochmann, Inorg. Chem., 2017, 56, 5728–5740 CrossRef CAS PubMed.
- J. Angulo, I. Díaz, J. J. Reina, G. Tabarani, F. Fieschi, J. Rojo and P. M. Nieto, ChemBioChem, 2008, 9, 2225–2227 CrossRef CAS PubMed.
- V. Jayalakshmi and N. R. Krishna, J. Magn. Reson., 2002, 155, 106–118 CrossRef CAS PubMed.
- M. Hirose, K. Yamato, S. Endo, R. Saito, T. Ueno, S. Hirai, H. Suzuki, M. Abei, Y. Natori and I. Hyodo, Oncoscience, 2014, 1, 830–843 Search PubMed.
- T. Noguchi, S. Oishi, K. Honda, Y. Kondoh, T. Saito, T. Kubo, M. Kaneda, H. Ohno, H. Osada and N. Fujii, Bioorg. Med. Chem. Lett., 2013, 23, 3802–3805 CrossRef CAS PubMed.
- L. Gu, H. Zhang, T. Liu, S. Zhou, Y. Du, J. Xiong, S. Yi, C.-K. Qu, H. Fu and M. Zhou, Cancer Cell, 2016, 30, 623–636 CrossRef CAS PubMed.
- D. J. Foley, A. Nelson and S. P. Marsden, Angew. Chem., Int. Ed., 2016, 55, 13650–13657 CrossRef CAS PubMed.
- D. Reed, Y. Shen, A. A. Shelat, L. A. Arnold, A. M. Ferreira, F. Zhu, N. Mills, D. C. Smithson, C. A. Regni, D. Bashford, S. A. Cicero, B. A. Schulman, A. G. Jochemsen, R. K. Guy and M. A. Dyer, J. Biol. Chem., 2010, 285, 10786–10796 CrossRef CAS PubMed.
- M. Wade, Y. C. Li and G. M. Wahl, Nat. Rev. Cancer, 2013, 13, 83 CrossRef CAS PubMed.
- M. Bista, D. Smithson, A. Pecak, G. Salinas, K. Pustelny, J. Min, A. Pirog, K. Finch, M. Zdzalik, B. Waddell, B. Wladyka, S. Kedracka-Krok, M. A. Dyer, G. Dubin and R. K. Guy, PLoS One, 2012, 7, e37518 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectra and processed biophysical assay data. See DOI: 10.1039/c9sc00059c |
| This journal is © The Royal Society of Chemistry 2019 |