
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(Z)-α-Boryl-crotylboron reagents via Z-selective alkene isomerization and application to stereoselective syntheses of (E)-δ-boryl-syn-homoallylic alcohols†
Shang
Gao‡
,
Jichao
Chen‡
and
Ming
Chen
*
Department of Chemistry and Biochemistry, Auburn University, Auburn, AL 36849, USA. E-mail: mzc0102@auburn.edu
First published on 26th February 2019
Abstract
Stereoselective synthesis of (Z)-α-boryl-crotylboronate is developed. Ni-catalyzed Z-selective alkene isomerization of α-boryl substituted homoallylboronate provided the targeted (Z)-crotylboronate with high selectivity. Stereoselective addition of the novel crotylboron reagent to aldehydes gave (E)-δ-boryl-substituted syn-homoallylic alcohols with excellent diastereoselectivities. The vinyl boronate unit in the products can be directly used for a subsequent C–C bond-forming transformation as illustrated in the synthesis of the C1–7 fragment of the natural products nannocystin A and nannocystin Ax.
Introduction
1,1-Bismetallic crotylation reagents, such as A (Scheme 1), are an important class of molecules that have recently attracted considerable attention. In contrast to the traditional crotyl organometallics,1 addition of these 1,1-bismetallic crotylation reagents to carbonyl compounds (e.g., aldehydes) will produce homoallylic alcohol products (i.e., B or C) with a functionalized alkene group that can directly engage in a C–C bond-formation event, for example, a cross-coupling reaction. In the case of Met1 ≠ Met2, reagent A is chiral and reactions of carbonyl compounds with A typically proceed through chirality transfer. The enantiomeric excess of the alcohol products will largely depend on the optical purity of the starting agent A. Additionally, depending on the different electronic properties and reactivities of the metal substituents, either δ-substituted homoallylic alcohol B or C can be produced selectively. Owing to their versatile reactivities, several types of 1,1-bismetallic crotylation reagents have been developed in the past three decades, including B/Si,2 B/Sn,3 Si/Sn,4 Si/Si,5 and Sn/Sn-substituted crotylation reagents.6 Importantly, many of these reagents have been successfully applied to the syntheses of bioactive natural products, which highlights the synthetic utilities of these reagents.7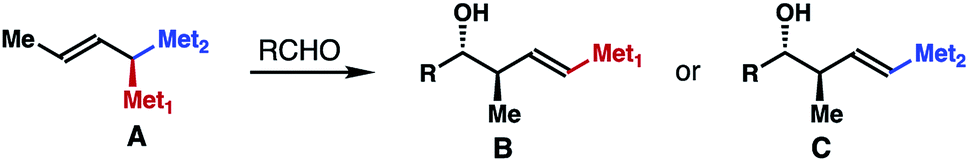 | ||
| Scheme 1 1,1-Bismetallic crotylation reagents. | ||
One subset of 1,1-bismetallic crotylation reagents is α-boryl substituted crotylboronates 2a and 2b (Scheme 2; Met1, Met2 = Bpin). An attractive feature of boronates 2 is that they are achiral, and their reactions with carbonyl compounds should proceed by way of the well-established, six-membered transition state8 to give δ-boryl-substituted homoallylic alcohols. In spite of their apparent synthetic potential, the synthesis of (E)-reagent 2a has only been disclosed recently.9 The Murakami9a,b and Cho9c groups independently showed that (E)-crotylboronate 2a can be generated via transition-metal catalyzed alkene transposition from the homoallylic bisboronate precursor 1 (Scheme 2). Addition of 2a to aldehydes provided δ-boryl-substituted (Z)-anti-homoallylic alcohols (anti-1,2-oxaborinan-3-enes D after intramolecular cyclization) with high selectivities. On the other hand, reactions of (Z)-reagent 2b with aldehydes should form δ-boryl-substituted (E)-syn-homoallylic alcohols 3 (Scheme 2) that would be highly useful in the construction of polyketide natural products. However, methods that could efficiently produce reagent 2b are still not available. Therefore, the development of methods that can allow access to such a reagent and δ-boryl-substituted (E)-syn-homoallylic alcohols 3 would be desirable. With our continuing efforts in developing novel allylboron reagents,10 we have developed and reported herein stereoselective synthesis of (Z)-α-boryl crotylboronate 2b and studies on crotylboration of aldehydes with reagent 2b.
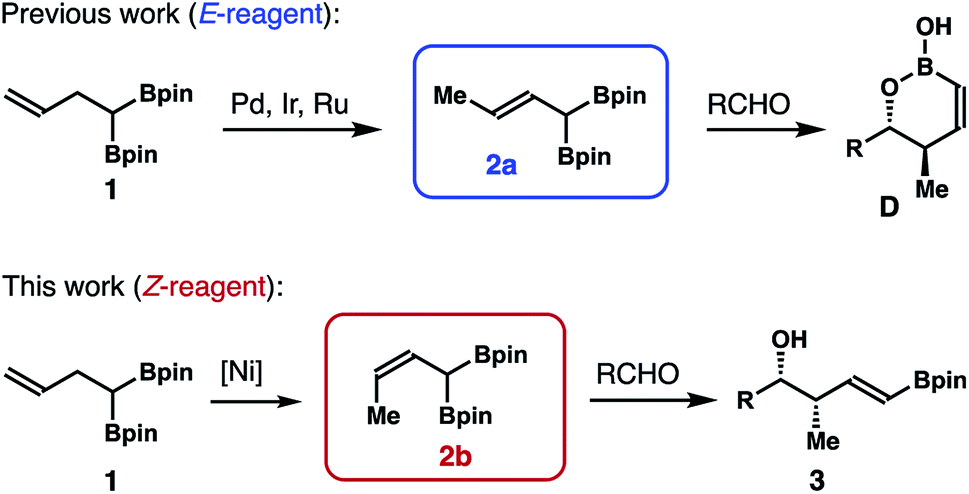 | ||
| Scheme 2 Recent development of α-boryl substituted crotylboron reagents. | ||
Results and discussion
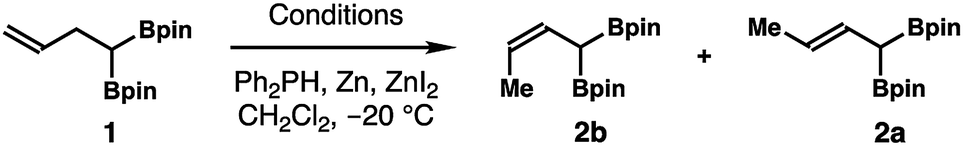
We envisaged a Z-selective alkene isomerization approach to access (Z)-α-boryl crotylboronate 2b from homoallylic bisboronate precursor 1 given its ready availability (Scheme 2). It has been shown by Hilt and co-workers that terminal alkenes can undergo transition metal-catalyzed olefin isomerization to give (Z)-2-alkene isomers with moderate to high selectivity.11 Inspired by their studies, we decided to pursue a Ni-catalyzed isomerization of 1,1-di(boryl)but-3-ene 112 to prepare (Z)-crotylboronate reagent 2b. As shown in Table 1, in the presence of 10 mol% of NiCl2 and dppp, 5 mol% Ph2PH, and 20 mol% of Zn and ZnI2, isomerization of homoallylboronate 1 did not form any product in CH2Cl2 at −20 °C for 24 h (entry 1, Table 1). However, when NiCl2 was replaced by NiBr2, the isomerization reaction occurred to give a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inseparable mixture of 2b and 2a in 70% yield, favouring the Z-isomer 2b (entry 2). Encouraged by the initial success, reactions with several Ni catalysts were examined next. The reaction with Ni(OAc)2 as the catalyst gave a 2:1 mixture of 2b and 2a in low yield (entry 3). An improved Z/E ratio (6:1) was achieved when Ni(acac)2 was employed as the catalyst (entry 4). A similar Z/E ratio (7:1) was obtained with NiCl2·glyme as the catalyst, albeit in a low yield (entry 5). Intriguingly, reactions with preformed Ni catalysts, Ni(dppp)Cl2 or Ni(dppe)Cl2, gave inferior results (entries 6 and 7). When NiBr2·diglyme and dppp were used as the catalyst/ligand combination, a 7:1 mixture of 2b and 2a was obtained in 58% yield (entry 8). Gratifyingly, when 1,2-dichloroethane was used as the solvent, isomerization of homoallylic bisboronate 1 gave an excellent Z/E ratio (2b:2a > 20:1) in the presence of NiBr2·diglyme and dppp. Reagent 2b was isolated in 70% yield (entry 9). A 2 mmol-scale reaction produced (Z)-crotylboronate 2b in 74% yield (entry 10).
:1 inseparable mixture of 2b and 2a in 70% yield, favouring the Z-isomer 2b (entry 2). Encouraged by the initial success, reactions with several Ni catalysts were examined next. The reaction with Ni(OAc)2 as the catalyst gave a 2:1 mixture of 2b and 2a in low yield (entry 3). An improved Z/E ratio (6:1) was achieved when Ni(acac)2 was employed as the catalyst (entry 4). A similar Z/E ratio (7:1) was obtained with NiCl2·glyme as the catalyst, albeit in a low yield (entry 5). Intriguingly, reactions with preformed Ni catalysts, Ni(dppp)Cl2 or Ni(dppe)Cl2, gave inferior results (entries 6 and 7). When NiBr2·diglyme and dppp were used as the catalyst/ligand combination, a 7:1 mixture of 2b and 2a was obtained in 58% yield (entry 8). Gratifyingly, when 1,2-dichloroethane was used as the solvent, isomerization of homoallylic bisboronate 1 gave an excellent Z/E ratio (2b:2a > 20:1) in the presence of NiBr2·diglyme and dppp. Reagent 2b was isolated in 70% yield (entry 9). A 2 mmol-scale reaction produced (Z)-crotylboronate 2b in 74% yield (entry 10).
|
|
|||
|---|---|---|---|
| Entry | Conditions |
2b:2ab |
Yieldc (%) |
| a Reaction conditions: boronate 1 (0.2 mmol, 1.0 equiv.), catalyst (10 mol%), ligand (10 mol%), Ph2PH (5 mol%), Zn (20 mol%), ZnI2 (20 mol%), CH2Cl2 (0.5 mL), −20 °C. b The Z/E ratios were determined by 1H NMR analysis of the crude reaction products. c Yields of isolated products are listed. d DCE was used as the solvent. e The reaction was conducted on a 2 mmol scale in DCE. dppp: 1,3-bis(diphenylphosphino)propane; dppe: 1,2 bis(diphenylphosphino)ethane. | |||
| 1 | NiCl2, dppp | N.D. | N.R. |
| 2 | NiBr2, dppp | 5:1 |
70 |
| 3 | Ni(OAc)2, dppp | 2:1 |
38 |
| 4 | Ni(acac)2, dppp | 6:1 |
76 |
| 5 | NiCl2·glyme, dppp | 7:1 |
36 |
| 6 | Ni(dppp)Cl2 | 3:1 |
56 |
| 7 | Ni(dppe)Cl2 | 3:1 |
64 |
| 8 | NiBr2·diglyme, dppp | 7:1 |
58 |
| 9d | NiBr2·diglyme, dppp | >20:1 |
70 |
| 10e | NiBr2·diglyme, dppp | >20:1 |
74 |
After obtaining (Z)-α-boryl-crotylboronate 2b, we conducted subsequent studies on aldehyde crotylboration with reagent 2b. In initial experiments, treatment of benzaldehyde with 1.3 equiv. of reagent 2b in toluene for 12 h provided (E)-δ-boryl-syn-homoallylic alcohol 3a in 90% yield. The olefin geometry in product 3a was assigned as E based on 1H NMR analysis of the coupling constant of olefinic protons. The stereochemical relationship of 3a was assigned as syn after comparing to the literature data.9a,b
The scope of an aldehyde that participates in this reaction was explored, and the results are summarized in Scheme 3. In general, the reaction worked well with a broad spectrum of aldehydes, including aromatic, heteroaromatic and α,β-unsaturated aldehydes. Reactions of 2b with aromatic aldehydes at ambient temperature in toluene gave alcohol products 3a–h in 78–94% yields. Alkenyl or alkynyl aldehydes reacted with 2b to furnish homoallylic alcohols 3i–k in 58–91% yields. Importantly, a variety of heteroaromatic aldehydes also participated in the reaction to provide alcohols 3l–r in 67–91% yields. Formation of other isomeric products was not observed in any of these reactions.
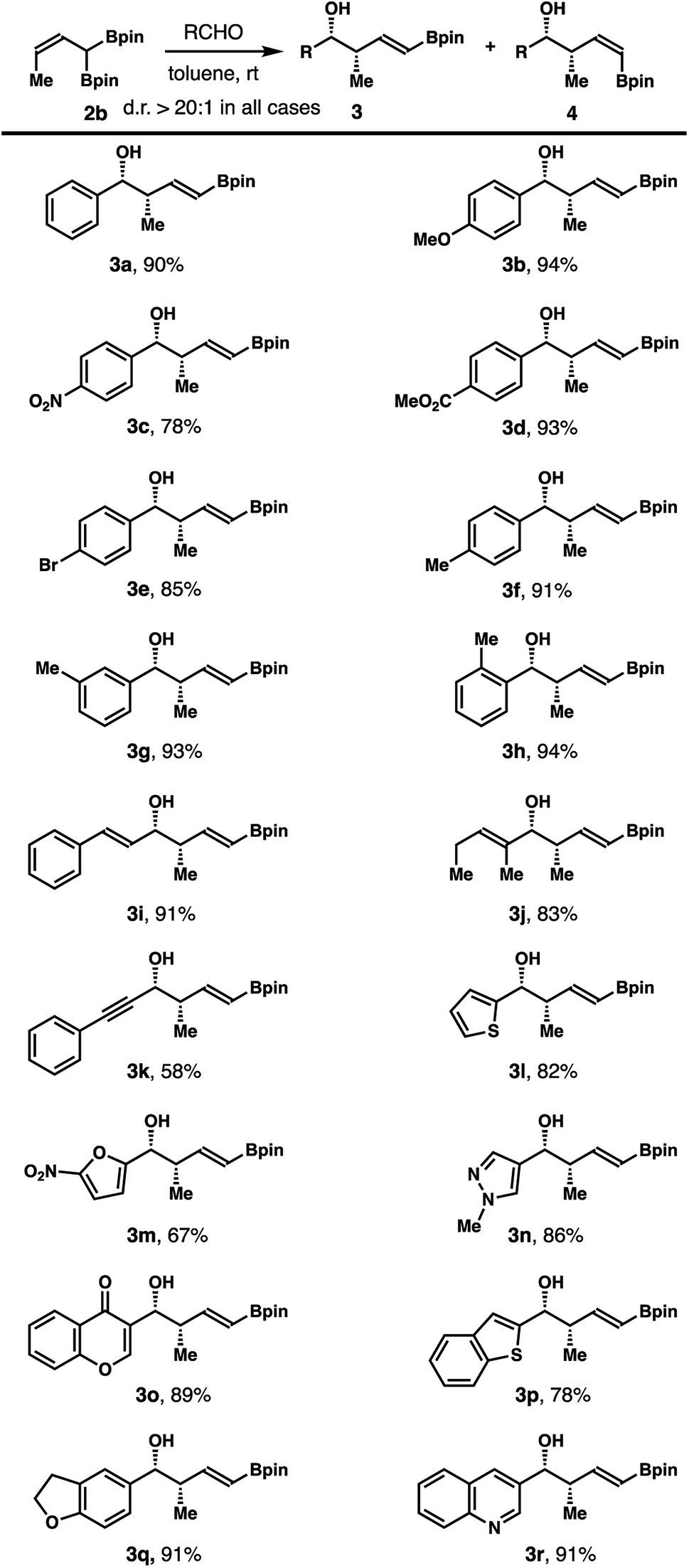 | ||
| Scheme 3 Scope of aromatic, heteroaromatic and α,β-unsaturated aldehydes for the reactions with (Z)-α-boryl-crotylboronate 2b. (a) Reaction conditions: crotylboronate 2b (0.13 mmol, 1.3 equiv.), aldehyde (0.1 mmol, 1.0 equiv.), toluene (0.3 mL), rt. (b) The diastereoselectivities and E/Z selectivities were determined by 1H NMR analysis of the crude reaction products. (c) Yields of isolated products are listed. | ||
Reactions of aliphatic aldehydes with boronate 2b were examined next. As shown in Scheme 4, aliphatic aldehydes including primary alkyl aldehydes, β-branched alkyl aldehydes, and secondary alkyl aldehydes all reacted with reagent 2b in toluene at ambient temperature to give homoallylic alcohols 3s–z in 51–92% yield with excellent diastereoselectivities and E/Z selectivities in all cases.
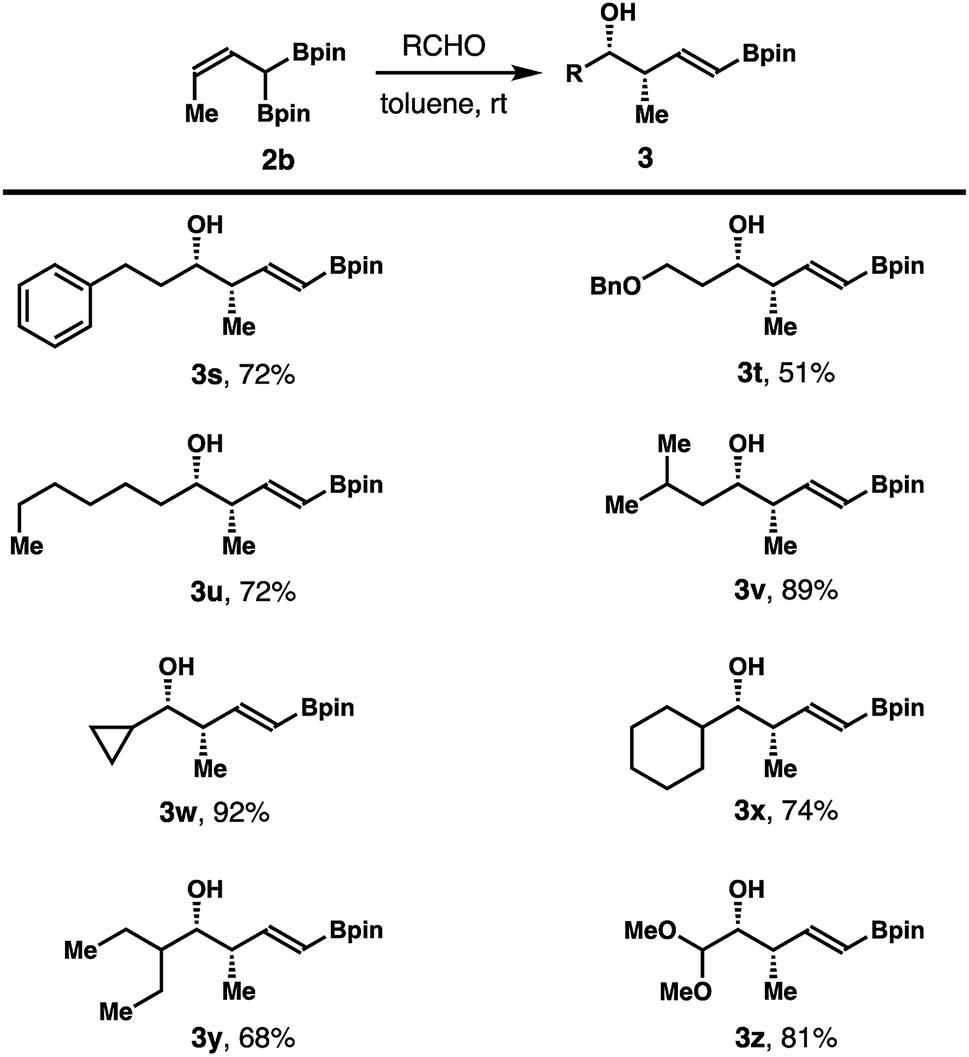 | ||
| Scheme 4 Scope of aliphatic aldehydes for the reactions with (Z)-crotylboronate 2b. (a) Reaction conditions: allyl boronate 2b (0.13 mmol, 1.3 equiv.), aldehyde (0.1 mmol, 1.0 equiv.), toluene (0.3 mL), rt. (b) The diastereoselectivities and E/Z selectivities were determined by 1H NMR analysis of the crude reaction products. (c) Yields of isolated products are listed. | ||
The alkene isomerization and crotylation reaction sequence can be conducted in one pot. As illustrated in Scheme 5, alkene isomerization in the presence of benzaldehyde at −20 °C for 24 h gave product 3a in 64% yield as a single isomer. Detectable amounts of other isomers were not formed from this one-pot procedure.
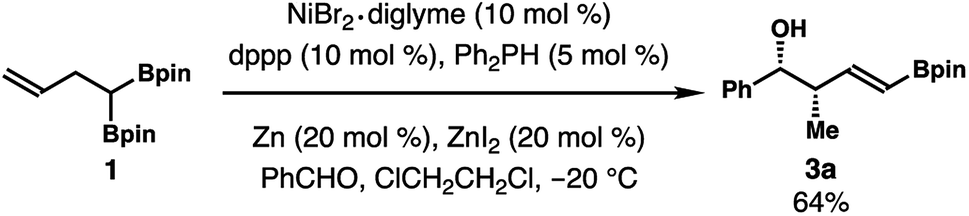 | ||
| Scheme 5 One-pot alkene isomerization and aldehyde allylboration. | ||
The high E-selectivity of this reaction can be rationalized by the following transition state analysis. Among the two competing transition states (TS-1 and TS-2; Scheme 6) that lead to the formation of products 3 and 4, TS-2 suffers from a severe A1,3 allylic strain13 between the pseudo-axially oriented –Bpin group and the methyl group (shown in red in TS-2). In contrast, the A1,3 allylic strain in TS-1 is only between the methyl group and the H atom (shown in light blue in TS-1). Although a gauche interaction may also be involved in TS-1, it is apparent that the A1,3 allylic strain between the –Bpin and methyl groups is severe enough to overcome the gauche interactions. As a result, crotylboration of aldehydes with reagent 2b proceeded through the lower energy transition state TS-1 to give product 3 with high selectivity.
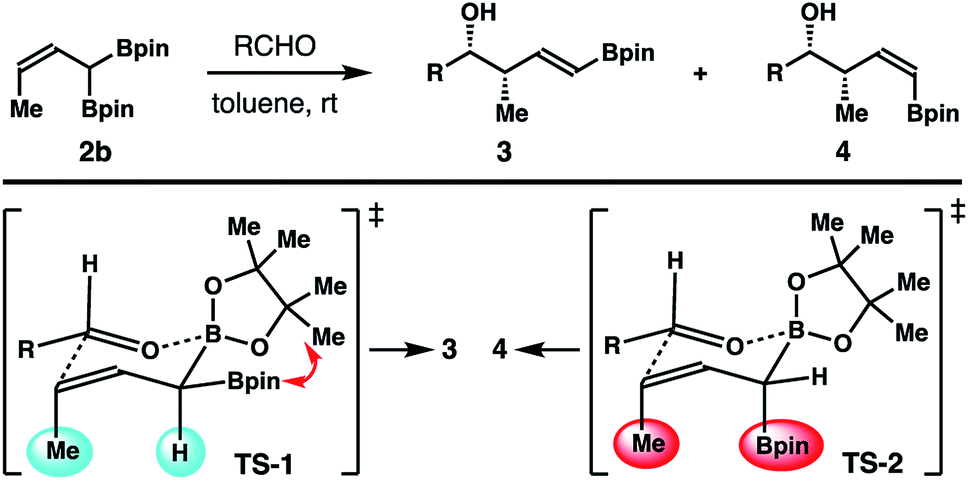 | ||
| Scheme 6 Transition state analyses for selective formation of homoallylic alcohols 3 from crotylboronate 2b. | ||
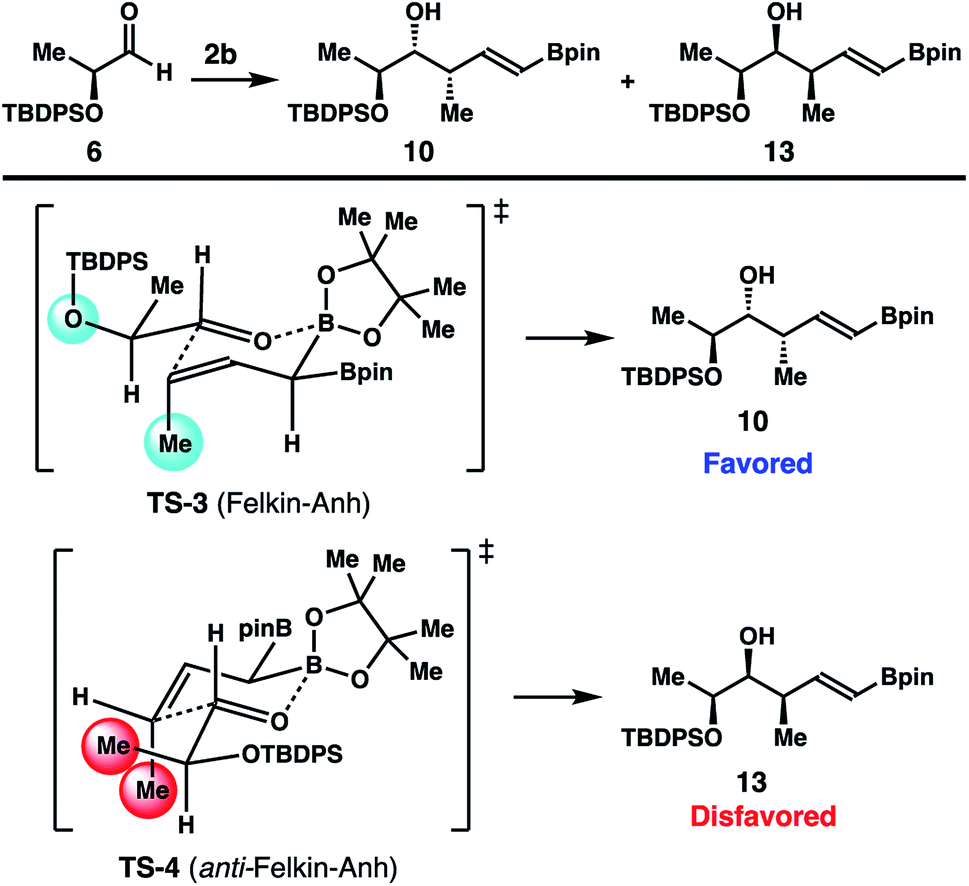
Studies on reactions of crotylboron reagent 2b with several chiral aldehydes (5–8) were also conducted. As illustrated in Scheme 7, the reaction of crotylboronate 2b with racemic 2-phenylpropionaldehyde (5) gave product 9 in 78% yield with 4.5:1 diastereoselectivity. The enantioenriched, lactate-derived aldehyde 6 reacted with reagent 2b to provide an 18:1 mixture, with isomer 10 as the major product in 93% yield. Addition of reagent 2b to N-Boc-L-prolinal (7) generated alcohol 11 in 74% yield with excellent diastereoselectivity (dr > 20:1). Finally, the reaction of reagent 2b with a more advanced chiral, nonracemic aldehyde 8 delivered isomer 12 as the only product (dr > 20:1). Homoallylic alcohol 12 was obtained in 82% yield after purification. The stereochemistry of 9 and 11 was assigned by comparing to the literature data after protodeboronation.14 The absolute configuration of the newly formed secondary hydroxyl groups of 10 and 12 was assigned by Mosher ester analysis.15 Importantly, the mild reaction conditions and high diastereoselectivities of these reactions with chiral aldehydes augur well for further application of reagent 2b in the syntheses of complex natural products and medicinally relevant agents.
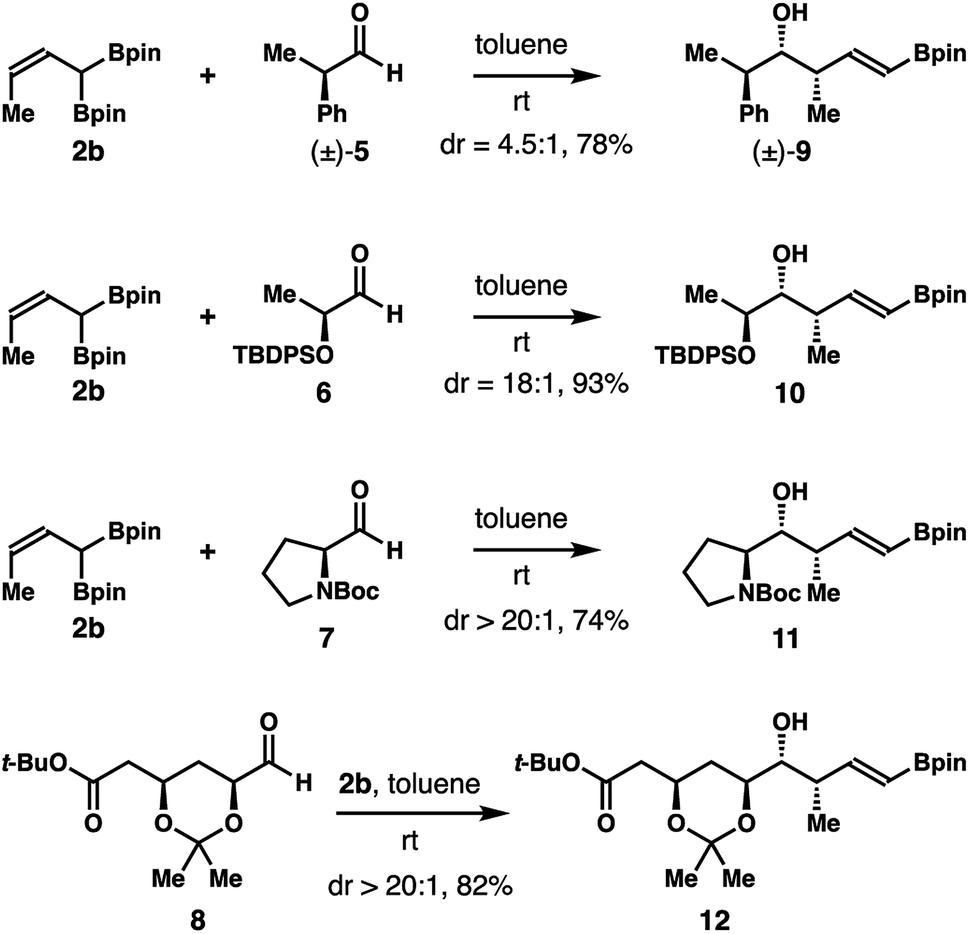 | ||
| Scheme 7 Diastereoselective crotylboration of chiral aldehydes with (Z)-crotylboronate 2b. | ||
A stereochemical model for the high diastereoselectivities in the reactions with enantioenriched aldehydes 6–8 is delineated in Scheme 8. The reaction of aldehyde 6 with reagent 2b could proceed through two potential transition states (TS-3 and TS-4; Scheme 8) to produce two alcohol products, 10 and 13. TS-3 operates under Felkin–Anh16 control to give homoallylic alcohol 10, while the competing transition state TS-4 is under anti-Felkin–Anh control to furnish the diastereomeric alcohol product 13. Upon close examination of the two transition states, it is apparent that TS-4 suffers from an unfavourable gauche-pentane interaction17 between the methyl group of aldehyde 6 and the methyl group of reagent 2b (shown in red in TS-4). In contrast, TS-3 operates under favourable Felkin–Anh control and only with minimal gauche-pentane interactions (shown in light blue in TS-3) between the methyl group of reagent 2b and the oxygen atom of aldehyde 6 (with the large TBDPS group pointing away from the methyl group of reagent 2b). Therefore, the reaction with aldehyde 6 proceeded through the favourable transition state, TS-3, to give product 10 with high diastereoselectivity. Based on this analysis, when the substituent of the aldehyde substrate is sterically much more demanding than a methyl group (e.g., aldehydes 7 and 8), TS-4 is more destabilized relative to TS-3 because of more severe gauche-pentane interactions. Consequently, reactions with these aldehydes should generate Felkin–Anh controlled products with higher selectivities. This prediction is fully consistent with the results obtained from the reactions of aldehydes 7 and 8.
 | ||
| Scheme 8 Transition state analyses for the reaction of chiral aldehyde 6 with crotylboronate 2b. | ||
The products (e.g., 3) generated from the reaction of reagent 2b with aldehydes contain a vinyl boronate group, which can be used directly for a variety of subsequent transformations.18 To further demonstrate the synthetic utility of this method, synthesis of the C1–7 fragment of the natural products nannocystin A and nannocystin Ax was carried out.19,20 As shown in Scheme 9, Pd-catalyzed Suzuki coupling21 of free alcohol 3a with vinyl bromide 1422 provided compound 15, the C1–7 fragment of nannocystin A and nannocystin Ax, in 70% yield (prepared in two steps from commercially available benzaldehyde).
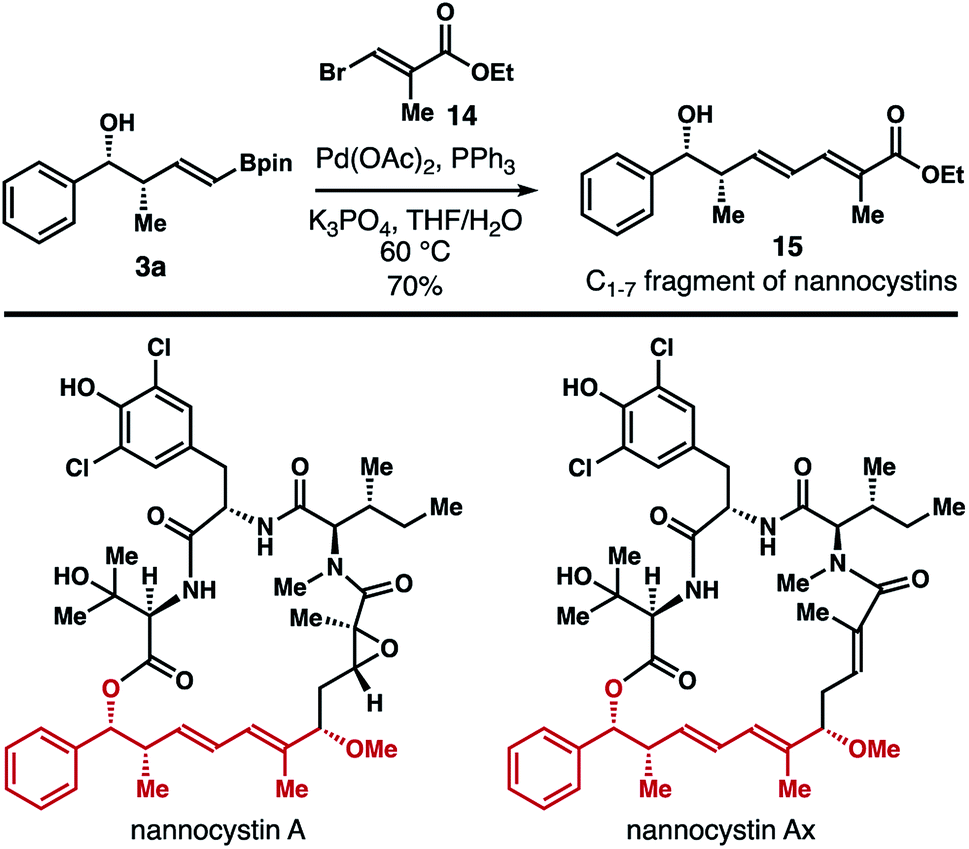 | ||
| Scheme 9 Synthesis of the C1–7 fragment of nannocystin A and nannocystin Ax. | ||
Conclusions
In summary, we developed a Ni-catalyzed, (Z)-selective olefin isomerization approach to synthesize a novel (Z)-α-boryl-crotylboron reagent 2b. Under optimized conditions, boronate 2b was obtained in good yield with exclusive (Z)-selectivity. Subsequent allylboration of aldehydes with reagent 2b gave (E)-δ-boryl-syn-homoallylic alcohols 3 in high yields with excellent diastereoselectivities. Reactions with several enantioenriched aldehydes proceeded under Felkin–Anh control to give homoallylic alcohol products with high diastereoselectivities. The vinyl boronate in products 3 can be directly used for subsequent C–C bond-forming transformations as illustrated in the synthesis of the C1–7 fragment of the natural products nannocystins A and Ax. Studies on asymmetric crotylation using reagent 2b are currently on-going.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by Auburn University is gratefully acknowledged.Notes and references
- (a) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS; (b) S. E. Denmark and N. G. Almstead, Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, p. 299 Search PubMed; (c) S. R. Chemler and W. R. Roush, Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, p. 403 Search PubMed; (d) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed; (e) H. Lachance and D. G. Hall, Org. React., 2008, 73, 1 Search PubMed; (f) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed; (g) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed; (h) C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2 CrossRef CAS PubMed.
- (a) Y. Yamamoto, H. Yatagai and K. Maruyama, J. Am. Chem. Soc., 1981, 103, 3229 CrossRef CAS; (b) D. J. S. Tsai and D. S. Matteson, Organometallics, 1983, 2, 236 CrossRef CAS; (c) Y. Yamamoto, K. Maruyama, T. Komatsu and W. Ito, J. Org. Chem., 1986, 51, 886 CrossRef CAS; (d) M. Shimizu, H. Kitagawa, T. Kurahashi and T. Hiyama, Angew. Chem., Int. Ed., 2001, 40, 4283 CrossRef CAS; (e) L. Carosi, H. Lachance and D. G. Hall, Tetrahedron Lett., 2005, 46, 8981 CrossRef CAS; (f) M. Chen and W. R. Roush, Org. Lett., 2013, 15, 1662 CrossRef CAS PubMed; (g) M. Chen and W. R. Roush, Tetrahedron, 2013, 69, 5468 CrossRef CAS PubMed.
- (a) M. Chen, D. H. Ess and W. R. Roush, J. Am. Chem. Soc., 2010, 132, 7881 CrossRef CAS PubMed; (b) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2011, 133, 5744 CrossRef CAS PubMed; (c) P. S. Stewart, M. Chen, W. R. Roush and D. H. Ess, Org. Lett., 2011, 13, 1478 CrossRef CAS PubMed; (d) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2012, 134, 3925 CrossRef CAS PubMed.
- (a) M. Lautens, A. H. Huboux, B. Chin and J. Downer, Tetrahedron Lett., 1990, 131, 5829 CrossRef; (b) M. Lautens and P. H. M. Delanghe, Angew. Chem., Int. Ed., 1994, 33, 2448 CrossRef; (c) M. Lautens, R. N. Ben and P. H. M. Delanghe, Tetrahedron, 1996, 52, 7221 CrossRef CAS.
- (a) B. Princet, G. Anselme and J. Pornet, Synth. Commun., 1999, 29, 3329 CrossRef CAS; (b) B. Princet, G. Anselme and J. Pornet, J. Organomet. Chem., 1999, 5921, 34 CrossRef; (c) B. Princet, H. Gardes-Gariglio and J. Pornet, J. Organomet. Chem., 2000, 604, 186 CrossRef CAS; (d) D. M. Hodgson, S. F. Barker, L. H. Mace and J. R. Moran, Chem. Commun., 2001, 153 RSC; (e) D. R. Williams, Á. I. Morales-Ramos and C. M. Williams, Org. Lett., 2006, 8, 4393 CrossRef CAS PubMed; (f) L. Li, X. Ye, Y. Wu, Z. Song, Z. Yin and Y. Xu, Org. Lett., 2013, 15, 1068 CrossRef CAS PubMed; (g) Y. Chu, Q. Pu, Z. Tang, L. Gao and Z. Song, Tetrahedron, 2017, 73, 3707 CrossRef CAS; (h) Z. Chu, K. Wang, L. Gao and Z. Song, Chem. Commun., 2017, 53, 3078 RSC.
- (a) H. Wakamatsu, M. Nishida, N. Adachi and M. Mori, J. Org. Chem., 2000, 65, 3966 CrossRef CAS PubMed; (b) H. J. Reich and J. W. Ringer, J. Org. Chem., 1988, 53, 455 CrossRef CAS.
- (a) H. Sun, J. R. Abbott and W. R. Roush, Org. Lett., 2011, 13, 2734 CrossRef CAS PubMed; (b) M. Chen and W. R. Roush, Org. Lett., 2012, 14, 426 CrossRef CAS PubMed; (c) M. Chen and W. R. Roush, Org. Lett., 2012, 14, 1880 CrossRef CAS PubMed; (d) M. Chen and W. R. Roush, J. Org. Chem., 2013, 78, 3 CrossRef CAS PubMed; (e) A. Grisin and P. A. Evans, Chem. Sci., 2015, 6, 6407 RSC; (f) Y.-H. Zhang, R. Liu and B. Liu, Chem. Commun., 2017, 53, 5549 RSC; (g) L. Kämmler and M. E. Maier, J. Org. Chem., 2018, 83, 4554 CrossRef PubMed.
- H. E. Zimmerman and M. D. Traxler, J. Am. Chem. Soc., 1957, 79, 1920 CrossRef CAS.
- (a) T. Miura, J. Nakahashi and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 6989 CrossRef CAS PubMed; (b) T. Miura, J. Nakahashi, W. Zhou, Y. Shiratori, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2017, 139, 10903 CrossRef CAS PubMed; (c) J. Park, S. Choi, Y. Lee and S. H. Cho, Org. Lett., 2017, 19, 4054 CrossRef CAS PubMed.
- (a) M. Wang, S. Khan, E. Miliordos and M. Chen, Org. Lett., 2018, 20, 3810 CrossRef CAS PubMed; (b) S. Gao and M. Chen, Org. Lett., 2018, 20, 6174 CrossRef CAS PubMed; (c) M. Wang, S. Khan, E. Miliordos and M. Chen, Adv. Synth. Catal., 2018, 360, 4634 CrossRef CAS; (d) S. Gao, M. Wang and M. Chen, Org. Lett., 2018, 20, 7921 CrossRef CAS PubMed.
- F. Weber, A. Schmidt, P. Röse, M. Fischer, O. Burghaus and G. Hilt, Org. Lett., 2015, 17, 2952 CrossRef CAS PubMed.
- S. A. Murray, J. C. Green, S. B. Tailor and S. J. Meek, Angew. Chem., Int. Ed., 2016, 55, 9065 CrossRef CAS PubMed.
- (a) R. W. Hoffmann, Chem. Rev., 1989, 89, 1841 CrossRef CAS. For recent examples: (b) M. Althaus, A. Mahmood, J. R. Suárez, S. P. Thomas and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 4025 CrossRef CAS PubMed; (c) L. T. Kliman, S. N. Mlynarski, G. E. Ferris and J. P. Morken, Angew. Chem., Int. Ed., 2012, 51, 521 CrossRef CAS PubMed; (d) B. Potter, A. A. Szymaniak, E. K. Edelstein and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 17918 CrossRef CAS PubMed.
- (a) H. Brinkmann and R. W. Hoffmann, Chem. Ber., 1990, 123, 2395 CrossRef CAS; (b) G. Niel, F. Roux, Y. Maisonnasse, I. Maugras, J. Poncet and P. Jouin, J. Chem. Soc., Perkin Trans. 1, 1994, 1275 RSC; (c) F. Roux, I. Maugras, J. Poncet, G. Niel and P. Jouin, Tetrahedron, 1994, 50, 5345 CrossRef CAS.
- (a) J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512 CrossRef CAS; (b) I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS; (c) T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451 CrossRef CAS PubMed.
- (a) M. Cherest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 18, 2199 CrossRef; (b) N. T. Anh and O. Eisenstein, Nouv. J. Chim., 1977, 1, 61 Search PubMed.
- (a) W. R. Roush, J. Org. Chem., 1991, 56, 4151 CrossRef CAS; (b) M. Chen and W. R. Roush, J. Am. Chem. Soc., 2012, 134, 3925 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS PubMed; (c) C. Wang, T. Tobrman, Z. Xu and E.-i. Negishi, Org. Lett., 2009, 11, 4092 CrossRef CAS PubMed; (d) R. E. Shade, A. M. Hyde, J.-C. Olsen and C. A. Merlic, J. Am. Chem. Soc., 2010, 132, 1202 CrossRef CAS PubMed; (e) C. Körner, P. Starkov and T. D. Sheppard, J. Am. Chem. Soc., 2010, 132, 5968 CrossRef PubMed.
- Isolation of nannocystins: (a) H. Hoffmann, H. Kogler, W. Heyse, H. Matter, M. Caspers, D. Schummer, C. Klemke-Jahn, A. Penarier, G. Bauer, L. Debussche and M. Brönstrup, Angew. Chem., Int. Ed., 2015, 54, 10145 CrossRef CAS PubMed; (b) P. Krastel, S. Roggo, M. Schirle, N. T. Ross, F. Perruccio, P. Aspesi, T. Aust, K. Buntin, D. Estoppey, B. Liechty, F. Mapa, K. Memmert, H. Miller, X. Pan, R. Riedl, C. Thibaut, J. Thomas, T. Wagner, E. Weber, X. Xie, E. K. Schmitt and D. Hoepfner, Angew. Chem., Int. Ed., 2015, 54, 10149 CrossRef CAS PubMed.
- Syntheses of nannocystins: (a) L. Liao, J. Zhou, Z. Xu and T. Ye, Angew. Chem., Int. Ed., 2016, 55, 13263 CrossRef CAS PubMed; (b) J. Huang and Z. Wang, Org. Lett., 2016, 18, 4702 CrossRef CAS PubMed; (c) Z. Yang, X. Xu, C.-H. Yang, Y. Tian, X. Chen, L. Lian, W. Pan, X. Su, W. Zhang and Y. Chen, Org. Lett., 2016, 18, 5768 CrossRef CAS PubMed; (d) Q. Liu, P. Hu and Y. He, J. Org. Chem., 2017, 82, 9217 CrossRef CAS PubMed; (e) Y.-H. Zhang, R. Liu and B. Liu, Chem. Commun., 2017, 53, 5549 RSC; (f) C. Poock and M. Kalesse, Org. Lett., 2017, 19, 4536 CrossRef CAS PubMed; (g) Z. Meng, L. Souillart, B. Monks, N. Huwyler, J. Herrmann, R. Müller and A. Fürstner, J. Org. Chem., 2018, 83, 6977 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- Y. Wang and W.-M. Dai, Eur. J. Org. Chem., 2014, 2014, 323 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00226j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |