
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein-induced low molecular weight hydrogelator self-assembly through a self-sustaining process†
Jennifer
Rodon Fores
 a,
Miryam
Criado-Gonzalez
abc,
Marc
Schmutz
a,
Christian
Blanck
a,
Pierre
Schaaf
*abc,
Fouzia
Boulmedais
a and
Loïc
Jierry
*a
a,
Miryam
Criado-Gonzalez
abc,
Marc
Schmutz
a,
Christian
Blanck
a,
Pierre
Schaaf
*abc,
Fouzia
Boulmedais
a and
Loïc
Jierry
*a
aUniversité de Strasbourg, CNRS, Institut Charles Sadron (UPR22), 23 rue du Loess, 67034 Strasbourg Cedex 2, BP 84047, France. E-mail: schaaf@unistra.fr; loic.jierry@ics-cnrs.unistra.fr
bInstitut National de la Santé et de la Recherche Médicale, INSERM Unité 1121, 11 rue Humann, 67085 Strasbourg Cedex, France
cUniversité de Strasbourg, Faculté de Chirurgie Dentaire, 8 rue Sainte Elisabeth, 67000 Strasbourg, France
First published on 11th March 2019
Abstract
Controlling how, when and where a self-assembly process occurs is essential for the design of the next generation of smart materials. Along this route, enzyme-assisted self-assembly is a powerful tool developed during the last decade. Here we introduce another strategy allowing for spatiotemporal control over peptide self-assemblies. We use a Fmoc-peptide precursor in dynamic equilibrium with its low molecular weight hydrogelator (LMWH) through a reversible disulfide bond. In the absence of proteins, no self-assembly of the hydrogelator is observed. In the presence of proteins, their interactions with the precursor initiate a self-assembly process of the hydrogelator around them. This self-assembly displaces the equilibrium between precursor and LMWH according to Le Chatelier's principle, producing new hydrogelators available to pursue the self-assembly growth. One thus establishes a self-sustaining cycle fuelled by the self-assembly itself until full consumption of the LMWH. For proteins in solutions this process can lead to a supramolecular hydrogel whereas for proteins deposited on a surface, the gel growth is initiated exclusively from the surface.
Introduction
Self-assembly processes are the heart of supramolecular chemistry. Relying on reversible non-covalent bonds, they allow the emergence of new types of materials with dynamic responses towards external stimuli and thus able to adapt to the surrounding environment.1 In the quest to design materials possessing high performance features similar to those found in biological tissues, an absolute prerequisite for success is to control self-assembly processes both spatially and temporally.2 Within this framework, the use of (bio)catalysts to trigger the localization of the self-assembly of low molecular weight hydrogelators (LMWH) has proven to be highly interesting.3 Indeed, enzymes can catalyze the transformation of non-assembling entities into self-assembling ones, rapidly and with high specificity.4 This approach called enzyme-assisted self-assembly (EASA) was introduced by Xu5 and extended by Ulijn and others6,7 and a large number of reports describing the use of EASA based on a short list of enzymes able to produce hydrogelators mainly through bond cleavage or bond formation have been published. Another approach based on a dynamic covalent library of peptides has also been developed thanks to enzymes such as thermolysin, able to both condense amino acids and hydrolyze the resulting peptides.3a,8 Compared to other triggers that modify the environment (pH, temperature or ionic strength for instance), enzymes appear up to now as the only tool capable to both control and direct the self-assembly process in an efficient way thanks to a range of methods to localize enzymes precisely in space. In spite of the lack of works dedicated to the elucidation of the self-assembly initiation step, it is well established that hydrogelators self-assemble leading to nanofibers: their resulting entanglement (or others nanostructures) affords the architectures of the supramolecular hydrogel that have been already carried out to a large scope of promising applications. Here, we introduce a new strategy to control spatiotemporally self-assembling processes. It is based on the simultaneous presence of proteins possessing or not catalytic activity, LMWH and its precursor, both being in dynamic equilibrium through a reversible bond (here a disulfide bond). The interaction of the precursor with the protein initiates a self-assembly process which leads to the establishment of a self-sustaining cycle where continuous hydrogelator generation is induced by the self-assembly process itself (Fig. 1a). This approach appears closely related to 2D dynamic covalent chemistry where adsorption of a product out of a combinatorial library displaces chemical reactions by harvesting a final product otherwise present in small amounts.9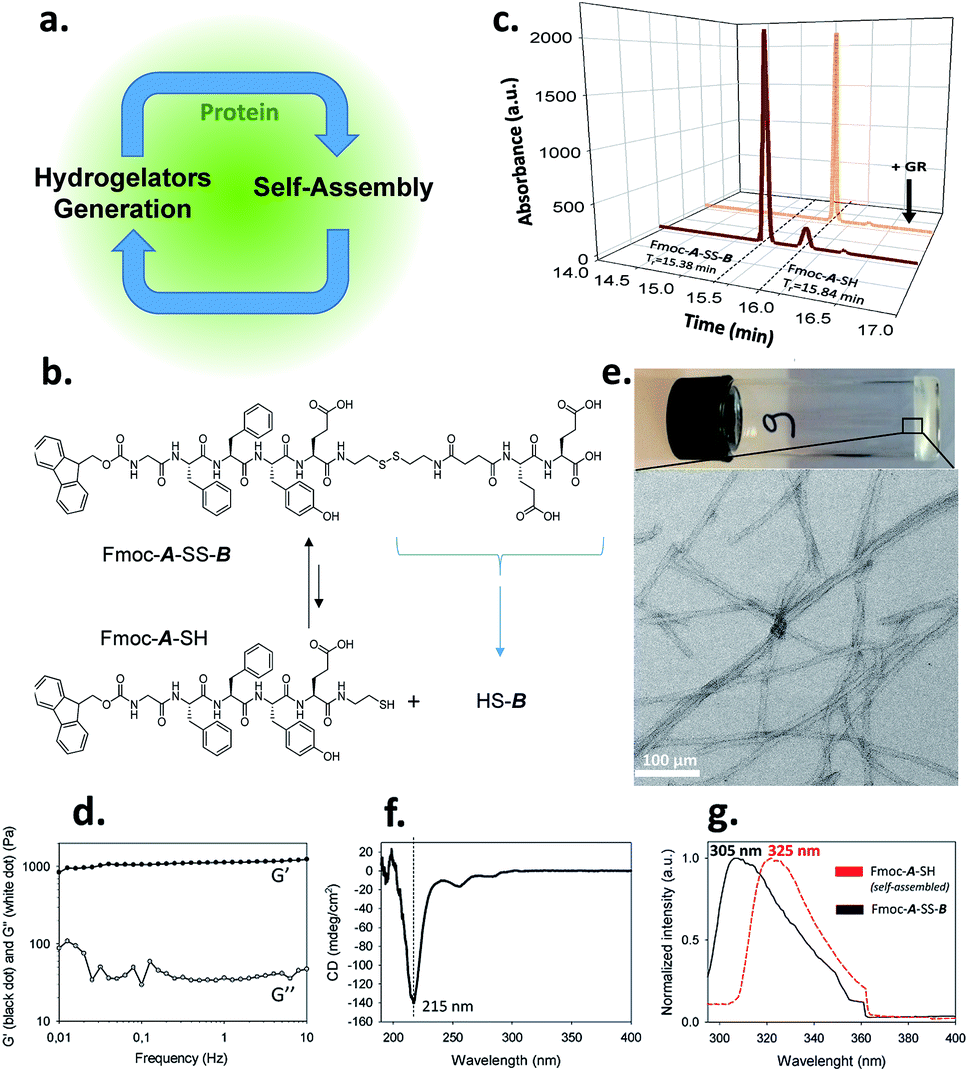 | ||
| Fig. 1 (a) Protein-assisted self-assembly's concept; (b) chemical equilibrium between Fmoc-A-SS-B and Fmoc-A-SH; (c) HPLC analysis of a solution of Fmoc-A-SS-B before (black curve) and after the addition of GR (orange curve); (d) G′ and G′′ measurements (log scales), (e) TEM image, (f) CD spectra and (g) normalized emission of fluorescence excimer intensity of Fmoc groups from Fmoc-A-SH based hydrogel. | ||
Results and discussion
We use an original peptide as LMWH, Fmoc-GFFYE-NH-(CH2)2-SH, called Fmoc-A-SH, inspired from literature (Fig. 1b).10 The fluorenylmethyloxycarbonyl (Fmoc) N-protected group has been added at the N-terminal position to enhance the ability of the corresponding hydrogelator Fmoc-A-SH to self-assemble.4,11 When the thiol group of this peptide is protected through a disulfide bridge, the resulting Fmoc-A-SS-B (Fmoc-GFFYE-NH-(CH2)2-S-S-(CH2)2-NHCO-(CH2)2-CO-EE-OH) is not able to self-assemble because of the highly negatively charged B sequence. When 10 mg mL−1 (7.3 mmol) of Fmoc-A-SS-B is dissolved in PBS buffer (pH 7.4) at 25 °C, no gelation occurs, even after 48 hours, as expected. High performance liquid chromatography (HPLC) monitoring the chemical composition of this solution over time shows that a thermodynamic equilibrium between Fmoc-A-SS-B and Fmoc-A-SH is quickly established (less than 2 minutes). At equilibrium, 10% of Fmoc-A-SH is present and 90% of Fmoc-A-SS-B constitutes the major compound (Fig. 1c). Of course, Fmoc-A-SS-A-Fmoc and B-SS-B are also both present in the mixture: Fmoc-A-SS-A-Fmoc is present at trace-level and B-SS-B is not measured through our HPLC's UV-detector.To get a supramolecular gel from a Fmoc-A-SS-B solution (10 mg mL−1), the disulfide bridge must be reduced and glutathione reductase (GR, 1 mg mL−1) was selected to play this role. Because the initiation of gelation should depend on the sufficient production of Fmoc-A-SH (to reach its critical gelation concentration), we followed its formation by HPLC as soon as GR was introduced in the vial. Gelation is qualitatively observed by a simple vial inversion test. Fmoc-A-SS-B disappears over the first minutes and correlatively Fmoc-A-SH forms (Fig. 1c). Roughly two minutes after addition of GR, 100% of the hydrogelator Fmoc-A-SH is formed and a gel is obtained having an elastic modulus G′ close to 1 kPa (Fig. 1d and S1 in ESI†). The gel is underpinned by a network of entangled nanofibers (Ø ∼ 15 nm) as observed by transmission electron microscopy (TEM) after negative staining (Fig. 1e and S2 in ESI†). Circular dichroism (CD) shows a β-sheet peptide assembly from Fmoc-A-SH (Fig. 1f) in which Fmoc groups are stacking one another, thus exhibiting a characteristic fluorescence emission of excimers at 325 nm when excited at 280 nm (Fig. 1g).
To demonstrate the catalytic role of GR in the gelation process, another enzyme unable to cleave the disulfide bridge of Fmoc-A-SS-B, alkaline phosphatase (AP), was used instead of GR and the same HPLC monitoring study was realized. As anticipated, no gel was formed even after 60 minutes following the addition of AP (1 mg mL−1) in the Fmoc-A-SS-B solution (10 mg mL−1). Yet, the HPLC monitoring revealed the formation of Fmoc-A-SH, more slowly than with GR, but reaching nevertheless a full chemical transformation (from Fmoc-A-SS-B) after one hour (Fig. S3 in ESI†). In order to verify if this unexpected result can be extended to other proteins than AP, we brought a freshly prepared Fmoc-A-SS-B solution in contact with bovine serum albumin (BSA, 1 mg mL−1). In spite of a slower kinetic of Fmoc-A-SH formation than for AP, Fmoc-A-SS-B ultimately almost fully disappeared after 1 h as measured by HPLC but no gel formed even 24 hours later (Fig. S3 in ESI†). The faster reduction of Fmoc-A-SS-B catalyzed by GR being perhaps involved in the efficiency of the gelation process, 1 equivalent of the strong reductive agent (tris(2-carboxyethyl)phosphine) (TCEP) was added to a Fmoc-A-SS-B solution to check this hypothesis: HPLC highlights an instantaneous full conversion into Fmoc-A-SH (Fig. S3 in ESI†) but no gelation was observed, even after one hour. A complete conversion into Fmoc-A-SH has also been observed by HPLC when 1 molar equivalent of dithiothreitol (DTT) or glutathione (GSH) was used instead of TCEP without gel formation as for TCEP (Fig. S4 in ESI†). Using higher Fmoc-A-SS-B and protein concentrations (both 30 mg mL−1) a gel was formed in the presence of BSA or AP whereas no gel was formed in contact with TCEP (Fig. S4 in ESI†).
Of course, self-assembly of Fmoc-A-SH may occur in solution without leading to hydrogel formation. Thus, to check whether self-assembly takes place in the four experiments described above, we used complementary analytical tools: TEM, fluorescence emission and infrared spectroscopies, CD, nuclear magnetic resonance (NMR) and dynamic light scattering (DLS). For all these studies, lower concentrations of peptide and protein were used: solutions of 1 mg mL−1 of Fmoc-A-SS-B and 1 mg mL−1 of one of the three proteins (GR, AP or BSA) were used except for DLS measurements where concentrations of protein were lower. At such small concentrations, no gelation from the Fmoc-A-SS-B solution occurs even in presence of GR (1 mg mL−1). TEM analysis of each four solutions mixing Fmoc-A-SS-B and GR (sol. 1), AP (sol. 2), BSA (sol. 3) and TCEP (sol. 4) showed nanostructures formation except for sol. 4 when TCEP was added. Typical images can be found in Fig. 2. GR and AP lead to counter clockwise twisted nanofibers: in the presence of GR, fibers are slightly twisted and their diameters are typically 15 nm whereas AP leads to highly twisted fibers that are 5 nm in diameter. Nanofibers are several hundred micrometers long in both cases (sol. 1 and 2). For BSA (sol. 3), wide platelets, several hundreds of nanometers long, are typically observed. Therefore, despite full conversion of Fmoc-A-SS-B in Fmoc-A-SH (confirmed by HPLC analysis) in all four solutions, the self-assembly of Fmoc-A-SH starts only in the presence of one of the three proteins GR, AP and BSA. In addition, we thus find that depending on the initial self-assembly conditions, the resulting self-assembled nanostructure can have various morphologies.12 Investigating the origin of these different morphologies is out of the scope of this article. Conclusions from TEM analysis have been confirmed by fluorescence emission measurements done on sol. 1, 2, 3 and 4 when excited at 280 nm. This wavelength allows to observe the fluorescence emission band of the Fmoc groups at 305 nm when Fmoc-A-SS-B is dissolved in solution (1 mg mL−1) and then to follow its shifting evolution to 320–325 nm over 60 minutes (Fig. 3). This shift is only noticed for sol. 1, 2 and 3 and is the characteristic signature of the Fmoc excimer formation found in the Fmoc-containing peptides stacked through π–π interactions.3d,11 The absence of shift for sol. 4 proves also the absence of self-assembled structures in this case. CD and IR measurements confirmed the presence of β-sheets resulting from Fmoc-A-SH self-assembly in sol. 1, 2 and 3 exclusively (when a protein is present) and not in sol. 4 (Fig. S5 in ESI†). The necessary participation of GR, AP or BSA in the self-assembly initiation of Fmoc-A-SH was also examined through DLS when each of these proteins was brought in contact with Fmoc-A-SS-B (1 mg mL−1). In the absence of Fmoc-A-SS-B, the size of each protein is close to 1 nm. One minute after the addition of Fmoc-A-SS-B (1 mg mL−1), the apparent size of the scattering species increases rapidly. Indeed, as shown in Table S1 and Fig. S6,† one observes a shift of the scattering peak towards higher diameters. It must be noticed that the given sizes are obtained by applying the models of a Brownian sphere and should not be taken as representing physically the size of the scattering object. They reflect their average hydrodynamic size. They nevertheless show that the self-assembly leads to a continuous increase of this size starting from that of the enzymes. This indicates that the self-assembly process should start at the enzymes. When 1 equivalent of TCEP was added to Fmoc-A-SS-B, no diffusive species were measured, even after one hour.
