
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-stabilized diazoalkane synthesis via the oxidation of free hydrazones by iodosylbenzene and application in in situ MIRC cyclopropanation†
Emmanuelle M. D.
Allouche
and
André B.
Charette
 *
*
Department of Chemistry, Université de Montréal, P. O. Box 6128 Station Downtown, Montreal, Quebec, H3C 3J7 Canada. E-mail: andre.charette@umontreal.ca
First published on 19th February 2019
Abstract
Electron-rich alkyl diazo compounds are powerful reagents in organic synthesis, but the risks associated with their toxicity and instability often limit their uses. Herein we describe an efficient, easy-to-handle and safe batch protocol for the in situ generation and cyclopropanation of these highly reactive non-stabilized diazoalkanes through the oxidation of free hydrazones using iodosylbenzene. Numerous substituted cyclopropanes have been synthesized using this methodology, including various gem-dimethylcyclopropanes of particular interest in medicinal chemistry.
Introduction
Diazo compounds are very powerful and versatile reagents that are often used for the synthesis of complex molecules.1 Driven by the release of molecular nitrogen, they are key intermediates to highly reactive carbenes, carbenoids or carbocations, thus enabling formal C–H, C–C and X–H bond insertions, while their intrinsic nucleophilicity also allows various cycloaddition reactions.2 However, this versatility comes at a price, as their hazardousness often discourages the full exploitation of their chemical potential.3,4 Among the different categories of diazo compounds, the non-stabilized ones bearing aliphatic substituents are particularly notorious due to their instability and toxicity.Classic methodologies such as the decomposition of N-arylsulfonylhydrazones5 (Bamford–Stevens reaction6) or the photolysis of 1,3,4-oxadiazolines7 have been applied to the synthesis of these compounds, but present various drawbacks. While the harsh conditions required to decompose N-tosylhydrazones are hardly compatible with the desired non-stabilized diazo compounds,5 the use of 1,3,4-oxadiazolines implies an additional preparation step and the use of a specific set-up.7 The oxidation of free hydrazones has also been investigated in batch and continuous flow systems by us and others, and allows the synthesis of the desired compounds under milder reaction conditions while being more atom-economical. In these methodologies, however, stoichiometric amounts of transition metals such as Ag2O8 or the toxic HgO9 and Pb(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 were needed. Alternatively, organic oxidants have also been used, although few examples of non-stabilized diazoalkanes were described and in generally lower yields than their stabilized analogs.11 Among these organic oxidants, hypervalent iodine(III) compounds appear to be efficient, although the diazo compounds generated this way cannot be recovered as they do react right away with the carboxylate counter-anion when classical reagents as PhI(OAc)2 or PhI(OCOCF3)2 are used.12 To overcome this, the Myers group used PhIF2 to oxidize TBS-protected hydrazones, avoiding the neutralization of the diazo compound therefore generated (Scheme 1a).13 The TBS hydrazones were used in this case as it was believed that the low yields generally observed were due to the instabilities of the diazo compounds and also the starting free alkylhydrazones under the reaction conditions.14 Although effective, this strategy is quite demanding, as it requires the preparation of an iodine reagent that cannot be isolated and stored due to its low stability.15 Finally, the oxidation step requires the use of a large excess of base which can be an issue with base-sensitive functionalities.
10 were needed. Alternatively, organic oxidants have also been used, although few examples of non-stabilized diazoalkanes were described and in generally lower yields than their stabilized analogs.11 Among these organic oxidants, hypervalent iodine(III) compounds appear to be efficient, although the diazo compounds generated this way cannot be recovered as they do react right away with the carboxylate counter-anion when classical reagents as PhI(OAc)2 or PhI(OCOCF3)2 are used.12 To overcome this, the Myers group used PhIF2 to oxidize TBS-protected hydrazones, avoiding the neutralization of the diazo compound therefore generated (Scheme 1a).13 The TBS hydrazones were used in this case as it was believed that the low yields generally observed were due to the instabilities of the diazo compounds and also the starting free alkylhydrazones under the reaction conditions.14 Although effective, this strategy is quite demanding, as it requires the preparation of an iodine reagent that cannot be isolated and stored due to its low stability.15 Finally, the oxidation step requires the use of a large excess of base which can be an issue with base-sensitive functionalities.
 | ||
| Scheme 1 (a) Esterification reaction of carboxylic acids with diazoalkanes generated in situ by the oxidation of N-tert-butyldimethylsilylhydrazones with (difluoroiodo)benzene. (b) This work: an easy-to-handle procedure for the in situ generation and cyclopropanation of a broad range of mono- and bis-alkyl diazo compounds. quant. = quantitative yield. | ||
In constant research of easy-to-handle, safe and greener methodologies, we envisioned that the development a general batch preparation of a large range of diazoalkanes from free-hydrazones would unleash their synthetic potential.
Results and discussion
We deemed that iodosylbenzene would be the perfect oxidizing reagent as its reduction would only generate one equivalent of the benign iodobenzene and water as byproducts (Scheme 1b). The group of Cai reported the successful oxidation of the benzophenone-derived hydrazone with this reagent for the development of a one-pot nickel(II)-catalyzed cyclopropanation.16 While this hydrazone is known to be stable and the corresponding semi-stabilized diazo compound is known to be more resilient, mono- and bis-alkyl hydrazones are prone to decompose into azines under the reaction conditions. We envisioned that a slow addition of a solution of the free hydrazone to a suspension of the oxidant would allow us to prevent its degradation. In order to quantify the amount of diazo formed, we would then quench the reaction mixture with dimethyl acetylenedicarboxylate to form the cycloaddition product. During our first attempt and using the dimethyl hydrazone 1a, we were pleased to observe 69% of the desired product 2a using only 1 equivalent of PhIO and 1.5 equivalents of the alkyne at −20 °C in dichloromethane and without any base, meaning that at least that much of 2-diazopropane has been formed after only 5 minutes (Scheme 2).17 | ||
| Scheme 2 First attempt of 2-diazopropane generation with iodosylbenzene as the oxidant. The reaction was run on a 0.2 mmol scale. Target concentration = 0.1 M. Yield of the diazo generated based on the 1H NMR yield of the [3 + 2] cycloadduct 2a with dimethyl acetylenedicarboxylate (triphenylmethane was used as the internal standard). | ||
As diazoalkanes have pronounced nucleophilicity,18 we thought that this promising strategy would be a practical way to synthesize highly substituted cyclopropanes by in situ Michael-induced ring-closure (MIRC) cyclopropanation of α,β-unsaturated carbonyl compounds, similar to the Corey–Chaykovsky reaction.19,20Gem-dimethylcyclopropanes21 are of particular interest as they are frequently found in many natural and non-natural products of clinical interest, the gem-dimethyl moiety being known to improve many biological properties.22 Thus, initial investigations focused on dimethyl hydrazone 1a for the generation and cyclopropanation of 2-diazopropane. A slow addition of a solution of 1.5 equivalents of 1a to a suspension of 1.5 equivalents of iodosylbenzene and 1 equivalent of isobutyl methacrylate would allow the slow generation of the desired diazo compound and its direct cyclopropanation, avoiding possible side reactions and accumulation of dangerous species. In our first attempt at −20 °C, we were able to witness the formation of the desired diazo compound but also its consumption by the apparition and fading of a pink colour. Once the addition over, the reaction was allowed to warm up to room temperature for 30 minutes, causing gas evolution. All those observations suggested that the diazo was formed and reacted with the alkene at low temperature via a [3 + 2] cyclization, followed at higher temperature by a ring contraction through nitrogen extrusion (Scheme 3), in contrast to the above-stated Corey–Chaykovsky reaction.23 After quenching, extraction and subsequent evaporation of the volatiles, 91% of the desired cyclopropane 3a was observed by 1H NMR (Table 1, Entry 1). Running the whole reaction at room temperature appeared to be beneficial, as the desired product was isolated in a quantitative yield (Table 1, Entry 2). Concerned about the environmental impact of our methodology, we decided to try our reaction in ethyl acetate, recognized as a green solvent. Here again, we were happy to isolate the desired cyclopropane 3a in quantitative yield when dry ethyl acetate was used and the reaction was run under argon (Table 1, Entry 3). We then tried to push the user-friendly aspect of this methodology even further by using from-the-bottle ethyl acetate and running the reaction without an inert atmosphere. We were delighted to isolate the desired product in a lower but still very good 81% yield (Table 1, Entry 4), showing the relative robustness of this methodology. Despite our attempts, however, lowering the equivalents of either iodosylbenzene or hydrazone always resulted in lower yields.24
 | ||
| Scheme 3 Proposed mechanism for the cyclopropanation formation through a 1,3-dipolar cycloaddition pathway followed by nitrogen extrusion. EWG = electron withdrawing group. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T 1 (°C) | T 2 (°C) | Yield (%) |
| a Determined by 1H NMR (triphenylmethane was used as the internal standard). b Isolated yield. c The reaction was run with from-the-bottle ethyl acetate under air. | ||||
| 1 | CH2Cl2 | −20 °C | −20 °C to rt | 91%a |
| 2 | CH2Cl2 | rt | rt | quant.b |
| 3 | EtOAc | rt | rt | quant.b |
| 4c | EtOAc | rt | rt | 81%b |
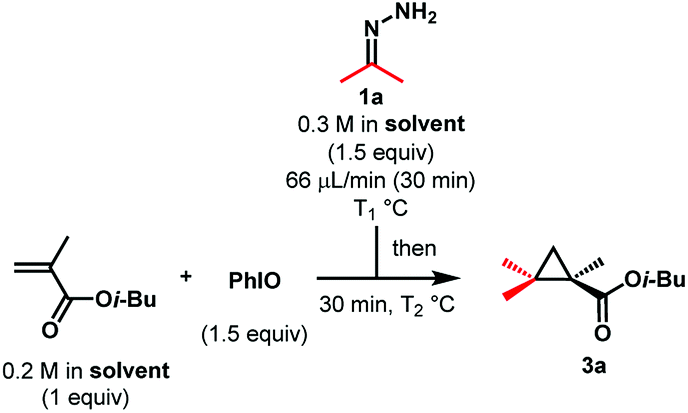
With these optimized conditions in hand, we investigated the scope of the reaction using hydrazone 1a on various Michael acceptors (Scheme 4). The 1-fluoro-2-2-gem-dimethyl cyclopropane 4a was obtained in an excellent 86% yield, although its brominated analogue 5a gave a much more moderate yield of 54% due to the propensity of the starting bromoalkene to quickly polymerize. It is noteworthy that despite this reaction being effective in ethyl acetate, these two entries were run in dichloromethane, as the corresponding products happen to be quite volatile. Enolizable positions are well tolerated under our neutral conditions as shown by the quantitative yield obtained for compound 6a. Tertiary amines and secondary amides do not harm the reaction either as compounds 7a and the amino-acid derivatives 8a and 9a were isolated in quantitative yields too. Compound 8a has also been synthesized on a 2.4 mmol scale (6-fold scale-up), under air and with from-the bottle ethyl acetate, the desired cyclopropane being isolated in a still good 79% yield.24 After showing that nitrile-containing molecules were viable substrates as compound 10a was isolated in a good 84% yield, we decided to investigate α,β-disubstituted Michael acceptors. While the reaction proceeded in a moderate 52% yield when a linear substrate was used (compound 12a), more constrained systems such as chromenone and coumarin derivatives gave the corresponding cyclopropanes 13a and 15a in quantitative yields. Once again, replacing the ester functionality by a nitrile had no effect on the reaction outcome (compound 14a). Ketone-containing Michael acceptors gave lower yields, as illustrated by compounds 16a and 17a being isolated in 52 and 51% yields, respectively. Unsurprisingly, these entries show that non-conjugated alkenes remain untouched. Interestingly, only 27% of the desired compound 11a was observed by 1H NMR when the simple tert-butyl acrylate was used and despite complete consumption of the starting material. In this case, the corresponding pyrazole was observed, indicating that steric bulk is an important factor for the ring contraction through nitrogen extrusion under these reaction conditions (Scheme 3).
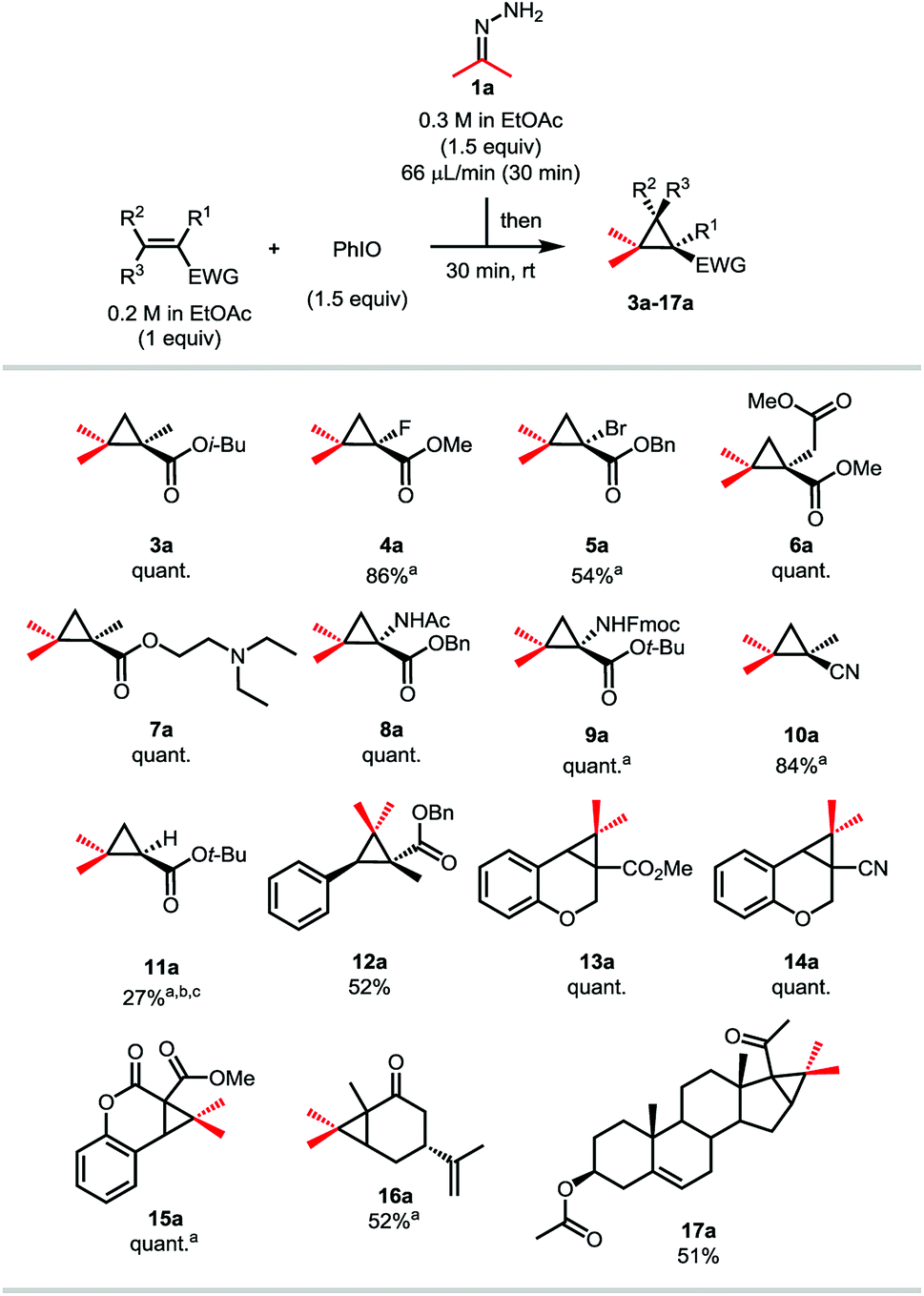 | ||
| Scheme 4 Scope of the Michael acceptors to produce gem-dimethylcyclopropanes. The reactions were run on a 0.4 mmol scale. Target concentration = 0.1 M. EWG = electron withdrawing group. quant. = quantitative yield. aThe reactions were run in CH2Cl2 instead of EtOAc. b1H NMR yield on the crude mixture. cFull conversion of the starting olefin. | ||
Having demonstrated that a wide variety of diverse pharmacologically potent gem-dimethylcyclopropanes can easily be accessed using the one-pot diazo-generation/MIRC cyclopropanation strategy, we then decided to focus our attention on the diazo partner. As stated previously, the bigger advantage of this methodology would be to allow the batch synthesis of a multitude of different unstable diazo compounds and their participation in subsequent reactions without having to worry about their accumulation or their stability. Isobutyl methacrylate and benzyl 2-acetamidoacrylate were chosen as model substrates, as the latter potentially allows the synthesis of unnatural cyclopropyl amino acids (Scheme 5). Hydrazones generating symmetrical diazo compounds were attempted at first, and we observed that substituting the previous hydrazone 1a with phenyl rings does not have a significant impact on the reaction, the desired product 8b being isolated in a 96% yield. A multitude of spiro compounds were then successfully synthesized, being of particular interest in drug discovery.25 The spiro[2,3]hexane derivative 3c was isolated in a moderate 71% yield, while the spiro[2,4]heptane, [2,5]octane and [2,6]nonane, compounds 3d, 8e and 3j, were obtained quantitatively. As proven by compounds 8f and 3g, substitution on the cyclohexyl hydrazone does not affect the efficiency of the reaction. Compound 8f is of particular interest as the 4,4-dimethylcyclohexane moiety is an important motif for medical chemistry applications.22c Hydrazones derived from heterocycles can be used as well, as shown by the excellent yields obtained for products 8h and 3i. Similarly to the cyclobutyl hydrazone, the related cyclooctyl gave a still acceptable but lower yield of 61% of the corresponding product 8k. These lower yields are believed to be due to the higher instability of the diazo compounds.26 Non-symmetrical hydrazones were then tested. 2-diazo-3-methylbutane was efficiently generated, the product 3l being isolated in a quantitative yield, although as a mixture of isomers presenting a low diastereoselective ratio. Diazo compounds bearing derivatizable functional groups were also successfully generated and cyclopropanated. Compounds 3m and 3n bearing a TBS protected alcohol or a terminal alkene were isolated in excellent 97 and 98% yields, respectively. Low diastereoselectivities were however also observed in both cases. Among all the different hydrazone categories, the monoalkyl ones are known to be the least stable,8b,13,14 and therefore usually give lower yields.8b Thanks to our dropwise addition and fast oxidation, the diazo intermediate was generated in a very efficient way from isobutylhydrazone, the [3 + 2] product 17o being isolated in a good 78% yield. Similar to product 11a in the previous scope, we think that steric repulsion on this compound is not sufficient to provoke the nitrogen extrusion at room temperature (Scheme 3). In order to display the versatility of the iodosylbenzene reagent, we extended our methodology to the more stable aryl- and heteroaryldiazomethanes.27 We were pleased to obtain excellent to quantitative yields for compounds 8p, 8q, 8r and 8s, all of them however displaying moderate diastereoselectivities.
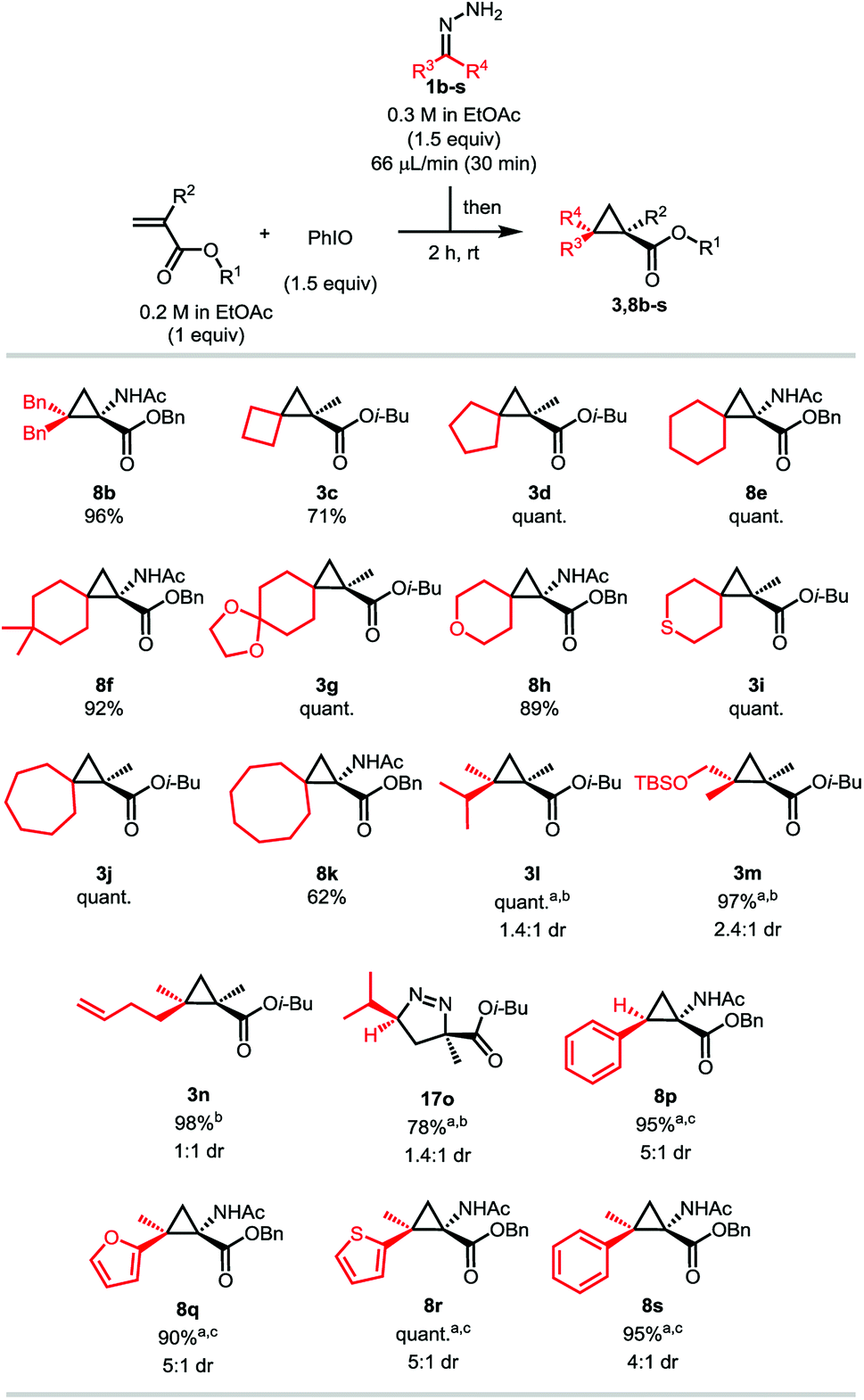 | ||
| Scheme 5 Scope of the hydrazones. The reactions were run on a 0.4 mmol scale. Target concentration = 0.1 M. quant. = quantitative yield. aThe major diastereomer is represented. bYields on the isolated mixture of both combined diastereomers. Diastereomeric ratio determined by 1H NMR on the isolated mixture. cCombined yields of the two diastereomers. Diastereomeric ratio determined according to the isolated yields of both diastereomers. | ||
Conclusions
In summary, we have developed an efficient, facile and safe batch preparation of a broad range of highly unstable mono- and bis-alkyl diazo compounds starting from free hydrazones and using a benign hypervalent iodine reagent. Applying this methodology to an in situ MIRC cyclopropanation reaction, these green and atom-economical conditions not only offer an easy access to gem-dimethyl cyclopropanes, but also to a large range of new highly substituted moieties. We were able to demonstrate a remarkably broad reaction scope and high functional-group tolerance, on top of very easy, simple and user-friendly setups and manipulations. These cyclopropanations are however not the only possible application: the neutral conditions of our oxidation step added to the quite inert nature of the few by-products generated–water and iodobenzene–promise the development of various other in situ processes, therefore finally unleashing their potential without having to ponder over the associated risks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-06438, the Canada Foundation for Innovation Leaders Opportunity Funds 227346, the Canada Research Chair Program CRC-227346, the FRQNT Centre in Green Chemistry and Catalysis (CGCC) Strategic Cluster RS-171310, and Université de Montréal. E. M. D. A. is grateful to Université de Montréal for postgraduate scholarships.Notes and references
- M. Regitz and G. Maas, Diazo Compounds, Academic Press, 1986, pp. 3–64 Search PubMed.
- (a) M. P. Doyle, M. A. McKervey and Y. Tao, in Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley-VCH, Weinheim, 1998, pp. 1–65 Search PubMed; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed; (c) K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233–3244 CrossRef CAS PubMed.
- (a) T. J. de Boer and H. J. Backer, Org. Synth., 1956, 36, 16–18 CrossRef CAS; (b) T. Sammakia, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001, pp. 1–7 Search PubMed; (c) C. D. Gutsche, Org. React., 2004, 364–430 Search PubMed.
- (a) E. B. Lewinn, Am. J. Med. Sci., 1949, 218, 556–562 CrossRef CAS PubMed; (b) R. Schoental, Nature, 1960, 188, 420–421 CrossRef CAS PubMed; (c) C. E. Lewis, J. Occup. Environ. Med., 1964, 6, 91–92 CAS; (d) P. A. Bray and R. K. Sokas, J. Occup. Environ. Med., 2015, 57, 15–16 CrossRef PubMed.
- (a) D. F. Taber and P. Guo, J. Org. Chem., 2008, 73, 9479–9481 CrossRef CAS PubMed; (b) D. F. Taber, P. Guo and N. Guo, J. Am. Chem. Soc., 2010, 132, 11179–11182 CAS; (c) J. Barluenga, N. Quinones, M. Tomas-Gamasa and M.-P. Cabal, Eur. J. Org. Chem., 2012, 2312–2317 CrossRef CAS; (d) D. M. Allwood, D. C. Blakemore and S. V. Ley, Org. Lett., 2014, 16, 3064–3067 CrossRef CAS PubMed; (e) A. R. Reddy, C.-Y. Zhou, Z. Guo, J. Wei and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 14175–14180 CrossRef CAS PubMed; (f) A. R. Reddy, F. Hao, K. Wu, C.-Y. Zhou and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 1810–1815 CrossRef CAS PubMed.
- W. R. Bamford and T. S. Stevens, J. Chem. Soc., 1952, 4735–4740 CAS.
- (a) M. W. Majchrzak, M. Bekhazi, I. Tse-Sheepy and J. Warkentin, J. Org. Chem., 1989, 54, 1842–1845 CrossRef CAS; (b) A. Greb, J.-S. Poh, S. Greed, C. Battilocchio, P. Pasau, D. C. Blakemore and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 16602–16605 CrossRef CAS PubMed.
- (a) D. E. Applequist and H. Babad, J. Org. Chem., 1962, 27, 288–290 CrossRef CAS; (b) P. Rullière, G. Benoit, E. M. D. Allouche and A. B. Charette, Angew. Chem., Int. Ed., 2018, 57, 5777–5782 CrossRef PubMed.
- A. C. Day, P. Raymond, R. M. Southam and M. C. Whiting, J. Chem. Soc. C, 1966, 467–469 RSC.
- (a) T. L. Holton and H. Schechter, J. Org. Chem., 1995, 60, 4725–4729 CrossRef CAS; (b) A. J. Wommack and J. S. Kingsbury, J. Org. Chem., 2013, 78, 10573–10587 CrossRef CAS PubMed.
- (a) M. I. Javed and M. Brewer, Org. Lett., 2007, 9, 1789–1792 CrossRef CAS PubMed; (b) C. Perusquía-Hernández, G. R. Lara-Issasi, B. A. Frontana-Uribe and E. Cuevas-Yañez, Tetrahedron Lett., 2013, 54, 3302–3305 CrossRef.
- D. H. R. Barton, J. C. Jaszberenyi, W. Liu and T. Shinada, Tetrahedron, 1996, 52, 14673–14688 CrossRef CAS For other iodine reagents used for oxidation of hydrazones into diazo compounds, see: (a) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef CAS PubMed; (b) S. M. Nicolle and C. J. Moody, Chem.–Eur. J., 2014, 20, 4420–4425 CrossRef CAS PubMed; (c) D. Rackl, C.-J. Yoo, C. W. Jones and H. M. L. Davies, Org. Lett., 2017, 19, 3055–3058 CrossRef CAS PubMed.
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222–12223 CrossRef CAS PubMed.
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 5436–5445 CrossRef CAS PubMed.
- G. Nikonov, Benzene, (Difluoroiodo)-, e-EROS Encyclopedia of Reagents for Organic Synthesis, 2011 Search PubMed.
- H. Liu, Y. Wei and C. Cai, New J. Chem., 2016, 40, 674–678 RSC.
- The mass balance loss has been attributed to the eventual degradation of 2-diazopropane prior to the quench with dimethyl acetylenedicarboxylate..
- T. Bug, M. Hartnagel, C. Schlierf and H. Mayr, Chem.–Eur. J., 2003, 9, 4068–4076 CrossRef CAS PubMed.
- (a) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1962, 84, 867–868 CrossRef CAS; (b) E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353–1364 CrossRef CAS.
- For selected examples of in situ generation and cyclopropanation of other types of diazo compounds, see: (a) V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcellini, Angew. Chem., Int. Ed., 2001, 40, 1433–1436 CrossRef CAS; (b) V. K. Aggarwal, J. de Vicente and R. V. Bonnert, Org. Lett., 2001, 3, 2785–2788 CrossRef CAS PubMed; (c) R. P. Wurz and A. B. Charette, Org. Lett., 2002, 4, 4531–4533 CAS; (d) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938–941 CrossRef CAS PubMed; (e) B. Morandi and E. M. Carreira, Science, 2012, 335, 1471–1474 CAS; (f) K. J. Hock, R. Spitzner and R. M. Koenigs, Green Chem., 2017, 19, 2118–2122 RSC; (g) Z. Liu, X. Zhang, G. Zanoni and X. Bi, Org. Lett., 2017, 19, 6646–6649 CrossRef CAS PubMed; (h) E. M. D. Allouche, A. Al-Saleh and A. B. Charette, Chem. Commun., 2018, 54, 13256–13259 CAS; (i) Y. Zheng, X. Yu, S. Lv, P. K. Mykhailiuk, Q. Ma, L. Hai and Y. Wu, RSC Adv., 2018, 8, 5114–5118 RSC.
- For selected examples of gem-dimethylcyclopropanations, see: (a) E. J. Corey and M. Jautelat, J. Am. Chem. Soc., 1967, 89, 3912–3914 CAS; (b) A. B. Charette and N. Wilb, Synlett, 2002, 1, 176–178 Search PubMed; (c) J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 130, 14098–14102 Search PubMed.
- (a) M. J. Durán-Peña, J. M. Botubol Ares, J. R. Hanson, I. G. Collado and R. Hernández-Galán, Nat. Prod. Rep., 2015, 32, 1236–1248 Search PubMed; (b) T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed; (c) T. T. Talele, J. Med. Chem., 2018, 61, 2166–2210 CrossRef CAS PubMed.
- The formation of cyclopropanes using diazo compounds in non-metal-catalyzed reactions is believed to proceed through a 1,3-dipolar cycloaddition (formation of the pyrazoline) followed by dinitrogen extrusion. For mechanistic considerations, see: (a) R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984 Search PubMed; (b) G. Mass, in Heterocyclic Compounds: 1,3-Dipolar Cycloaddition, ed. A. Padwa and W. H. Pearson, John Wiley and Sons, Inc., New York, 2002, vol. 59, pp. 541–621 Search PubMed; (c) Y. Nakano, M. Hamaguchi and T. Nagai, J. Org. Chem., 1989, 54, 1135–1144 CrossRef CAS.
- See ESI† for more details..
- G. Müller, T. Berkenbosch, J. C. Benningshof, D. Stumpfe and J. Bajorath, Chem.–Eur. J., 2017, 23, 703–710 CrossRef PubMed.
- D. E. Applequist and D. E. McGreer, J. Am. Chem. Soc., 1960, 82, 1965–1972 CrossRef CAS.
- For MIRC cyclopropanations of aryl and aryl-alkyl diazo compounds generated under continuous flow conditions, see: N. M. Roda, D. N. Tran, C. Battilocchio, R. Labes, R. J. Ingham, J. M. Hawkins and S. V. Ley, Org. Biomol. Chem., 2015, 13, 2550–2554 RSC . For MIRC cyclopropanations of aryl diazo compounds generated in situ from N-tosylhydrazones, see: L. A. Adams, V. K. Aggarwal, R. V. Bonnert, B. Bressel, R. J. Cox, J. Shepherd, J. de Vicente, M. Walter, W. G. Whittingham and C. L. Winn, J. Org. Chem., 2003, 68, 9433–9440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05558k |
| This journal is © The Royal Society of Chemistry 2019 |