
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing 13C-labeled myo-inositol to interrogate inositol phosphate messengers by NMR†
Robert K.
Harmel‡
 ab,
Robert
Puschmann‡
ab,
Minh
Nguyen Trung
ab,
Adolfo
Saiardi
c,
Peter
Schmieder
a and
Dorothea
Fiedler
*ab
ab,
Robert
Puschmann‡
ab,
Minh
Nguyen Trung
ab,
Adolfo
Saiardi
c,
Peter
Schmieder
a and
Dorothea
Fiedler
*ab
aLeibniz-Forschungsinstitut für Molekulare Pharmakologie, Robert-Rössle-Straße 10, 13125 Berlin, Germany. E-mail: fiedler@fmp-berlin.de
bInstitute of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany
cMedical Research Council Laboratory for Molecular Cell Biology, University College London, London, UK
First published on 23rd April 2019
Abstract
Inositol poly- and pyrophosphates (InsPs and PP-InsPs) are an important group of metabolites and mediate a wide range of processes in eukaryotic cells. To elucidate the functions of these molecules, robust techniques for the characterization of inositol phosphate metabolism are required, both at the biochemical and the cellular level. Here, a new tool-set is reported, which employs uniformly 13C-labeled compounds ([13C6]myo-inositol, [13C6]InsP5, [13C6]InsP6, and [13C6]5PP-InsP5), in combination with commonly accessible NMR technology. This approach permitted the detection and quantification of InsPs and PP-InsPs within complex mixtures and at physiological concentrations. Specifically, the enzymatic activity of IP6K1 could be monitored in vitro in real time. Metabolic labeling of mammalian cells with [13C6]myo-inositol enabled the analysis of cellular pools of InsPs and PP-InsPs, and uncovered high concentrations of 5PP-InsP5 in HCT116 cells, especially in response to genetic and pharmacological perturbation. The reported method greatly facilitates the analysis of this otherwise spectroscopically silent group of molecules, and holds great promise to comprehensively analyze inositol-based signaling molecules under normal and pathological conditions.
Introduction
Small molecule messengers are key components in cellular decision-making processes.1 Among those, inositol-based signaling molecules occupy a privileged node and include both water-soluble, diffusible messengers (best characterized by inositol 1,4,5-trisphosphate, InsP3, a calcium releasing factor), as well as the lipid-anchored phosphatidylinositol phosphates (PtdInsPs).2–4 Within the class of soluble inositol phosphates (InsPs) much research has recently focused on a unique, highly phosphorylated subgroup, termed the inositol pyrophosphates (PP-InsPs).5–7 In these molecules high-energy diphosphate groups are attached to the inositol scaffold by the action of two classes of evolutionarily conserved small molecule kinases, the inositol hexakisphosphate kinases (IP6Ks) and the diphosphoinositol pentakisphosphate kinases (PPIP5Ks).8–11 Altered activity of these kinases has been linked to hearing loss, decrease in tumor cell motility, life-span extension and insulin hypersensitivity.12–16 Consequently, the enzymes involved in InsP and PP-InsP synthesis and turnover are becoming sought-after targets for treating wide-spread diseases, such as diabetes and cancer. While genetic studies have provided key insights into InsP and PP-InsP function, many questions regarding the dynamic interconversion and turnover of these messengers, as well as the regulation of the enzymes involved in their biosynthesis remain unanswered.Tritiated [3H]myo-inositol has been used extensively to incorporate radioactive labels into cellular InsP species, and to facilitate their detection after resolution by strong anion exchange chromatography (SAX-HPLC) (Fig. 1a).17 Use of this method has led to many seminal findings, but the requirement for radioactivity and the limited compatibility with certain cell lines restricts its applicability. A parallel approach circumvented the use of radioactivity by analyzing HPLC fractions in the presence of a metal-dependent dye, but has remained sparsely used.17 More recently, a method based on gel electrophoresis was reported.18 Here, the extracted InsPs and PP-InsPs have to be enriched over TiO2 beads, and are then resolved on a high-percentage polyacrylamide gel (PAGE) (Fig. 1a).19 Although the independence from radioactive tracers has allowed more laboratories to employ this approach, the lack of an InsP-specific analytical handle requires a more elaborate sample preparation and is limited to the detection of only the most highly phosphorylated InsPs.
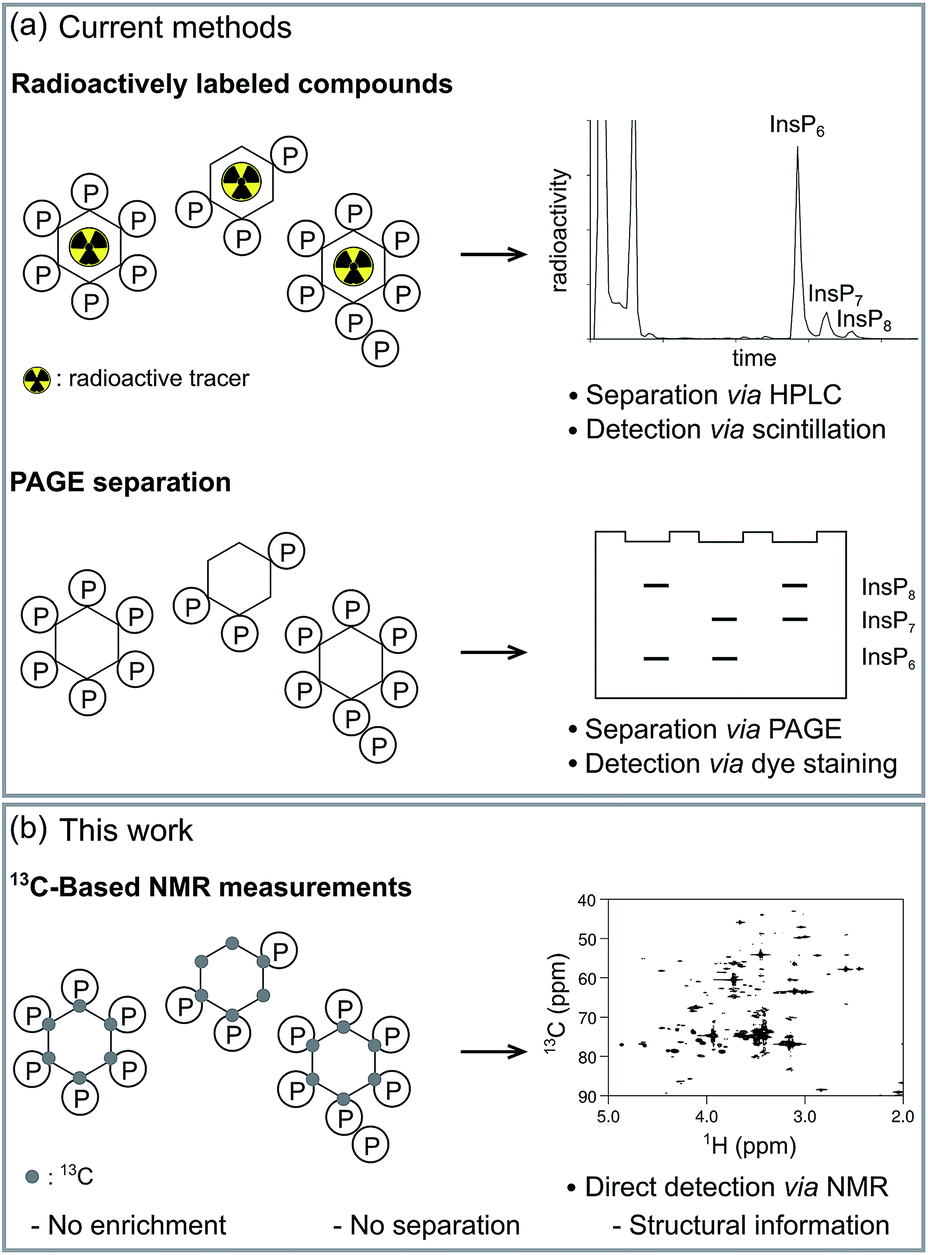 | ||
| Fig. 1 Methods for the analysis of inositol polyphosphates. (a) Radioactively labeled InsPs can be resolved via SAX-HPLC and the amount of radioactivity in each fraction is analyzed. Alternatively, InsPs are enriched with TiO2-beads from complex mixtures, subsequently resolved via PAGE, and detected by staining with toluidine blue. (b) 13C-labeled InsPs can be directly measured in complex samples using NMR spectroscopy. | ||
While all methods mentioned above have been instrumental to date, they do not provide structural information about the InsPs and PP-InsPs, such as the clear distinction of the structural isomers of PP-InsPs. The methods also lack the ability to monitor conversion of InsP species in real time in vitro, thereby forfeiting informative kinetic insight contained within biochemical experiments. Furthermore, because of the various separation and resolution steps, direct measurements in complex samples, such as cell extracts, have not been possible so far.
NMR spectroscopy can provide detailed information about the chemical and structural environment of a nucleus solely based on the chemical shift. For example, two-dimensional 1H,31P-NMR has been used to elucidate the structure of PP-InsPs.20,21 Since experiments utilizing the 3J(1H,31P) coupling are of limited sensitivity due to the inefficient magnetization transfer via these couplings, high analyte concentrations were necessary. In addition, the low chemical shift dispersion and the broad lines of 31P nuclei can limit the information content.20,21 By contrast, the chemical shift dispersion of the NMR-active nucleus 13C is superior to 31P and the sensitivity of two-dimensional experiments is much better, due to an efficient magnetization transfer via1J(1H,13C) one-bond couplings. Since 13C has a natural abundance of only approx. 1%, isotopic labeling of selected compounds should provide targeted information on these molecules in complex environments. Indeed, labeling of small molecules and metabolites with 13C has been applied widely to elucidate various metabolic pathways and processes.22–24 Consequently, 13C-labeled myo-inositol (1), and the corresponding labeled InsPs and PP-InsPs, should also enable NMR measurements at low, biologically relevant analyte concentrations and within intricate samples (Fig. 1b).
Here, we report high yielding syntheses of 13C-labeled myo-inositol and 13C-labeled inositol polyphosphates and their subsequent applications. We employ the 13C-labeled InsPs to monitor in vitro activity of IP6Ks in real time using NMR spectroscopy and consequently characterize the kinetic parameters of IP6Ks. Moreover, we demonstrate metabolic labeling of two mammalian cell lines with [13C6]myo-inositol, and subsequent detection of the in vivo generated [13C6]InsP species within a complex cell extract. These findings highlight the utility and applicability of 13C-labeled InsPs/PP-InsPs to improve our understanding of their biochemistry and metabolism, and to interrogate and elucidate their cellular functions.
Results and discussion
Enzymatic synthesis of [13C6]myo-inositol on a gram scale
To analyze inositol-based signaling molecules by NMR we first sought to prepare pure [13C6]myo-inositol on a large scale. The approaches to obtain 13C-labeled myo-inositol reported to date have either relied on expensive [13C6]glucose-6-phosphate as a starting material (7000€ per g) or – when starting from [13C6]glucose – were low yielding (9.9% and 6.7%, respectively).25–27 In addition, all procedures lacked a scalable purification strategy for the product, precluding the isolation of large and pure amounts of product. Consequently, these methods have only been applied on a small scale. To circumvent these limitations, and to subsequently synthesize sufficient quantities of [13C6]InsPs, we pursued a 3-step enzymatic synthesis that uses affordable [13C6]glucose (80€ per g) as a starting material and employed a chemical purification strategy at the end (Fig. 2).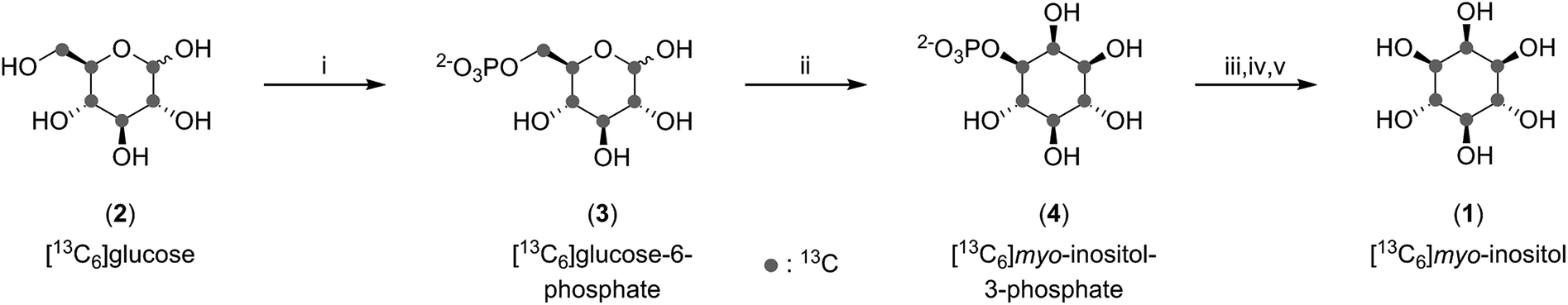 | ||
| Fig. 2 Enzymatic synthesis of [13C6]myo-inositol followed by derivatization and purification. (i) MOPS pH 6.5, creatine phosphate, ATP, DTT, MgCl2, hexokinase, creatine kinase in H2O at rt. (ii) Tris pH 8.0, NAD+, NaCl, MgCl2, inositol phosphate synthase in H2O at 80 °C. (iii) Glycine pH 9.8, ZnCl2, alkaline phosphatase in H2O at 35 °C. (iv) Ac2O in pyridine at 120 °C. (v) NaOMe in MeOH at rt. | ||
As a first step, [13C6]glucose (2) was phosphorylated at the 6-position using commercially available hexokinase in a modified literature procedure.28 Next, we optimized and scaled up the isomerization reaction of [13C6]glucose-6-phosphate (3) to [13C6]myo-inositol-3-phosphate (4) published by Saiardi and coworkers.25 Subsequently, 1 could be obtained by dephosphorylation using commercially available alkaline phosphatase. While the desired product was clearly visible in the 13C-NMR spectrum, the isolated yield exceeded 100%, indicative of contamination with salts and buffer components. Because purification of 1 by ion-exchange, normal-phase, and reverse-phase chromatography proved difficult, we applied a chemical derivatization strategy in which the product mixture containing 1 was treated with an excess of acetic anhydride in pyridine. The resulting [13C6]myo-inositol hexakisacetate (S1) could be purified by normal-phase chromatography, and subsequent saponification provided gram amounts of 1 in excellent purity with a 50% overall yield (with respect to [13C6]glucose). In comparison to previous syntheses, our approach provides a simpler and shorter route with greatly improved yield.
Chemical synthesis of [13C6]InsP5, [13C6]InsP6 and enzymatic synthesis of [13C6]5PP-InsP5
With large quantities of pure 1 at hand, we wanted to prepare the 13C-labeled inositol polyphosphates [13C6]InsP5 (5), [13C6]InsP6 (6) and [13C6]5PP-InsP5 (7), since InsP5 and InsP6 are the most abundant soluble inositol phosphates in mammalian cells, and 5PP-InsP5 is the best characterized inositol pyrophosphate messenger.6,29 The chemical syntheses of 5 and 6 were conducted according to published procedures for the unlabeled compounds, and both 5 and 6 were isolated in good yield (Fig. 3a). The HMQC spectra of 5 and 6 displayed four resonances, due to the plane of symmetry in these molecules (Fig. 3b and c). Importantly, the spectra depicted in Fig. 3 were recorded at an InsP concentration of 5 μM, using only 32 scans, and illustrate the high sensitivity provided by labeling with 13C.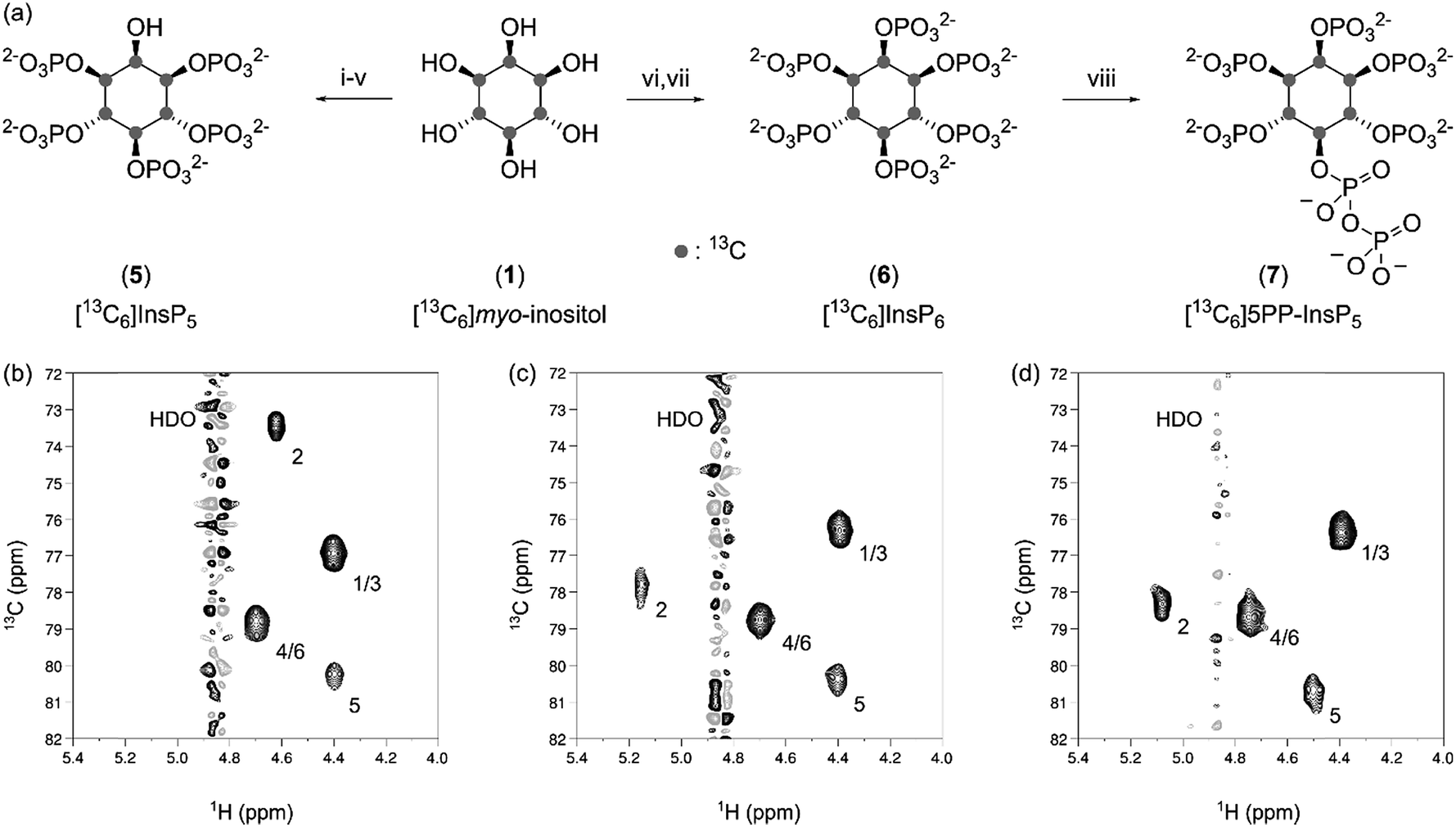 | ||
| Fig. 3 Synthesis and characterization of [13C6]inositol polyphosphates. (a) Reagents and conditions: (i) CSA, trimethyl orthobenzoate, TEA in DMSO at 80 °C; 85% yield (ii) TFA in H2O at rt; quantitative yield (iii) 5-phenyltetrazole, xylyl phosphoramidite, mCPBA in DCM at rt; 79% yield (iv) Pd(OH)2/C in MeOH/H2O at rt; 88% yield (v) NH3 (aq.) at 60 °C; 65% yield (vi) xylyl phosphoramidite, 1H-tetrazole, mCPBA in DCM at rt; 38% yield (vii) Pd(OH)2/C in MeOH/H2O at rt; 83% yield (viii) MES pH 6.4, NaCl, ATP, creatine phosphate, MgCl2, DTT, IP6KA in H2O at 37 °C; quantitative yield (b) 1H,13C-HMQC spectrum of [13C6]InsP5 at 5 μM. (c) 1H,13C-HMQC spectrum of [13C6]InsP6 at 5 μM. (d) 1H,13C-HMQC spectrum of [13C6]5PP-InsP5 at 5 μM. The positions of the carbon atoms and the solvent signal are indicated. | ||
Compared to the syntheses of 5 and 6, obtaining [13C6]5PP-InsP5 (7) by chemical means would be more challenging. Although synthetic access to 5PP-InsP5 has been improved significantly in recent years, the syntheses still require several synthetic steps, and provide an overall yield of 6–8% starting from myo-inositol.30,31 Therefore, we focused our efforts on an enzymatic synthesis utilizing [13C6]InsP6 (6) as a substrate, to conserve as much labeled material as possible. Specifically, we used ehIP6KA, an IP6K from Entamoeba histolytica, to produce 7 in one step from 6, as this enzyme has the highest reported activity of all known IP6Ks.32 Within 45 minutes we observed full conversion of 6 using an ATP regenerating system (Fig. 3a and S1†).
The isolation of enzymatically generated PP-InsPs previously relied on SAX-HPLC separation, or purification via PAGE.33,34 While these methods indeed afforded the desired materials, the methods could only be conducted on a small scale, and typically resulted in contamination of the product with large amounts of salt. To overcome these limitations, a purification protocol was developed in which the addition of magnesium ions caused precipitation of the PP-InsP.35,36 Subsequent metal chelation chromatography provided [13C6]5PP-InsP5 (7) as an ammonium salt with excellent purity and quantitative isolated yield (Fig. 3a and d).
Overall, the enzymatic synthesis, coupled to a new precipitation protocol, can deliver large quantities of 5PP-InsP5 in a short amount of time and constitutes a significant improvement in the accessibility of 5PP-InsP5 to researchers across disciplines.
In situ measurement of IP6K activity
While optimizing the enzymatic synthesis of 7, we noted that the progression of the reaction could be followed readily by NMR, even in the presence of high concentrations of non-deuterated buffer and ATP (Fig. S1†). Specifically, the diagnostic signals for the protons at the 2-position of the inositol ring of [13C6]InsP6 and [13C6]5PP-InsP5 were baseline-separated in the 1H-dimension (5.05 ppm for 6, 4.95 ppm for 7) and showed no overlap with signals from other reaction components within the mixture (Fig. S1†). The ability to resolve the 1H-signals at the 2-position provided the opportunity to monitor the enzymatic reaction in a time-resolved fashion, using a pseudo-2D spin-echo difference experiment. This pulse sequence is similar to the 1H,13C-HMQC experiments and results in a series of one-dimensional spectra that only display resonances for 1H-nuclei that are bound to 13C-nuclei. By plotting the peak intensity of the 1H-nuclei at the 2-position against time (each experiment required only 75 seconds at a substrate concentration of 175 μM) the progress of the kinase reaction could be observed (Fig. S2†). The non-invasive nature of the NMR measurements allowed for continuous reaction monitoring, providing time-resolved data from one sample.Determination of kinetic parameters for IP6K1 using NMR
Considering our ability to reliably measure kinase activity by NMR, we next wanted to determine the kinetic parameters for human IP6K1. Measurement of the initial rates at constant InsP6 concentration (175 μM) and varying ATP concentrations (62.5 μM to 8 mM) provided the Michalis–Menten constant (KM) for ATP and the maximum velocity (Vmax) for IP6K1 (Fig. 4a).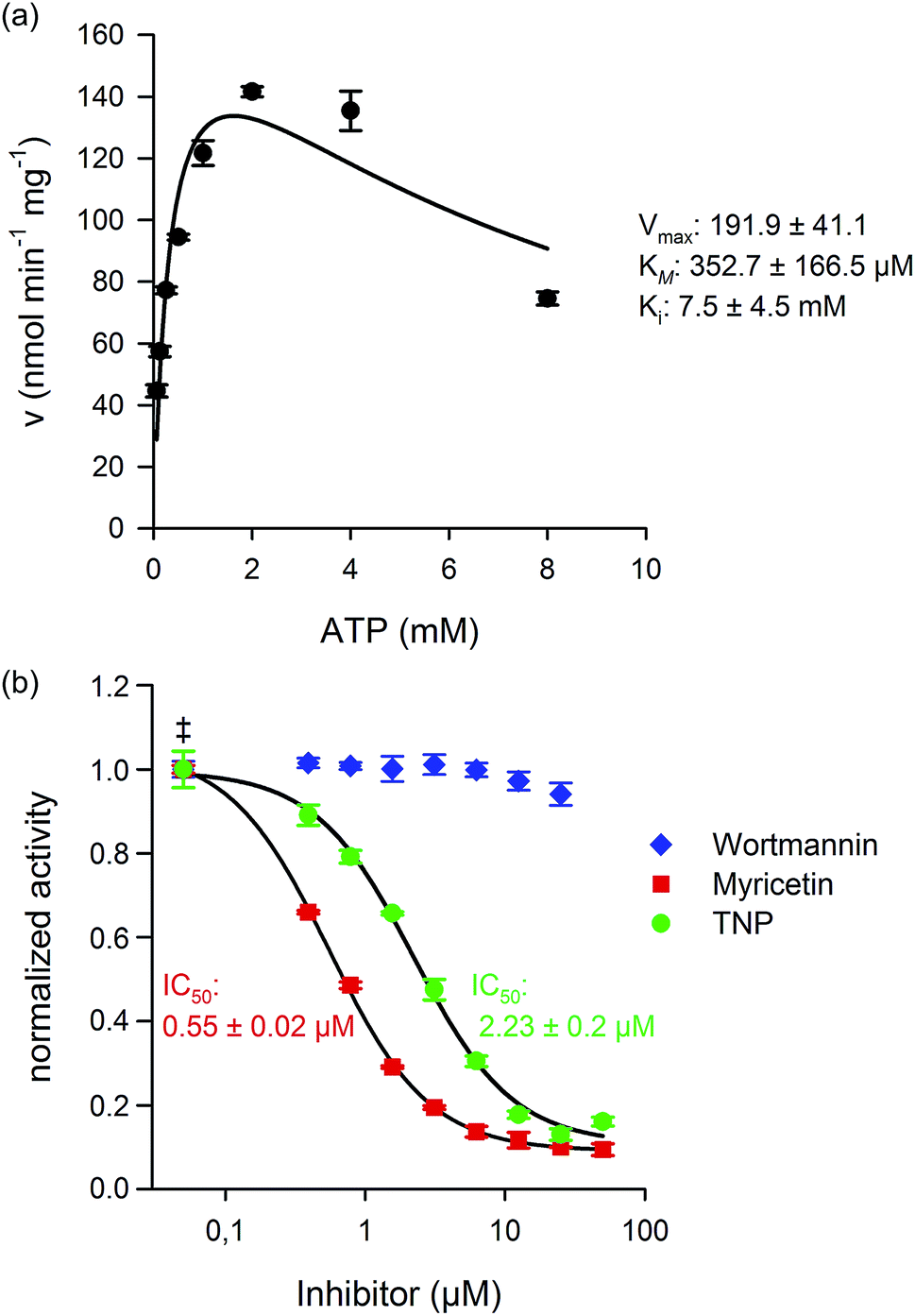 | ||
| Fig. 4 Kinetic characterization and inhibition of IP6K1. (a) Michaelis–Menten kinetics for IP6K1. The initial velocity was measured in triplicate at different ATP concentrations and fitted against a model for substrate inhibition. (b) The inhibitory effects of TNP (N2-(m-trifluorobenzyl)-N6-(p-nitrobenzyl)purine), Myricetin, and Wortmannin were tested and the IC50 values were determined. The ATP concentration was 2.5 mM for all inhibitor measurements (‡ enzyme activity at 0 μM inhibitor). | ||
The KM measured for ATP (353 ± 167 μM) was comparable to previous reports and is close to cellular ATP concentrations, which supports the proposition that the IP6Ks are uniquely sensitive to ATP availability.37,38 The Vmax value (192 ± 41 nmol min−1 mg−1) for IP6K1 was also within the range of published literature values, which varied greatly depending on the protein-tag and purification strategy (37.3 nmol min−1 mg−1 to 1410 nmol min−1 mg−1).37,39 Interestingly, we found that ATP concentrations above 2 mM led to substrate inhibition of IP6K1 (Ki 7.48 mM), which had not been quantified before (Fig. 4a).39 The robust measurements of InsP conversion by NMR spectroscopy has provided kinetic data with low experimental error, which, in turn, has allowed the observation of substrate inhibition by ATP.
Measuring the potency of IP6K inhibitors
IP6Ks regulate important aspects of metabolism and signaling in mammals and in pathogenic microorganisms like Trypanosoma brucei and therefore increased efforts to identify selective inhibitors have been made.38,40,41 Thus far, two molecules, TNP (N2-(m-trifluorobenzyl)-N6-(p-nitrobenzyl)purine), and myricetin have been shown to inhibit IP6K1 activity at low micromolar concentrations. The potency of the inhibitors was characterized using radiolabeled InsPs in the case of TNP, and an indirect ATP-consumption assay for myricetin.38,40 Given that we could directly measure product formation, we expanded the scope of our NMR method towards the characterization of IP6K1 inhibitors. Indeed, we found that TNP and myricetin inhibited IP6K1 with IC50 values of 2.25 μM and 0.6 μM, respectively, while wortmannin, a PI3K inhibitor, did not affect enzyme activity, even at high concentrations (Fig. 4b). While the NMR based method is not compatible with high-throughput screening to identify new kinase inhibitors, it provides a direct read-out of the substrate and the reaction product and is highly reliable. This type of assay can thus serve as a platform to develop suitable high-throughput screening approaches for the kinases and phosphatases involved in InsP metabolism and can ultimately provide the necessary validation for the discovered inhibitors.Metabolic labeling of mammalian cells with [13C6]myo-inositol
To date, the most widely used method to assay cellular InsP and PP-InsP levels employs metabolic labeling of the cells with tritiated, radioactive myo-inositol. After incorporation into the cellular InsP and PP-InsP pools, the labeled compounds are extracted, fractionated via SAX-HPLC chromatography, and analyzed using scintillation counting (Fig. 1a). We envisioned to replace [3H]myo-inositol with [13C6]myo-inositol and tested the metabolic labeling of the human colon cancer cell line HCT116. HCT116 cells were grown in the presence of 100 μM [13C6]myo-inositol and subsequently lysed using 1 M HClO4. The extract was neutralized, lyophilized, dissolved in D2O, and a 1H,13C-HMQC spectrum was recorded (Fig. 5a). The spectrum displayed several strong peaks in the characteristic chemical shift region for InsPs and PP-InsPs (1H: 5.0–3.6 ppm; 13C: 81–71 ppm, Fig. 5b). At high spectral resolution a characteristic triplet pattern emerged due to the coupling of neighboring 13C nuclei in the myo-inositol ring. Comparison to an extract prepared with [12C6]myo-inositol confirmed that these peaks were a result of labeling with [13C6]myo-inositol (Fig. S3†). To verify that the labeling success was not limited to HCT116 cells, we next treated human embryonic kidney cells (HEK293T) with [13C6]myo-inositol and observed robust labeling again (Fig. S4†).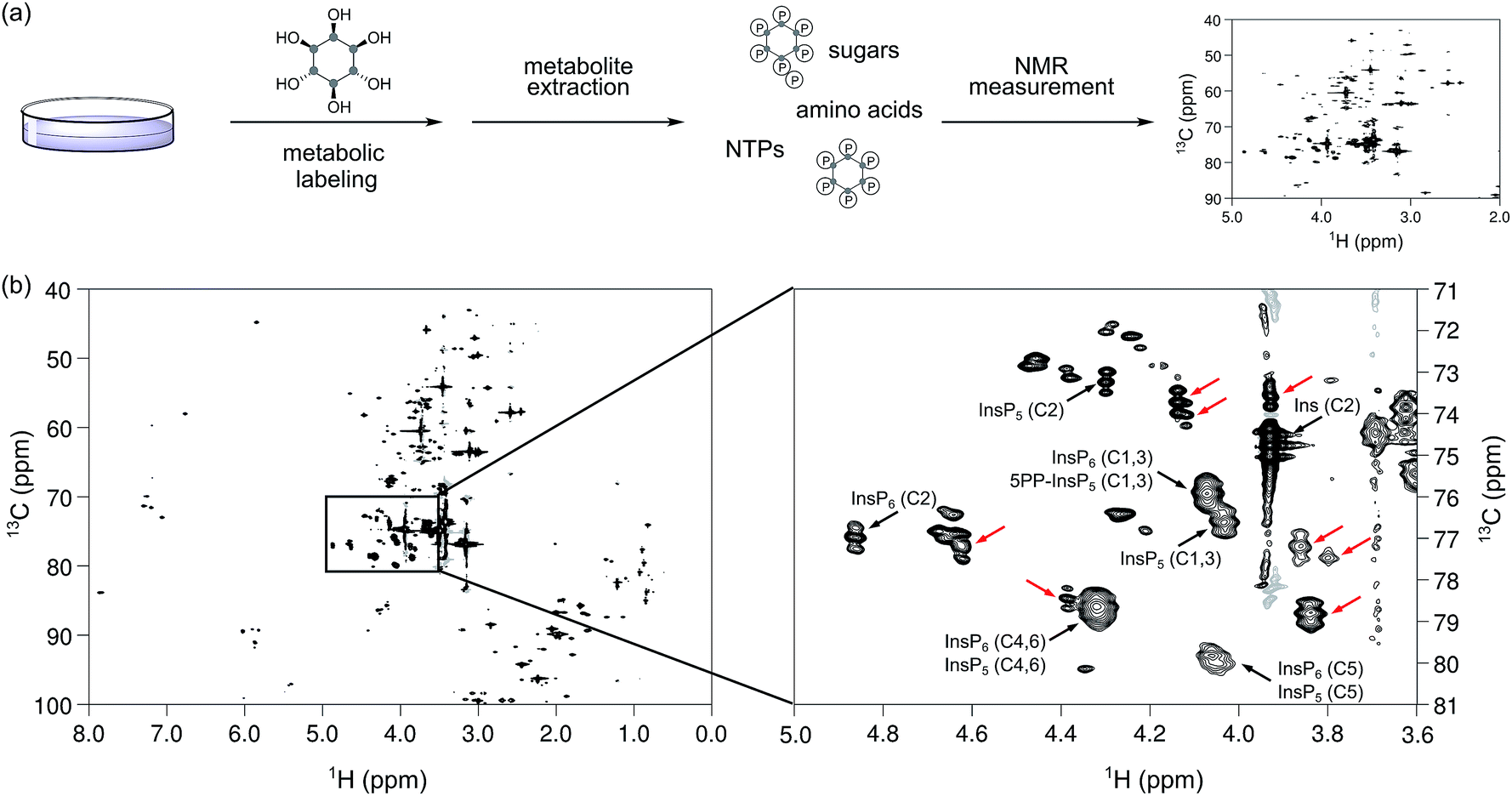 | ||
| Fig. 5 Metabolic labeling of mammalian cell line HCT116, followed by NMR analysis. (a) General workflow for the preparation of whole cell extracts for NMR spectroscopy. (b) Left: 1H,13C HMQC spectrum of an HCT116 extract. The peaks from 80 to 100 ppm in the F1 and between 0 and 3 ppm are folded into an empty region of the spectrum. Right: the inositol phosphate region of the spectrum is depicted in more detail. The identified InsPs are annotated while peaks that exhibit the expected splitting pattern in the carbon dimension but could not be attributed to either myo-inositol (Ins), InsP5, InsP6, nor 5PP-InsP5 are highlighted by red arrows. | ||
To annotate characteristic resonances for endogenous InsP5, InsP6 and 5PP-InsP5, we performed spike-in experiments with synthetic standards on the labeled extracts (Fig. S5†) and confirmed the presence of the above mentioned InsPs (as annotated in Fig. 5b). We further added a mixture containing synthetic InsP5, InsP6 and 5PP-InsP5 to an unlabeled extract from HCT116 and detected all signals at their expected chemical shifts (Fig. S6†).
For absolute quantification of InsP5, InsP6 and 5PP-InsP5, we prepared [13C6]myo-inositol labeled HCT116 extracts containing a known concentration of an internal standard (tetramethyl phosphonium bromide, PMe4Br). Resonances corresponding to the C2-position were integrated and referenced to a set of calibration curves (Fig. S6†). Assuming full replacement of the unlabeled InsP pools we calculated respective concentrations of 26.7 ± 2.3 μM InsP5, 29.4 ± 7.6 μM InsP6, and 1.9 ± 0.5 μM PP-InsP5, based on packed cell volume (Fig. 6a; Table S1†). The measured concentrations are consistent with previous publications with respect to InsP5 and InsP6, however, the amount of 5PP-InsP5 determined by NMR is at least 2-fold higher.29,42 These results underline the mild sample preparation and the quantitative nature of NMR spectroscopy and highlight its potential for future measurements of labeled cells or tissues.
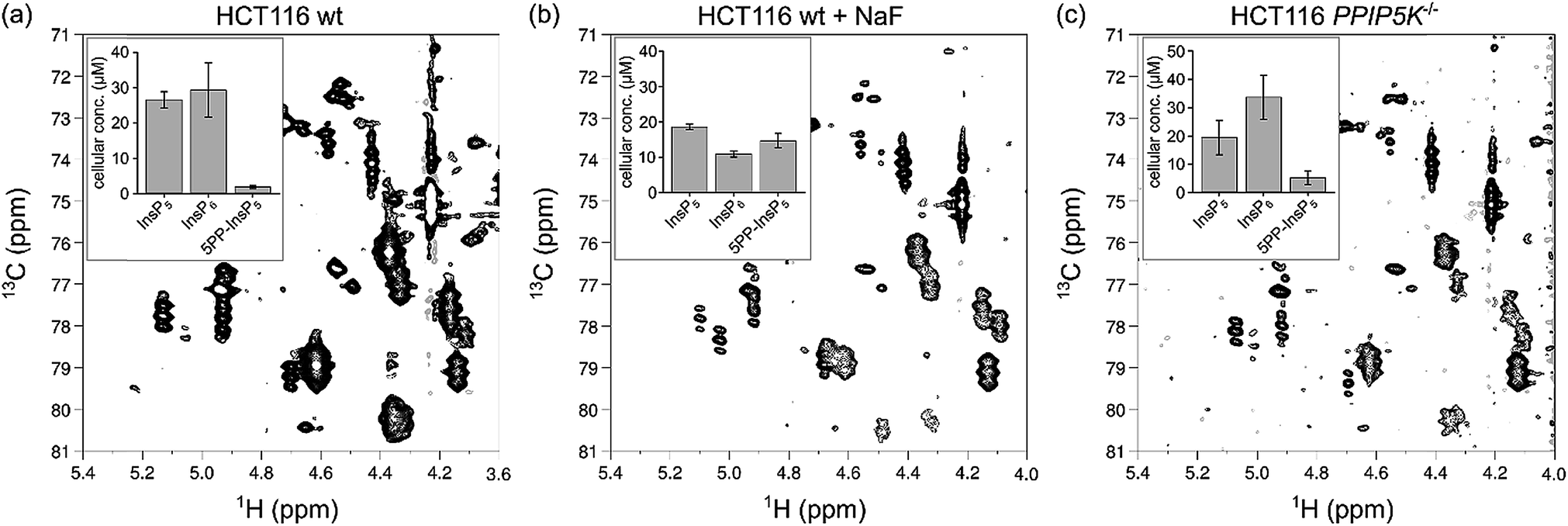 | ||
| Fig. 6 Changes in 5PP-InsP5 levels can be observed by NMR spectroscopy. (a) 1H,13C HMQC spectrum of a HCT116 wt cell extract. (b) 1H,13C HMQC spectrum of an extract of HCT116 wt cells treated with 10 mM NaF before extraction. (c) 1H,13C HMQC spectrum of HCT116 PPIP5K−/− cell extract. The inserts display triplicates of absolute cellular concentration of InsP5, InsP6 and 5PP-InsP5 based on packed cell volume. | ||
Metabolic labeling of cells with perturbed PP-InsP levels
The ability to quantify mixtures of InsP messengers prompted us to analyze the concentrations of InsP5, InsP6 and 5PP-InsP5 in HCT116 cells with perturbed PP-InsP metabolism. When HCT116 cells were treated for one hour with 10 mM sodium fluoride (NaF), an agent previously shown to elevate 5PP-InsP5 levels,43 the content of 5PP-InsP5 increased dramatically to an absolute concentration of 14.7 ± 2.0 μM (Fig. 6b; Table S1†). Elevated concentrations of 5PP-InsP5 were also reported in cells lacking PPIP5K1 and PPIP5K2.44 We therefore grew PPIP5K−/− cells and indeed, a general increase of 5PP-InsP5 was detected compared to HCT116 wt cells (Fig. 6c, Table S1†). Addition of NaF to this knock-out cell line resulted in extracts which contained more 5PP-InsP5 than InsP6 or InsP5 (Fig. S7, Table S1†). These trends of increasing 5PP-InsP5 concentrations had been observed before and the ratio of 5PP-InsP5 to InsP6 in all our samples was consistent with a previous report.44[13C6]myo-Inositol is further metabolized
To our surprise, the analysis of the mammalian extracts uncovered additional NMR signals that displayed a triplet-pattern in the carbon-dimension. These signals were absent in the corresponding samples prepared with unlabeled myo-inositol and must therefore stem from metabolic labeling with [13C6]myo-inositol (Fig. 5b and S3†). Interestingly, several of these signals increased upon NaF treatment (Fig. 6b). To determine whether these signals correspond to other InsP or PP-InsP species, the 13C-labeled, NaF treated HCT116 wt extract was incubated with TiO2 beads, which enrich phosphate containing molecules.19,45 In addition to retaining InsP5, InsP6 and 5PP-InsP5, a putative unsymmetrical InsP species (InsP1 or InsP2) could be observed (Fig. S8†). Nevertheless, most unassigned labeled resonances were not bound by the TiO2 beads. To test for conversion of myo-inositol to metabolically linked molecules, spectra of D-chiro-inositol, scyllo-inositol and glucuronic acid were recorded (Fig. S9†), none of which were superimposable with our unassigned labeled species.46,47 Lastly, we considered that the additional peaks may be attributed to PtdInsPs. However, after a phospholipid extraction, the aqueous layer still contained the unidentified resonances, strongly implying a non-lipid, hydrophilic character of these molecules (Fig. S9†). These observations suggest that the supplemented [13C6]myo-inositol has been diverted into an unknown metabolic pathway. Detection of these unanticipated species was only possible because the NMR-analysis was conducted on the crude extract and did not require a separation step. Elucidating the structure and function of these molecules connected to inositol metabolism will be of interest in the future.Conclusion
Inositol-containing molecules are ubiquitous in nature. Yet, their detection and quantification has proven to pose a formidable challenge, as these molecules typically lack a handle that would facilitate their analysis. Therefore, researchers in the field rely strongly on separation techniques and radioactive tools to answer the biological questions surrounding these metabolites.48 We now provide a new set of methods, which emanate from access to benign 13C-labeled compounds, in combination with commonly accessible NMR technology. This approach has enabled the detection and quantification of InsPs and PP-InsPs within complex mixtures. Specifically, the 13C-labeled compounds could be employed to biochemically characterize the enzymatic activity of IP6K1 in real time. In addition, the non-toxic nature of [13C6]myo-inositol allowed for metabolic labeling of mammalian cells, followed by detection of the generated InsP and PP-InsP species, without the need for separation or enrichment. Furthermore, the structural information gained by the NMR analysis ensured reliable assignment of the identity of the detected InsP species.In the future, the facile applicability of this method will allow for the interrogation of new, or more complex, biological questions. On the biochemical side, for example, characterization of InsP- and PP-InsP phosphatases will benefit greatly from the structural information provided by the NMR measurements, and the substrate specificity of these enzymes can be unequivocally determined. Additionally, new enzymes that have been, or will be, found to metabolize InsPs can be characterized more easily, and the reaction products can be assigned with certainty.
We also envision an important application for the labeled InsPs and PP-InsPs to gain better structural insights into their interactions with proteins. To date, the only known canonical binding domain for the highly phosphorylated inositols is the SPX-domain, but many other proteins physically interact with these metabolites.49 Instead of isotopically labeling these proteins to investigate their ligand-binding via NMR, applying a labeled ligand can have advantages. For example, using a 13C-labeled mannose trisaccharide has recently informed the binding mode between the sugar and its protein binding domain.50 By analogy, the 13C-labeled compounds reported here could help to obtain structural information on the conformation and the biochemical environment of the InsP ligands.
Closely related to InsP messengers are the lipid-anchored phosphatidyl inositols (PtdInsPs), which are crucial in signaling and occupy a pivotal role in health and disease.3 Following the procedure established here for metabolic labeling, but isolating and analyzing the membrane fractions instead, may provide important information on this group of messengers. Considering that the PtdInsPs and InsPs are metabolically linked, concomitant analysis of both groups of molecules could uncover unprecedented information on the coordination of these two messenger networks. While NMR suffers from an innate low sensitivity, advances using hyperpolarization methods in recent years have greatly enhanced the signal of 13C nuclei and could help to increase the sensitivity of our method in order to reliably detect inositol phosphates of low abundance, such as 1PP-InsP5 and InsP8, or PtdInsPs via NMR.51,52
Overall, [13C6]InsPs expand the analytical tool-box to interrogate inositol based messengers and their role and regulation at a biochemical and cellular level.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Lena von Oertzen for assistance with the cell culture and Stephen Shears for providing the IP6KA plasmid and the PPIP5K−/− cell line. R. K. H. and R. P. gratefully acknowledge funding from the Leibniz-Gemeinschaft (SAW-2017-FMP-1). A. S. is supported by the UK Medical Research Council (MRC) core support to the MRC/UCL Laboratory for Molecular Cell Biology University Unit (MC UU 12018/4).References
- M. F.-F. T. Alberts, J. Lewis, R. Roberts and W. Lefers, Molecular Biology of the cell, Garland Science, 2009 Search PubMed.
- M. J. Berridge, Physiol. Rev., 2016, 96, 1261–1296 Search PubMed.
- T. Balla, Physiol. Rev., 2013, 93, 1019–1137 Search PubMed.
- R. F. Irvine and M. J. Schell, Nat. Rev. Mol. Cell Biol., 2001, 2, 327–338 CrossRef CAS PubMed.
- S. B. Shears, J. Cell. Physiol., 2018, 233, 1897–1912 CrossRef CAS PubMed.
- S. G. Thota and R. Bhandari, J. Biosci., 2015, 40, 593–605 CrossRef CAS PubMed.
- M. S. C. Wilson, T. M. Livermore and A. Saiardi, Biochem. J., 2013, 452, 369–379 CrossRef CAS PubMed.
- A. Saiardi, H. Erdjument-Bromage, A. M. Snowman, P. Tempst and S. H. Snyder, Curr. Biol., 1999, 9, 1323–1326 Search PubMed.
- S. Mulugu, W. Bai, P. C. Fridy, R. J. Bastidas, J. C. Otto, D. E. Dollins, T. A. Haystead, A. A. Ribeiro and J. D. York, Science, 2007, 316, 106–109 Search PubMed.
- P. C. Fridy, J. C. Otto, D. E. Dollins and J. D. York, J. Biol. Chem., 2007, 282, 30754–30762 CrossRef CAS PubMed.
- J. H. Choi, J. Williams, J. Cho, J. R. Falck and S. B. Shears, J. Biol. Chem., 2007, 282, 30763–30775 CrossRef CAS PubMed.
- R. S. Jadav, D. Kumar, N. Buwa, S. Ganguli, S. R. Thampatty, N. Balasubramanian and R. Bhandari, Cell. Signalling, 2016, 28, 1124–1136 Search PubMed.
- Y. Moritoh, M. Oka, Y. Yasuhara, H. Hozumi, K. Iwachidow, H. Fuse and R. Tozawa, Sci. Rep., 2016, 6, 32072 Search PubMed.
- A. Chakraborty, M. A. Koldobskiy, N. T. Bello, M. Maxwell, J. J. Potter, K. R. Juluri, D. Maag, S. Kim, A. S. Huang, M. J. Dailey, M. Saleh, A. M. Snowman, T. H. Moran, E. Mezey and S. H. Snyder, Cell, 2010, 143, 897–910 Search PubMed.
- H. Chen, X. Sun, W. Ge, Y. Qian, R. Bai and S. Zheng, Oncotarget, 2017, 8, 95054–95065 Search PubMed.
- R. Yousaf, C. Gu, Z. M. Ahmed, S. N. Khan, T. B. Friedman, S. Riazuddin, S. B. Shears and S. Riazuddin, PLoS Genet., 2018, 14, e1007297 CrossRef PubMed.
- G. W. Mayr, Biochem. J., 1988, 254, 585–591 CrossRef CAS PubMed.
- O. Losito, Z. Szijgyarto, A. C. Resnick and A. Saiardi, PLoS One, 2009, 4, e5580 CrossRef PubMed.
- M. S. C. Wilson, S. J. Bulley, F. Pisani, R. F. Irvine and A. Saiardi, Open Biol., 2015, 5, 150014 CrossRef PubMed.
- T. Laussmann, R. Eujen, C. M. Weisshuhn and U. Thiel, Biochem. J., 1996, 315, 715–720 CrossRef CAS PubMed.
- T. Laussmann, K. M. Reddy, K. K. Reddy, J. R. Falck and G. Vogel, Biochem. J., 1997, 322, 31–33 CrossRef CAS PubMed.
- O. B. Ijare, D. S. Baskin, M. A. Sharpe and K. Pichumani, Anal. Biochem., 2018, 552, 110–117 Search PubMed.
- K. R. Keshari, D. M. Wilson, A. P. Chen, R. Bok, P. E. Z. Larson, S. Hu, M. Van Criekinge, J. M. Macdonald, D. B. Vigneron and J. Kurhanewicz, J. Am. Chem. Soc., 2009, 131, 17591–17596 Search PubMed.
- J. M. Park, M. Wu, K. Datta, S. C. Liu, A. Castillo, H. Lough, D. M. Spielman and K. L. Billingsley, J. Am. Chem. Soc., 2017, 139, 6629–6634 CrossRef CAS PubMed.
- A. Saiardi, C. Guillermier, O. Loss, J. C. Poczatek and C. Lechene, Surf. Interface Anal., 2014, 46, 169–172 CrossRef CAS PubMed.
- P. Sahai, M. Chawla and R. A. Vishwakarma, J. Chem. Soc., Perkin Trans. 1, 2000, 1283–1290 RSC.
- M. J. Moure, Y. Zhuo, G. J. Boons and J. H. Prestegard, Chem. Commun., 2017, 53, 12398–12401 RSC.
- L. G. Morin, Clin. Chem., 1977, 23, 1569–1575 CAS.
- C. J. Barker, J. Wright, P. J. Hughes, C. J. Kirk and R. H. Michell, Biochem. J., 2004, 380, 465–473 Search PubMed.
- H. Zhang, J. Thompson and G. D. Prestwich, Org. Lett., 2009, 11, 1551–1554 CrossRef CAS PubMed.
- I. Pavlovic, D. T. Thakor, J. R. Vargas, C. J. McKinlay, S. Hauke, P. Anstaett, R. C. Camunã, L. Bigler, G. Gasser, C. Schultz, P. A. Wender and H. J. Jessen, Nat. Commun., 2016, 7, 10622 CrossRef CAS PubMed.
- B. Löser, M. M. Nalaskowski, W. Fanick, H. Lin, E. Tannich and G. W. Mayr, Mol. Biochem. Parasitol., 2012, 181, 49–52 CrossRef.
- A. Saiardi, R. Bhandari, A. C. Resnick, A. M. Snowman and S. H. Snyder, Science, 2004, 306, 2101–2105 CrossRef CAS PubMed.
- O. Loss, C. Azevedo, Z. Szijgyarto, D. Bosch and A. Saiardi, J. Visualized Exp., 2011, 3027 Search PubMed.
- A. Hager, M. Wu, H. Wang, N. W. Brown, S. B. Shears, N. Veiga and D. Fiedler, Chem.–Eur. J., 2016, 22, 12406–12414 CrossRef CAS.
- N. Veiga, J. Torres, S. Domínguez, A. Mederos, R. F. Irvine, A. Díaz and C. Kremer, J. Inorg. Biochem., 2006, 100, 1800–1810 CrossRef CAS PubMed.
- T. Wundenberg, N. Grabinski, H. Lin and G. W. Mayr, Biochem. J., 2014, 462, 173–184 CrossRef CAS.
- M. Wormald, G. Liao, M. Kimos, J. Barrow and H. Wei, PLoS One, 2017, 12, e0188852 Search PubMed.
- S. M. Voglmaier, M. E. Bembenek, A. I. Kaplin, G. Dormán, J. D. Olszewski, G. D. Prestwich and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4305–4310 Search PubMed.
- U. Padmanabhan, D. E. Dollins, P. C. Fridy, J. D. York and C. P. Downes, J. Biol. Chem., 2009, 284, 10571–10582 CrossRef CAS PubMed.
- A. Saiardi, C. Azevedo, Y. Desfougères, P. Portela-Torres and M. S. C. Wilson, Adv. Biol. Regul., 2018, 67, 74–83 CrossRef CAS PubMed.
- C. Albert, S. T. Safrany, M. E. Bembenek, K. M. Reddy, K. K. Reddy, J. R. Falck, M. Bröcker, S. B. Shears and G. W. Mayr, Biochem. J., 1997, 327, 553–560 CrossRef CAS PubMed.
- H. Lin, P. C. Fridy, A. A. Ribeiro, J. H. Choi, D. K. Barma, G. Vogel, J. R. Falck, S. B. Shears, J. D. York and G. W. Mayr, J. Biol. Chem., 2009, 284, 1863–1872 CrossRef CAS.
- C. Gu, H.-N. Nguyen, D. Ganini, Z. Chen, H. J. Jessen, Z. Gu, H. Wang and S. B. Shears, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11968–11973 CrossRef CAS.
- M. Wilson and A. Saiardi, Bio-Protoc., 2018, 8, e2959 Search PubMed.
- R. J. Arner, K. S. Prabhu, J. T. Thompson, G. R. Hildenbrandt, A. D. Liken and C. C. Reddy, Biochem. J., 2001, 360, 313–320 CrossRef CAS.
- M. P. Thomas, S. J. Mills and B. V. L. Potter, Angew. Chem., Int. Ed., 2016, 55, 1614–1650 CrossRef CAS.
- M. S. C. Wilson and A. Saiardi, Top. Curr. Chem., 2017, 375, 14 CrossRef.
- M. Wu, L. S. Chong, D. H. Perlman, A. C. Resnick and D. Fiedler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6757–E6765 CrossRef CAS.
- G. Nestor, T. Anderson, S. Oscarson and A. M. Gronenborn, J. Am. Chem. Soc., 2017, 139, 6210–6216 CrossRef CAS.
- C. Kjeldsen, J. H. Ardenkjær-Larsen and J. O. Duus, J. Am. Chem. Soc., 2018, 140, 3030–3034 CrossRef CAS.
- M. L. Hirsch, N. Kalechofsky, A. Belzer, M. Rosay and J. G. Kempf, J. Am. Chem. Soc., 2015, 137, 8428–8434 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00151d |
| ‡ R. K. H. and R. P. contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |