
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled growth of imine-linked two-dimensional covalent organic framework nanoparticles†
Rebecca L.
Li
 a,
Nathan C.
Flanders
a,
Austin M.
Evans
a,
Woojung
Ji
a,
Ioannina
Castano
a,
Lin X.
Chen
ad,
Nathan C.
Gianneschi
abc and
William R.
Dichtel
*a
a,
Nathan C.
Flanders
a,
Austin M.
Evans
a,
Woojung
Ji
a,
Ioannina
Castano
a,
Lin X.
Chen
ad,
Nathan C.
Gianneschi
abc and
William R.
Dichtel
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL, 60208 USA. E-mail: wdichtel@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
cDepartment of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA
dChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 21st February 2019
Abstract
Covalent organic frameworks (COFs) consist of monomers arranged in predictable structures with emergent properties. However, improved crystallinity, porosity, and solution processability remain major challenges. To this end, colloidal COF nanoparticles are useful for mechanistic studies of nucleation and growth and enable advanced spectroscopy and solution processing of thin films. Here we present a general approach to synthesize imine-linked 2D COF nanoparticles and control their size by favoring imine polymerization while preventing the nucleation of new particles. The method yields uniform, crystalline, and high-surface-area particles and is applicable to several imine-linked COFs. In situ X-ray scattering experiments reveal the nucleation of amorphous polymers, which crystallize via imine exchange processes during and after particle growth, consistent with previous mechanistic studies of imine-linked COF powders. The separation of particle formation and growth processes offers control of particle size and may enable further improvements in crystallinity in the future.
Introduction
Covalent organic frameworks (COFs) are polymers with two-dimensional (2D) or three-dimensional (3D) periodic structures.1–4 Their straightforward design, thermal and chemical stability, crystallinity, and permanent porosity, show promise for applications including water purification,5,6 energy storage devices,7,8 and catalysis.9 However, methods to select and control the morphology, while achieving processability of imine-linked COFs remain underdeveloped, which precludes many of the above applications. Through empirical screening of reaction conditions, morphologies such as sea-urchin-shaped or hollow particles,10–12 core–shell structures,13,14 and tubes15 have been reported, but these structures are still isolated as insoluble microcrystalline powders, preventing further reprocessing. Developing methods to obtain COFs as thin films,16,17 few-layer structures18 and nanosized particles19,20 show promise for harnessing their structural precision for complex membranes, devices, and payload delivery.Colloidal COF nanoparticles are solution processable and amenable for in situ techniques to study their formation.19 Previously, we established nucleation and elongation growth regimes for boronate ester-linked 2D COF colloids by introducing monomers at different rates, which enabled the growth of single-crystal 2D COFs.20 COFs linked by imines and β-ketoenamines were reported as solution-stable suspensions more recently,21,22 and properties related to photothermal conversion,23 catalysis24 and chromatographic separations25 were explored. The promise of this form and its connection with controlling morphology and materials quality motivate the present study, in which we report solution-stable, imine-linked COF colloids without the need for templates or additives. The method produces high quality COF particles with uniform sizes and shapes and is shown to be general across three structures. Furthermore, we developed a seeded growth methodology derived from classical nucleation theory and inspired by boronate-ester COF studies that provided systematic size control.20In situ analysis of the size and crystallinity indicates that crystallization occurs more slowly than particle growth, which is consistent with previous mechanistic studies of imine-linked COF powders.26 This study of COF colloid formation probes mechanistic questions that are difficult to address when studying bulk powder samples and enables size and shape control for the first time.
Results and discussion
The condensation of 1,3,5-tris(4-aminophenyl)benzene (TAPB) and terephthaldehyde (PDA) in the presence of a transimination catalyst Sc(OTf)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 provides the TAPB–PDA COF, which was previously isolated as a polycrystalline, insoluble powder when prepared in a 4:1 mixture of 1,4-dioxane:mesitylene.28 When the polymerization was conducted in MeCN, a stable colloidal suspension of TAPB–PDA COF nanoparticles was obtained instead (Fig. 1a). The crystallinity of the colloids dispersed in their growth solvent was characterized using wide-angle X-ray scattering (WAXS) with peaks observed at q = 0.20, 0.35, and 0.40 Å−1, corresponding to the (100), (110), and (200) Bragg diffraction peaks, respectively, consistent with previously reported diffraction patterns.28 0.16 equivalents of Sc(OTf)3 per amine functional group provided the most crystalline materials as determined by the increased intensity and reduced full-width-at-half-max (FWHM) of the (100) Bragg diffraction peak (Fig. S1†). Dynamic light scattering (DLS) of these colloids provided an average diameter of 360 nm and a polydispersity less than 0.2 (Fig. S2†), which was consistent with atomic force microscopy (AFM) of drop-cast solutions which showed uniform particle height (Fig. S3†). Scanning electron microscopy (SEM) images revealed that these particles are spherical with rough surfaces (Fig. 1b and c). Transmission electron microscopy (TEM) images showed the dense stacking of sheet-like structures (Fig. 1d). Collectively, these diffraction and microscopy experiments demonstrate that crystalline COF nanoparticles with well-defined shape and size are formed and stabilized when prepared in MeCN.
27 provides the TAPB–PDA COF, which was previously isolated as a polycrystalline, insoluble powder when prepared in a 4:1 mixture of 1,4-dioxane:mesitylene.28 When the polymerization was conducted in MeCN, a stable colloidal suspension of TAPB–PDA COF nanoparticles was obtained instead (Fig. 1a). The crystallinity of the colloids dispersed in their growth solvent was characterized using wide-angle X-ray scattering (WAXS) with peaks observed at q = 0.20, 0.35, and 0.40 Å−1, corresponding to the (100), (110), and (200) Bragg diffraction peaks, respectively, consistent with previously reported diffraction patterns.28 0.16 equivalents of Sc(OTf)3 per amine functional group provided the most crystalline materials as determined by the increased intensity and reduced full-width-at-half-max (FWHM) of the (100) Bragg diffraction peak (Fig. S1†). Dynamic light scattering (DLS) of these colloids provided an average diameter of 360 nm and a polydispersity less than 0.2 (Fig. S2†), which was consistent with atomic force microscopy (AFM) of drop-cast solutions which showed uniform particle height (Fig. S3†). Scanning electron microscopy (SEM) images revealed that these particles are spherical with rough surfaces (Fig. 1b and c). Transmission electron microscopy (TEM) images showed the dense stacking of sheet-like structures (Fig. 1d). Collectively, these diffraction and microscopy experiments demonstrate that crystalline COF nanoparticles with well-defined shape and size are formed and stabilized when prepared in MeCN.
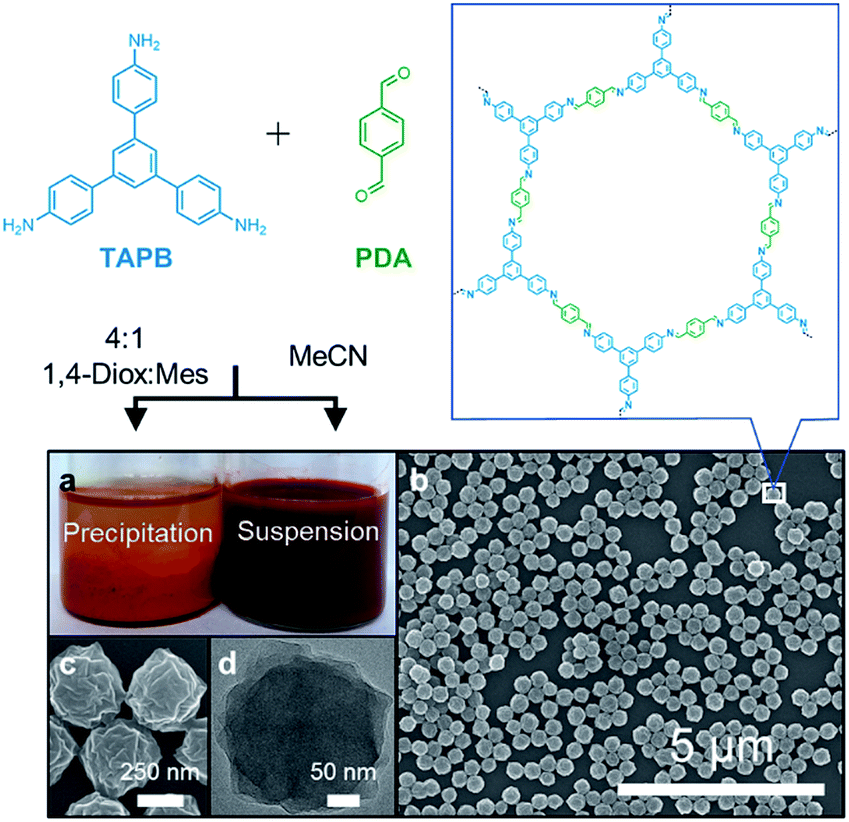 | ||
| Fig. 1 (a) Synthesis of imine-linked COFs with a typical solvent mixture of 4:1 1,4-dioxane:mesitylene yields insoluble polycrystalline precipitates, whereas MeCN produces stable colloidal suspensions. (b) SEM of drop-cast as synthesized colloidal suspensions (c) higher magnification SEM. (d) TEM of an individual COF nanoparticle. | ||
The colloids were precipitated by adding saturated aqueous NaCl, and the isolated solids were washed by Soxhlet extraction using MeOH for 12 hours and activated using supercritical CO2. The powder X-ray diffraction pattern (PXRD) of the isolated solids exhibited well-defined and relatively sharp (100), (110), (200), (210), (220), and (320) Bragg diffraction peaks (Fig. S4†), which matched well with an eclipsed model. The N2 adsorption isotherm of the precipitated colloids corresponded to a Brunauer–Emmett–Teller (BET) surface area of 2070 m2 g−1, from which nonlocal density functional theory analysis provided a narrow pore width distribution centered at 3.4 nm (Fig. S5–S7†). An infrared spectrum of the precipitated colloids showed the characteristic imine stretch at 1624 cm−1, along with no evidence of the carbonyl stretch of the PDA monomer at 1687 cm−1 and the amine stretches of the TAPB monomer at the range 3300–3500 cm−1, all of which match the reported spectra (Fig. S8†).28 Collectively, these bulk characterization techniques indicate that the precipitated COF nanoparticles are high-quality imine-linked COFs. Notably, once precipitated and isolated as solids, the colloids could not be re-dispersed in MeCN, indicating irreversible aggregation. However, the as-synthesized colloids were stable in solution for at least seven months, during which their size distribution remained almost unchanged (Fig. S9†).
The generality of the approach was tested by preparing other imine-linked 2D COF colloids. When TAPB was condensed with 4,4′-biphenyldicarbaldehyde (BDA) to synthesize a COF with larger pore volume than TAPB–PDA COF, the most crystalline colloids were also obtained using 0.16 equivalents of Sc(OTf)3 with features in the WAXS at q = 0.17, 0.29, 0.33 Å−1 corresponding to (100), (110) and (200) Bragg diffraction peaks, consistent with previous report (Fig. S10†).28 These colloids have an average diameter of 560 nm as measured using DLS and their morphology from SEM images showed spherical particles with spiked protrusions (Fig. S10†). We also prepared COF nanoparticles bearing functional side-chains by condensing TAPB with a derivative of PDA functionalized with azide-terminated diethylene glycol chains (1) to produce stable crystalline COF colloids. We previously incorporated this monomer into an imine-linked COF that exhibited high affinity and adsorption capacity for GenX and perfluorinated alkyl substances (PFAS) following the reduction of the azides to primary amines.5 Based on the in situ WAXS diffraction, 0.04 equivalents of Sc(OTf)3 per amine functional group afforded the most crystalline colloids with diffraction peaks at q = 0.2, 0.4 Å−1 corresponding to (100), (200) Bragg diffraction peaks, consistent with our previous report (Fig. S10†).5 DLS indicated an average diameter of 110 nm, and oblong particles in this size range were observed by SEM (Fig. S10†). These results demonstrate that MeCN stabilizes colloidal imine-linked 2D COF nanoparticles with different structures.
The size of the TAPB–PDA colloids was controlled by varying the initial monomer concentration. At a constant catalyst loading of 0.16 equivalents of Sc(OTf)3, the average diameter ranged from 200 nm to 330 nm, at initial TAPB concentrations of 0.53 mM to 4.25 mM (each employing 1.5 equiv. of PDA, Fig. S11†). The colloids remained spherical except at the lowest TAPB concentration of 0.53 mM, at which they exhibited a rougher surface morphology (Fig. S12†). In solvo WAXS diffraction of colloids showed a progression of decreasing crystallinity as starting TAPB concentrations are decreased from 4.25 mM to 0.53 mM. To eliminate the effect of low concentration on diffraction signal, colloids were isolated via centrifugation and packed into capillaries as wet solids for WAXS diffraction. Consistent with the PXRD pattern obtained from dried and activated colloid powders, WAXS diffraction of solids centrifuged from colloids all showed high order diffraction peaks including (110), (200), (210) Bragg diffraction peaks. However, the broadening of the (100) peak suggested that lower monomer concentration produces colloids with smaller crystalline domains (Fig. S13†). These findings indicate that both the particle size and average crystalline domain size decreases with the initial monomer concentration, which is inconsistent with a nucleation–elongation growth mechanism. These observations are more consistent with the initial formation of amorphous polymer nanoparticles that subsequently crystallize, which is consistent with previous mechanistic studies of imine-linked COF powders.26
In situ small-angle X-ray scattering (SAXS)/WAXS analysis of the imine-linked COF colloid formation provided a direct means to observe the different time scales of the polymerization and the crystallization processes. Condensation of TAPB and PDA at 0.16 equivalents of Sc(OTf)3 was carried out in a capillary with simultaneous SAXS/WAXS to determine the crystallinity and size of TAPB–PDA COF colloids as a function of reaction time (Fig. S14 and 15†). An average particle size of 283 nm in diameter was modelled from the SAXS region after 4 min (Fig. 2). However, no crystallinity was observed, as judged by the absence of the (100) diffraction peak. After 18 minutes, the average particle diameter had reached 390 nm, close to the final average diameter of 441 nm, and the FWHM of the (100) diffraction peak had narrowed by 50% relative to its first measurable value. At extended reaction times, as the particles reached their final size, this peak continued to sharpen, indicating that the average crystallite size continued to increase, presumably through continuous imine exchange processes. Fitting the particle size curve with respect to time revealed two time components in the kinetics of particle growth. An initial fast component with a time constant of 382 seconds suggests the rapid formation of amorphous colloidal polymer particles. In addition, the subsequent slow component with a time constant of 1274 seconds matches with reasonable agreement with the time constant of 1600 seconds determined from the fit of the FWHM change with time suggesting crystallization of amorphous polymer. These results demonstrate that the polymerization and the crystallization processes of imine-linked 2D COFs are separate processes that occur at different rates.
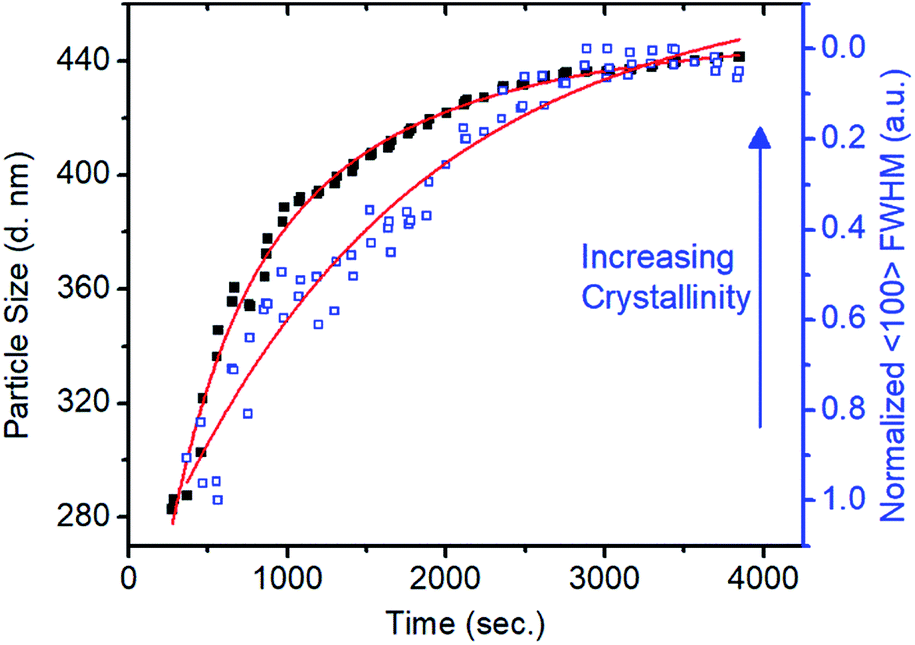 | ||
| Fig. 2 Relationship between particle size and crystallinity in terms of FWHM with respect to time. Curve fit (red) illustrates the different rate constants of the polymerization and crystallization processes. | ||
The polymerization conditions that generate crystalline materials limit nanoparticle size to a narrow range. Therefore, we developed a seeded growth approach to access larger particles along with excellent size control. Separate solutions of TAPB (8.5 mM) and PDA (12.7 mM) were added simultaneously to a solution of TAPB–PDA COF colloids using a syringe pump. Additional Sc(OTf)3 (4.08 mM) was included in the PDA solution such that the catalyst concentration and percent loading relative to the nitrogen-containing functional groups remained constant throughout the addition. Depending on the addition rate, two growth regimes were observed: the nucleation of new particles and the growth of existing particles. When monomers were added at a fast rate of 50 equiv. h−1, the average particle diameter as tracked by DLS first increased from 330 nm to 490 nm after two equivalents of monomers were added, but then decreased to 230 nm after six equivalents were added (Fig. 3a). A bimodal particle size distribution was observed after four equivalents of new monomers were added, whose number average distribution was dominated by newly nucleated particles, along with the original populations as a minor component (Fig. S16†). In contrast, when the rate of monomer addition was decreased to 1 equiv. h−1, the average diameter increased from 330 nm to 690 nm after six equivalents of monomers were added, and the size distributions remained monomodal (Fig. 3c and d). These measurements suggest that the added monomers attach to and enlarge the initial colloids instead of forming new colloids.
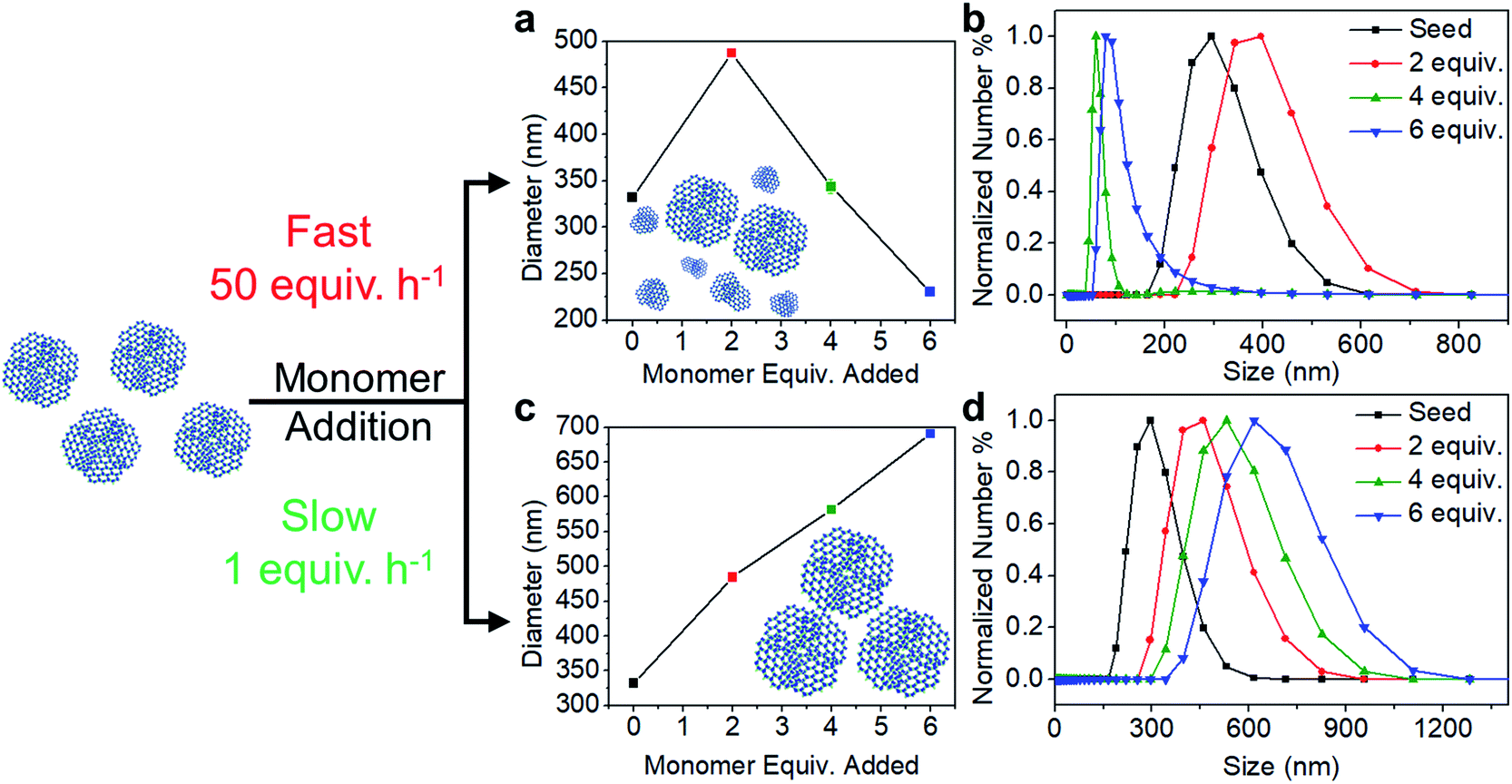 | ||
| Fig. 3 Trend in the average size of particles with monomer addition at a fast rate of 50 equiv. h−1 and at a slow rate of 1 equiv. h−1. (a, c) Average size of particles with additional equivalents of monomers (b, d) corresponding number percent distribution. | ||
The morphologies of particles subjected to different monomer addition rates differ dramatically, as observed by SEM. Micrographs of particles obtained from fast monomer addition (50 equiv. h−1) showed clear evidence of the formation of new particles. Smaller particles are observed along with larger particles, which correspond to the original seeds, after four equivalents of monomers were added. Even more new particles are observed in the micrographs when six equivalents of monomers were added (Fig. 4a–c). However, when the monomers were added at 1 equiv. h−1, the particles maintained their uniform spherical morphology without the appearance of additional species (Fig. 4d–f). Particles consistently enlarged in size after 2, 4, and 6 equivalents of monomers were added.
 | ||
| Fig. 4 SEM images of colloidal suspension at 50 equiv. h−1 (a–c) and 1 equiv. h−1 (d–f) of monomer addition. Aliquots were drop-casted after (a, d) 2 equivalents, (b, e) 4 equivalents, and (c, f) 6 equivalents of monomers were added. | ||
In solvo WAXS diffraction experiments also differentiate between the nucleation and growth regimes and are consistent with an amorphous to crystalline transition even in the seeded growth regime. With fast monomer addition (50 equiv. h−1) the drop in intensity to FWHM ratio of the (100) diffractions peak suggests the decrease in the average domain size of the COF colloids (Fig. 5a). This finding is consistent with DLS measurements and SEM images that indicate the formation of new particles with small crystalline domains. Under slow addition, the crystalline domain sizes remained unchanged as demonstrated by the similar FWHM of the (100) diffraction peak (Fig. 5b). Given that we see no evidence of the nucleation of new particles under these conditions, it is plausible that the monomers and oligomers initially attach to existing colloids as an amorphous shell, which subsequently crystallize with domain size similar to those obtained during the initial polymerization.
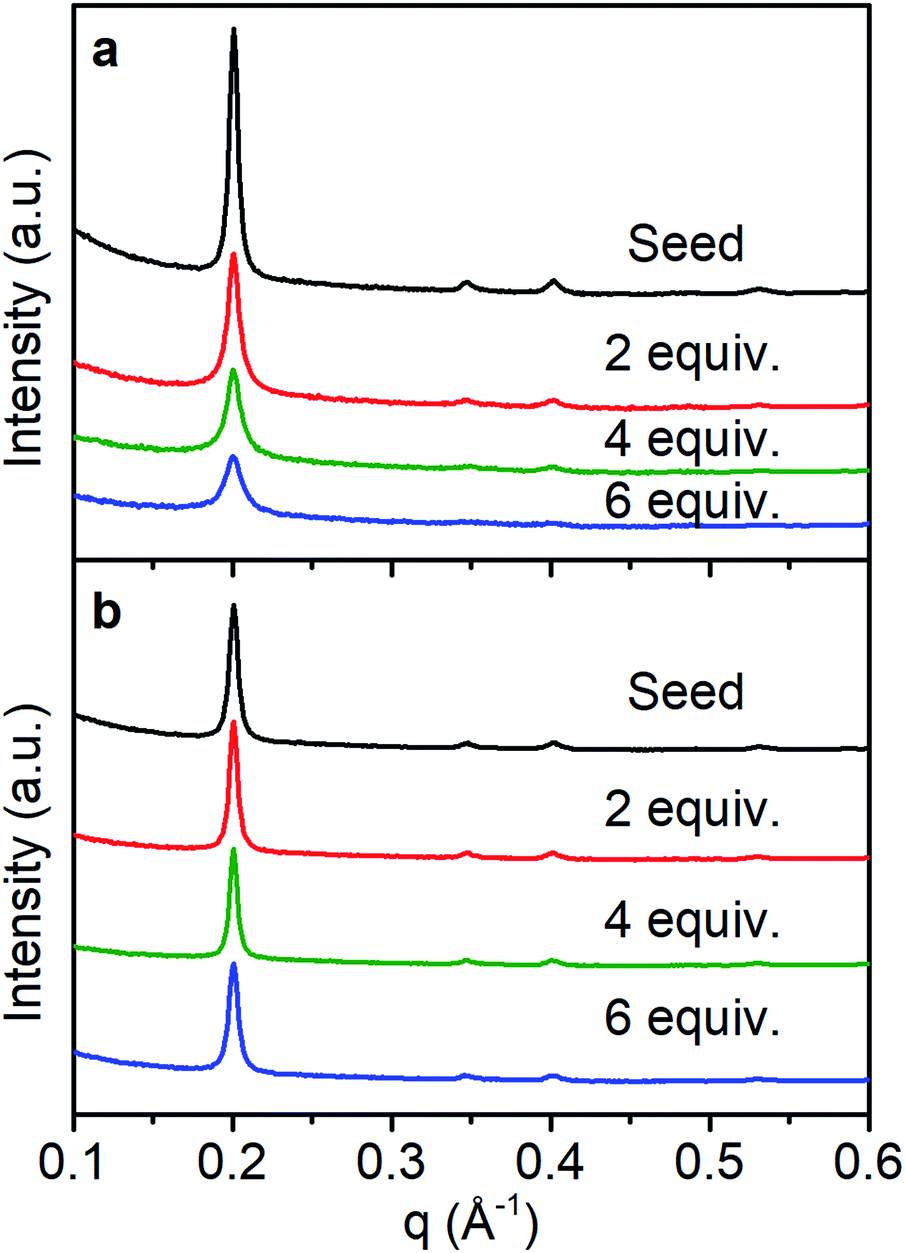 | ||
| Fig. 5 WAXS diffraction monitoring change in crystallinity as monomers are added with a rate of (a) 50 equiv. h−1, (b) 1 equiv. h−1. | ||
Conclusions
Imine-linked 2D COFs were synthesized as colloidal nanoparticles with uniform morphology, well-defined size, good crystallinity and BET surface areas over 2000 m2 g−1. In situ SAXS/WAXS experiments indicate that COF particles form faster than the framework crystallizes, highlighting an amorphous to crystalline transition in the homogeneous nucleation and growth stages of these nanoparticles. A seeded growth strategy with slow monomer addition provided a means to enlarge particles up to at least 690 nm. Imine-linked COF colloids represent a convenient form for solution processing, catalysis, payload delivery, among other potential uses for well-defined, porous materials that form stable solutions. Further improvements in the process, such as achieving seeds with single crystalline domains, might be used to further improve the quality of this versatile, emerging class of two-dimensional polymer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Army Research Office for a Multidisciplinary University Research Initiatives (MURI) award under grant number W911NF-15-1-0447. Parts of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and the Dow Chemical Company. This research used resources of the Advanced Photon Source and Center for Nanoscale Materials, both U.S. Department of Energy (DOE) Office of Science User Facilities operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC0206CH11357. A. M. E. is supported by the National Science Foundation Graduate Research Fellowship under Grant No. (DGE-1324585), the Ryan Fellowship and the Northwestern University International Institute for Nanotechnology. This work has also made use of the IMSERC, EPIC, and Keck II facility of NUANCE Center at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205), the MRSEC program (NSF DMR-1720139) at the Materials Research Center, the Keck Foundation, the State of Illinois and International Institute for Nanotechnology (IIN). N. C. F. and L. X. C. are partially supported by Basic Energy Science, CBG Division, US Department of Energy through Argonne National Laboratory under Contract No. DE-AC02-06CH11357. I. C. is partially supported by the Army Research Office under grant W911NF-18-1-0359 and by the National Science Foundation under Grant No. (DMR-1720139). Author contributions R. L. L., N. C. F., A. M. E., W. J., and W. R. D. performed and interpreted the colloid growth and characterizations. R. L. L., N. C. F., A. M. E., L. X. C., and W. R. D. performed and interpreted the in situ X-ray diffraction experiments. I. C. and N. C. G. performed and interpreted TEM experiments. All authors wrote and revised the manuscript.Notes and references
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef CAS PubMed.
- W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905 CrossRef CAS PubMed.
- S. Kim, C. Park, M. Lee, I. Song, J. Kim, M. Lee, J. Jung, Y. Kim, H. Lim and H. C. Choi, Adv. Funct. Mater., 2017, 27, 1700925 CrossRef.
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed.
- Z.-J. Yin, S.-Q. Xu, T.-G. Zhan, Q.-Y. Qi, Z.-Q. Wu and X. Zhao, Chem. Commun., 2017, 53, 7266–7269 RSC.
- Y. Peng, M. Zhao, B. Chen, Z. Zhang, Y. Huang, F. Dai, Z. Lai, X. Cui, C. Tan and H. Zhang, Adv. Mater., 2017, 30, 1705454 CrossRef PubMed.
- G. Zhang, M. Tsujimoto, D. Packwood, N. T. Duong, Y. Nishiyama, K. Kadota, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2018, 140, 2602–2609 CrossRef CAS PubMed.
- B. Gole, V. Stepanenko, S. Rager, M. Grüne, D. D. Medina, T. Bein, F. Würthner and F. Beuerle, Angew. Chem., Int. Ed., 2018, 57, 846–850 CrossRef CAS PubMed.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Díaz Díaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef CAS PubMed.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52 CrossRef CAS PubMed.
- Y. Zhao, L. Guo, F. Gándara, Y. Ma, Z. Liu, C. Zhu, H. Lyu, C. A. Trickett, E. A. Kapustin, O. Terasaki and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 13166–13172 CrossRef CAS PubMed.
- D. Rodriguez-San-Miguel, J. J. Corral-Perez, E. Gil-Gonzalez, D. Cuellas, J. Arauzo, V. M. Monsalvo, V. Carcelen and F. Zamora, CrystEngComm, 2017, 19, 4872–4876 RSC.
- J. Tan, S. Namuangruk, W. Kong, N. Kungwan, J. Guo and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 13979–13984 CrossRef CAS PubMed.
- D. Rodríguez-San-Miguel, A. Yazdi, V. Guillerm, J. Pérez-Carvajal, V. Puntes, D. Maspoch and F. Zamora, Chem.–Eur. J., 2017, 23, 8623–8627 CrossRef PubMed.
- C.-X. Yang, C. Liu, Y.-M. Cao and X.-P. Yan, Chem. Commun., 2015, 51, 12254–12257 RSC.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC.
- N. Giuseppone, J.-L. Schmitt, E. Schwartz and J.-M. Lehn, J. Am. Chem. Soc., 2005, 127, 5528–5539 CrossRef CAS PubMed.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional characterization. See DOI: 10.1039/c9sc00289h |
| This journal is © The Royal Society of Chemistry 2019 |