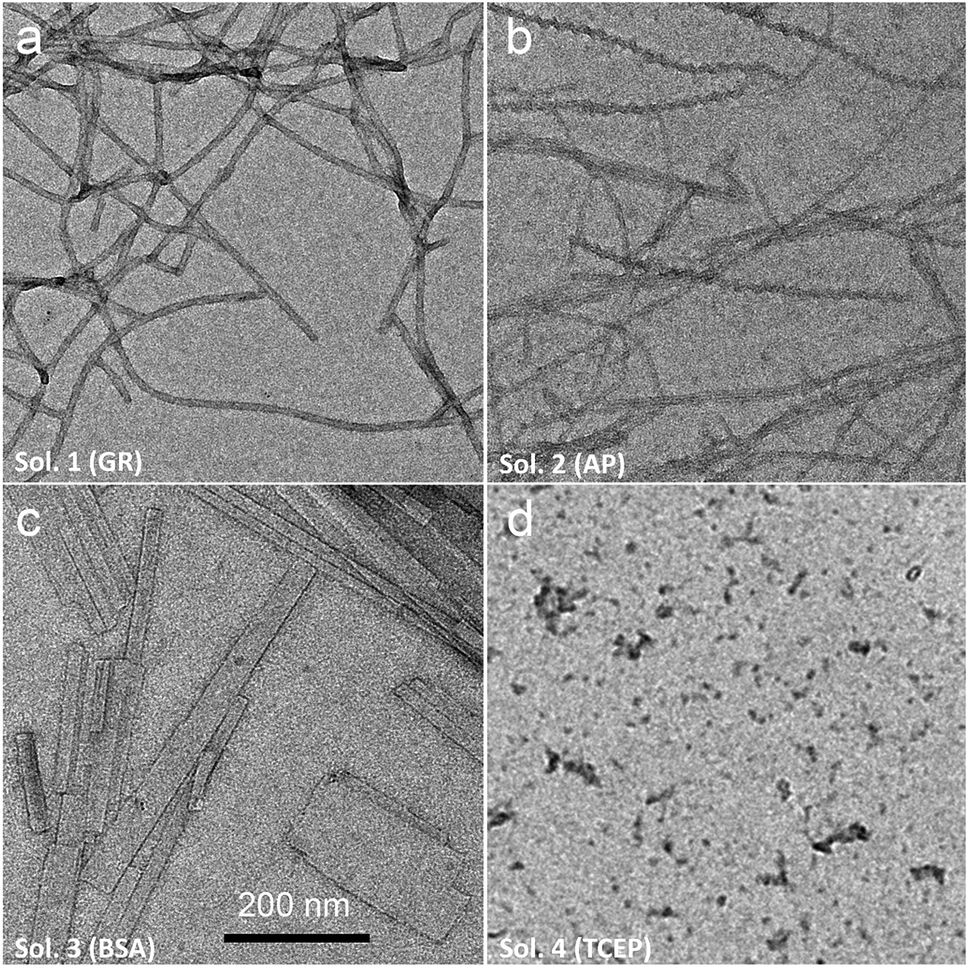 | ||
| Fig. 2 TEM analysis of Fmoc-A-SS-B solution (1 mg mL−1) after the addition of (a) GR (sol. 1), (b) AP (sol. 2), (c) BSA (sol. 3) to reach 1 mg mL−1 of biomacromolecule's final concentration or (d) 1 equivalent of TCEP (sol. 4). | ||
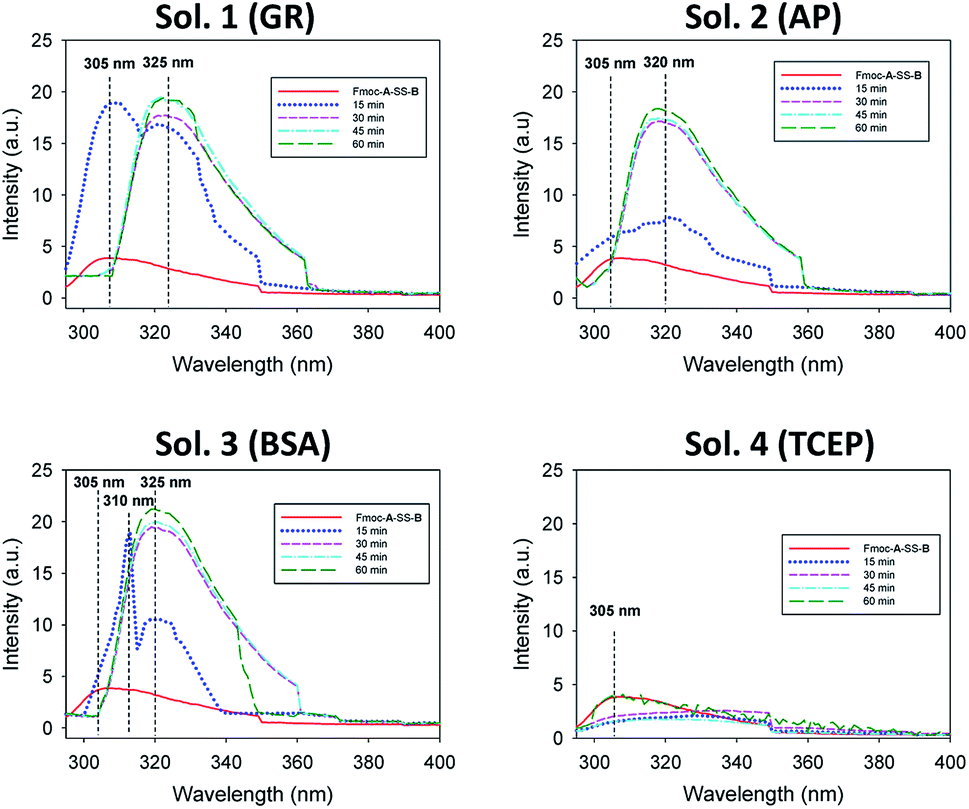 | ||
| Fig. 3 Evolution over 60 minutes of the emission fluorescence intensity of Fmoc-A-SS-B solution (1 mg mL−1) in presence of 1 mg mL−1 of GR (sol. 1), AP (sol. 2), BSA (sol. 3) or 1 equivalent of TCEP (sol. 4) when the solution is excited at 280 nm. | ||
From this set of experiments one concludes that (i) the self-assembly process of Fmoc-A-SH peptides is initiated from Fmoc-A-SS-B only in the presence of a protein or an enzyme even if this later does not play a catalytic role in the disulfide reduction; (ii) the equilibrium reaction between Fmoc-A-SS-B and Fmoc-A-SH is totally shifted towards Fmoc-A-SH only in the presence of one of the investigated proteins. Therefore one can propose the following self-assembly mechanism schematically represented in Fig. 4a: in the presence of proteins such as BSA or AP, Fmoc-A-SS-B or Fmoc-A-SH interact with them and initiate the self-assembly of Fmoc-A-SH, thus reducing the concentration of free Fmoc-A-SH in solution. This consumption of free Fmoc-A-SH disturbs the equilibrium which counteracts by providing Fmoc-A-SH from Fmoc-A-SS-B according to Le Chatelier's principle. Of course, because the self-assembly process of Fmoc-A-SH continues over time, a total displacement of the equilibrium takes place leading to the growth of nanofibers underpinning a supramolecular hydrogel (at high Fmoc-A-SS-B concentration, i.e. 30 mg mL−1).
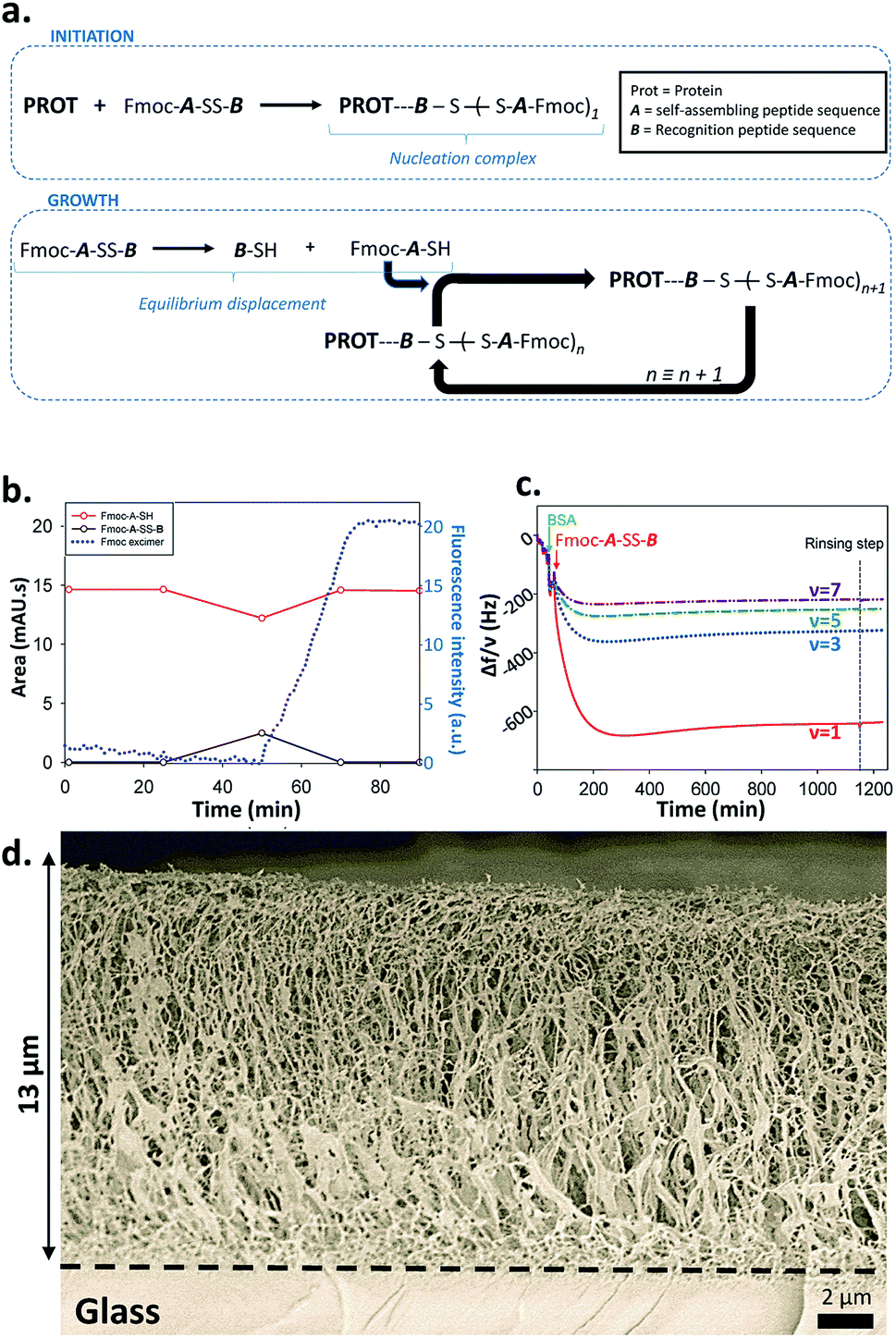 | ||
| Fig. 4 (a) Mechanism proposed for the protein-assisted self-assembly's concept; (b) HPLC monitoring and fluorescence emission intensity measurement at 320 nm (λex = 280 nm) of Fmoc-A-SH solution after the addition of BSA; (c) QCM-D monitoring of the self-assembly growth initiated from a substrate modified with AP; (d) Cryo-SEM image of a cross sectioned supramolecular hydrogel formed from a AP layer adsorbed onto a glass slide. | ||
In order to prove that it is the self-assembly process that is at the origin of the total equilibrium displacement in favor of Fmoc-A-SH we dissolved the lacking-Fmoc disulfide peptide A-SS-B in water (10 mg mL−1). This peptide is unable to self-assemble because of the absence of the Fmoc group. The thermodynamic equilibrium (disulphide/thiol) is established in a few minutes with 95% of A-SS-B and 5% of A-SH according to HPLC analysis. Addition of BSA or AP (1 mg mL−1) to this solution does not change the initial A-SS-B/A-SH ratio, thus proving our case (Fig. S7 in ESI†). On the other hand, addition of GR (1 mg mL−1) to this solution yields to an entire production of A-SH with no gel formation as expected, even 1 hour later.
What remains to be understood is how the self-assembly process is initiated by the presence of the protein and in particular if initiation takes place through interactions between the proteins with Fmoc-A-SH or Fmoc-A-SS-B. Indeed, the Fmoc group in Fmoc-A-SS-B (or Fmoc-A-SH) is an N-protecting group known to interact with proteins13 and to be involved in the self-assembly of a large number of LMWH thanks to hydrophobic effects.4,11 In addition, the peptide sequence B (Fig. 1b) is a highly negatively charged group that could also be able to interact with proteins. We brought a Fmoc-A-SS-B solution (10 mg mL−1) in contact with 1 equivalent of TCEP to generate 100% of Fmoc-A-SH that do not self-assemble as described before. When BSA (1 mg mL−1) was added to this Fmoc-A-SH solution, a gel was obtained but only one hour after the addition of BSA. To understand why the hydrogel formation takes one hour instead of being immediate, we monitored by HPLC the establishment of the natural thiol/disulphide equilibrium between Fmoc-A-SH and Fmoc-A-SS-B over time and in parallel we followed the fluorescence emission evolution of this mixture when excited at 280 nm. Both were tracked after the addition of BSA in the 100% Fmoc-A-SH solution (generated using TCEP) freshly prepared (Fig. 4b). We observe that 25 minutes after the addition of TCEP (and thus also BSA), the proportion of Fmoc-A-SH starts to decrease concomitantly with the growing formation of Fmoc-A-SS-B. This is due to the re-equilibration between these two species, thanks to the presence of dissolved oxygen in the solution. This evolution is reversed 25 minutes later (t = 50 min) leading to full disappearance of Fmoc-A-SS-B. Almost at the same time (50 minutes after the addition of TCEP), the shift of the fluorescence emission band of the Fmoc group from 305 nm to 325 nm is observed, proving the stacking of Fmoc-A-SH. The replacement of BSA by AP or GR in this experiment shows the same scenario (Fig. S8 in ESI†). One can thus conclude that the presence of Fmoc-A-SH is necessary but not sufficient to get the hydrogel formation. Both a few amount of Fmoc-A-SS-B and the presence of a protein are required to initiate the self-assembly process. The expected electrostatic interaction between the negatively charged B sequence of Fmoc-A-SS-B and positively charged patches onto proteins can be verified through the addition of a competitive negatively charged compounds. To mimic the three negative charges of B sequence due to the two glutamic acid residues and the carboxylic acid in C-terminal position, we used sodium citrate. Thus, 1, 10 and 100 equivalents of citrate were added into Fmoc-A-SS-B (30 mg mL−1) solutions before the addition of BSA (30 mg mL−1). In presence of 10 equivalent of sodium citrate, only a viscous liquid is obtained. When 100 equivalents are added, the gelation process is inhibited, due to the competitive interaction between sodium citrate and Fmoc-A-SS-B with BSA (Fig. S9†). This simple test highlights the necessary interaction between Fmoc-A-SS-B and proteins involving the carboxylate groups present in the B sequence of the peptide, to allow the hydrogelation process to start.
As reported recently by Martin, Thordarson and coworkers, hydrophobic N-terminal capping groups on short peptide hydrogelators are involved in the formation of pre-assembled structures present in solution.14 Because of the Fmoc group present in Fmoc-A-SS-B peptide, 1H NMR analysis of this peptide in D2O was investigated (Fig. S10†). These analyses confirm that Fmoc-A-SS-B is in a pre-aggregated state, probably in fast equilibrium with discrete molecules. This aggregation involved phenylalanine and tyrosine residues of Fmoc-A-SS-B but not the Fmoc group which is in agreement with the absence of fluorescence excimer measurements (that appears from Fmoc stacking), as mentioned above. 1H NMR (D2O) monitoring of Fmoc-A-SH generated in situ from Fmoc-A-SS-B in presence of TCEP shows that Fmoc-A-SH is maintained in an aggregated state, despite the reduction of the disulfide bridge (Fig. S11†). The introduction of a protein such as BSA into the aqueous solution of Fmoc-A-SS-B leads to a full disappearance of all 1H NMR spectra signals (Fig. S12†), suggesting that all Fmoc-A-SH peptide are involved in supramolecular assembly.14
The design of complex chemical systems controlling their nanoarchitecture at the molecular scale through a bottom up approach15 can be envisaged using our self-assembly process: thanks to the initiator role of the protein, it is possible to spatially control the self-assembly process and thus the gelation buildup localization near surfaces. We thus covered surfaces by BSA or AP using an adequate polyelectrolyte multilayer3d,e,16 and brought these modified surfaces in contact with Fmoc-A-SS-B solutions. The spatially localized self-assembly process can be followed by quartz crystal microbalance with dissipation (QCM-D). Contact of these surfaces covered by BSA or AP with Fmoc-A-SS-B solutions leads to a huge and rapid decrease of the different measured frequencies and concomitantly an increase of the corresponding dissipation (Fig. 4c and S13 in ESI†). Such QCM-D signal evolutions are typical of the formation of highly hydrated gels exceeding several micrometers in thickness.3d,e Cryo-SEM analysis of the peptide self-assemblies grown up on surfaces covered by BSA or AP were performed (Fig. 4d and S14 in ESI†). In both cases one observes a fibrillar network that grows exclusively from the surface, with fibers oriented perpendicularly to the surface from the bottom (surface) to the top of the gel.3c The self-assembly appears thus the same for BSA and AP. The assembly of Fmoc-A-SH located on surfaces was also monitored by fluorescence emission spectroscopy, following the characteristic excimer emission of Fmoc group at 325 nm (when excited at 280 nm) and the β-sheet secondary structure adopted in the peptide assembly was confirmed by IR spectroscopy in ATR mode (Fig. S15 in ESI†). Similar results were obtained when the surface was covered by GR instead of BSA and AP (Fig. S16 in ESI†). To check the hypothesis discussed previously that Fmoc-A-SS-B interacts first onto positive patches of proteins to initiate the self-assembly process, we have used positively charged multilayer ended by the polycation, poly-L-lysine (PLL), and a negatively charged multilayer ended by the polyanion, poly-L-glutamic acid (PGA). The QCM-D monitoring shows clearly that an important mass is adsorbed strongly onto a polycationic polypeptide layer and does not adsorb on a polyanionic layer when brought in contact with Fmoc-A-SS-B (Fig. S17†). The frequency decrease observed with poly-L-lysine deposited on surface is similar to those observed using proteins (Fig. 4c, S13 and S16†). This means that the presence of the polypeptide cationic layer can allows the initiation of the whole self-assembly process. The absence of tertiary structure in poly-L-lysine suggests also that the electrostatic interaction is the sine qua none condition for Fmoc-A-SS-B to interact with proteins/polypeptides.
Conclusions
We have found a chemical pair “LMWH-precursor ” where both partners are in dynamic equilibrium. Interestingly, the self-assembly of the LMWH (hydrogelator) takes place only when it is initiated by the interaction between the protein and the precursor. This aspect can be exploited upon for the spatial localization of the self-assembly process by specific localization of proteins as demonstrated herein. From the mechanistic point of view, the interaction between the precursor and the protein leads to a self-assembly that acts as a scavenging process of the hydrogelator, displacing thus the “LMWH-precursor” equilibrium towards the full generation of the LMWH. Our study also highlights that adsorption of hydrogelators on enzymes (in the case GR) is a sine qua non step required to initiate the self-assembly. Despite many reports in enzyme-assisted self-assembly's field, this aspect was not yet unambiguously demonstrated. Chemical equilibrium generating a combinatorial library of peptides thanks to the use of an enzyme able to catalyse both peptides formation and peptides hydrolysis has been already reported3a,8 but is singularly different from the approach described here. Indeed, here it is the self-assembly itself that displaces the dynamic equilibrium without the action of the protein, this later being necessary to initiate the self-assembly through its interaction with the precursor of the LMWH. In the field of complex functional nanoarchitectures design which is typically based on bottom up bioinspired strategies, we are convinced that this new self-assembly process of LMWH initiated by proteins will echo to the chemical research community and beyond, for instance in medicine and biology in which localized supramolecular hydrogelation in living systems shows currently a remarkable development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the following financial supports from Agence Nationale de la Recherche (MECHANOCAT ANR-15-CE29-0015-02 and EASA ANR-18-CE06-0025-02), International Center for Frontier Research in Chemistry (ICFRC-no. 56), LabEx Chimie des Systèmes Complexes (CSC, PSC-016), and funding from Institut Carnot MICA. Electron microscopy and characterization platform from Institut Charles Sadron are acknowledged for all Cryo-SEM/TEM analyses and DLS formation respectively.Notes and references
- J.-M. Lehn, in Supramolecular Chemistry, Wiley-VCH, Weinheim, 1995 CrossRef CAS PubMed; T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed; E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111 CrossRef PubMed; J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276 CrossRef PubMed.
- S. Mann, Angew. Chem., Int. Ed., 2008, 47, 5306 CrossRef CAS PubMed; J. M. Barral and H. F. Epstein, BioEssays, 1999, 21, 813 CrossRef PubMed; R. Li and D. F. Albertini, Nat. Rev. Mol. Cell Biol., 2013, 14, 141 CrossRef PubMed.
- (a) R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19 CrossRef CAS PubMed; (b) R. J. Williams, T. E. Hall, V. Glattauer, J. White, P. J. Pasic, A. B. Sorensen, L. Waddington, K. M. McLean, P. D. Currie and P. G. Hartley, Biomaterials, 2011, 32, 5304 CrossRef CAS PubMed; (c) A. G. L. Olive, N. H. Abdullah, I. Ziemecka, E. Mendes, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2014, 53, 4132 CrossRef CAS PubMed; (d) C. Vigier-Carrière, T. Garnier, D. Wagner, P. Lavalle, M. Rabineau, J. Hemmerlé, B. Senger, P. Schaaf, F. Boulmedais and L. Jierry, Angew. Chem., Int. Ed., 2015, 54, 10198 CrossRef PubMed; (e) J. Rodon Fores, M. L. M. Mendez, X. Mao, D. Wagner, M. Schmutz, M. Rabineau, P. Lavalle, P. Schaaf, F. Boulmedais and L. Jierry, Angew. Chem., Int. Ed., 2017, 56, 15984 CrossRef CAS PubMed; (f) D. Spitzer, V. Marichez, G. J. M. Formon, P. Besenius and T. M. Hermans, Angew. Chem., Int. Ed., 2018, 57, 11349 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440 CrossRef CAS; Z. Yang and B. Xu, Adv. Mater., 2006, 18, 3043 CrossRef.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070 CrossRef CAS PubMed; K. Thornton, A. M. Smith, C. L. R. Merry and R. V. Ulijn, Biochem. Soc. Trans., 2009, 37, 660 CrossRef PubMed; R. J. Williams, R. J. Mart and R. V. Ulijn, Biopolymers, 2010, 94, 107 CrossRef PubMed.
- X. Qin, W. Xie, S. Tian, J. Cai, H. Yuan, Z. Yu, G. L. Butterfoss, A. C. Khuong and R. A. Gross, Chem. Commun., 2013, 49, 4839 RSC; B.-H. Hu and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 14298 CrossRef CAS PubMed; S. C. Bremmer, A. J. McNeil and M. B. Soellner, Chem. Commun., 2014, 50, 1691 RSC; L. Chronopoulou, S. Lorenzoni, G. Masci, M. Dentini, A. R. Togna, G. Togna, F. Bordi and C. Palocci, Soft Matter, 2010, 6, 2525 RSC; Y. Liu, V. Javvaji, S. R. Raghavan, W. E. Bentley and G. F. Payne, J. Agric. Food Chem., 2012, 60, 8963 CrossRef PubMed.
- C. G. Pappas, R. Shafi, I. R. Sasseli, H. Siccardi, T. Wang, V. Narang, R. Abzalimov, N. Wijerathne and R. V. Ulijn, Nat. Nanotechnol., 2016, 11, 960 CrossRef CAS PubMed; S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789 CrossRef PubMed; S. K. M. Nalluri and R. V. Ulijn, Chem. Sci., 2013, 4, 3699 RSC; D. B. Rasale, S. Biswas, M. Konda and A. K. Das, RSC Adv., 2015, 5, 1529 RSC.
- J. Plas, D. Waghray, J. Adisoejoso, O. Ivasenko, W. Dehaen and S. De Feyter, Chem. Commun., 2015, 51, 16338 RSC; B. Bartolec, M. Altay and S. Otto, Chem. Commun., 2018, 54, 13096 RSC.
- C. Yang, H. Wang, D. Li and L. Wang, Chin. J. Chem., 2013, 31, 494 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37 CrossRef CAS.
- R. Marti-Centelles and B. Escuder, ChemNanoMat, 2018, 4, 798 CrossRef CAS; E. R. Draper, H. Su, C. Brasnett, R. J. Poole, A. Seddon, H. Cui and D. J. Adams, Angew. Chem., Int. Ed., 2017, 129, 10603 CrossRef; C. Colquhoun, E. R. Draper, R. Schweins, M. Marcello, D. Vadukul, L. C. Serpell and D. J. Adams, Soft Matter, 2017, 13, 1914 RSC; K. L. Morris, L. Chen, A. Rodger, D. J. Adams and L. C. Serpell, Soft Matter, 2015, 11, 1174 RSC; J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Z. Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11, 927 RSC.
- N. Javid, S. Roy, M. Zelzer, Z. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368 CrossRef CAS PubMed.
- A. D. Martin, J. P. Wojciechowski, A. B. Robinson, C. Heu, C. J. Garvey, J. Ratcliffe, L. J. Waddington, J. Gardiner and P. Thordarson, Sci. Rep., 2017, 43947 CrossRef PubMed.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195 CrossRef CAS PubMed; K. Ariga, Q. Ji, W. Nakanishi, J. P. Hill and M. Aono, Mater. Horiz., 2015, 2, 406 RSC; B. Yang, D. J. Adams, M. Marlow and M. Zelzer, Langmuir, 2018, 34, 15109 CrossRef PubMed.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00312f |
| This journal is © The Royal Society of Chemistry 2019 |