
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical fluoromethylation triggered lactonizations of alkenes under semi-aqueous conditions†
Sheng
Zhang
 *a,
Lijun
Li
a,
Jinjin
Zhang
a,
Junqi
Zhang
a,
Mengyu
Xue
a and
Kun
Xu
*ab
*a,
Lijun
Li
a,
Jinjin
Zhang
a,
Junqi
Zhang
a,
Mengyu
Xue
a and
Kun
Xu
*ab
aEngineering Technology Research Center of Henan Province for Photo- and Electrochemical Catalysis, College of Chemistry and Pharmaceutical Engineering, Nanyang Normal University, Nanyang, 473061, China. E-mail: shengzhang@nynu.edu.cn; xukun@nynu.edu.cn
bCollege of Life Science & Bioengineering, Beijing University of Technology, Beijing 100124, China
First published on 14th February 2019
Abstract
An electrochemical difluoromethylation triggered lactonization of alkenes was developed for the first time. This protocol employs readily prepared CF2HSO2Na as the difluoromethylating reagent, affording unprecedented CF2H-containing lactones in moderate yields. Moreover, with CF3SO2Na as the trifluoromethylating reagent, a wide array of CF3-containing lactones were obtained under additional supporting electrolyte- and catalyst-free conditions.
Introduction
The introduction of fluorine atoms into organic molecules has attracted increasing interest because the incorporation of fluorine-containing groups can significantly modify the properties of bioactive molecules.1 In contrast to various methods for trifluoromethylation of organic substrates,2,3 direct difluoromethylation is still underdeveloped,4 even though the difluoromethyl group (CF2H) is an intriguing structural motif in drug design.5 Among the existing methods for direct difluoromethylations, radical processes have played an important role in obtaining CF2H-containing compounds.6 It is noteworthy that there are many recent reports of photoinduced difluoromethylations of heterocycles7 and alkenes.8 However, expensive Ir- or Ru-based photoredox catalysts and synthetically challenging CF2H radical precursors are commonly required. Synthetic electrochemistry has the obvious advantage of generating radicals in a controllable way to minimize the possibilities of radical dimerizations, and can realize some transformations in ways that were previously difficult or inaccessible by traditional methods.9,10 In this context, Baran, Blackmond and co-workers disclosed an electrochemical difluoromethylation of heterocycles in a divided cell with zinc sulfinates as the difluoromethylating reagent and n-Bu4NClO4 as the supporting electrolyte (Scheme 1a).11 Recently, a breakthrough in electrochemical difluoromethylation of alkynes with CF2HSO2NHNHBoc was reported by Xu and co-workers with Et4NBF4 as the supporting electrolyte (Scheme 1b).12 Given the importance of the CF2H group in medicinal chemistry and the advantages of synthetic electrochemistry, the development of new electrochemical difluoromethylation reactions in a user-friendly single cell setup in the absence of an additional supporting electrolyte is attractive.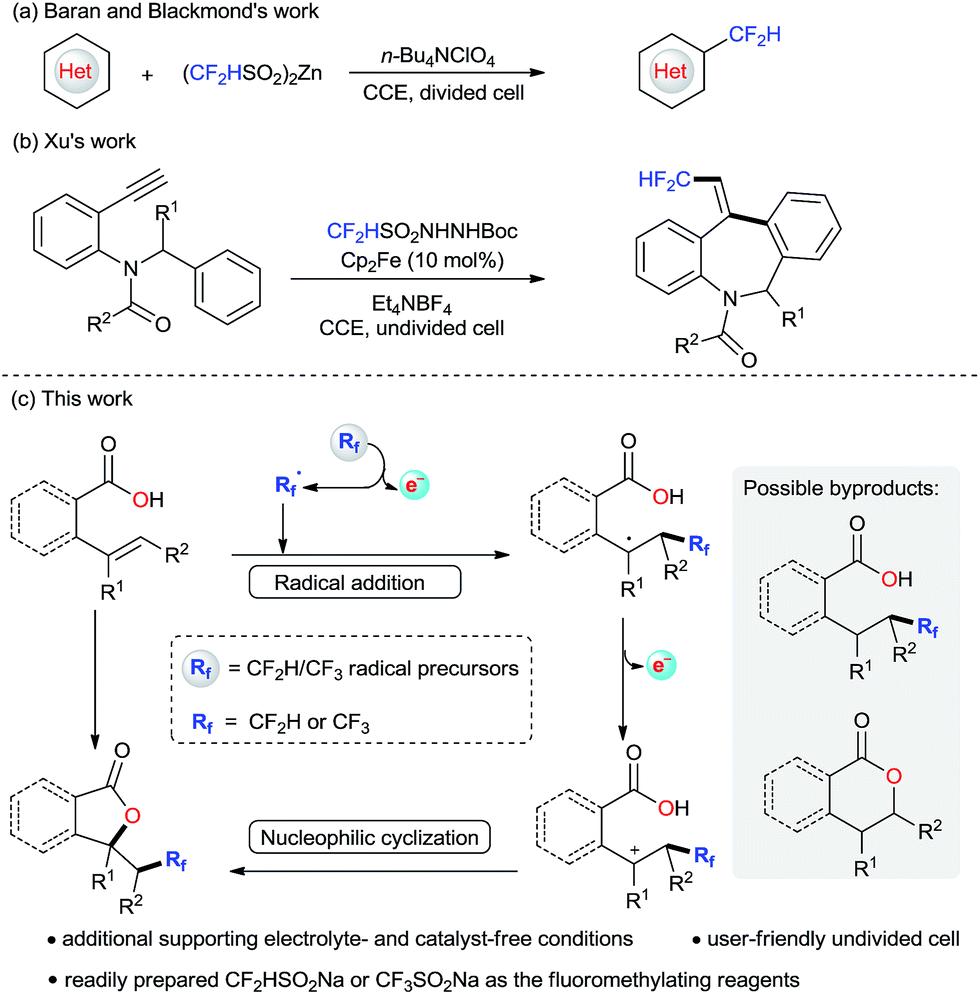 | ||
| Scheme 1 Electrochemical difluoromethylations. | ||
Lactones constitute useful building blocks in many pharmaceutically relevant molecules.13 In this regard, the construction of unprecedented CF2H-containing lactones may be beneficial for medicinal chemistry.14 We have been interested in electrochemical lactonizations; however, only C–O bonds were constructed for these transformations.15,16 Considering the powerfulness of radical alkene difunctionalizations for the enhancement of molecular complexity in a single preparative operation,17 we speculated that it might be possible to construct CF2H-containing lactones via an electrochemical difluoromethylation triggered lactonization of alkenes. The proposed synthetic pathway is shown in Scheme 1c. First, electrochemically generated fluoromethyl radical undergoes alkene addition to give a carbon radical intermediate. Further electrochemical oxidation gives a carbocationic intermediate, which undergoes subsequent nucleophilic cyclization to afford desired fluoromethylated lactones. While the proposed reaction pathway appears quite reasonable, its implementation proved to be challenging. First, the electrochemical oxidation of the carbon radical intermediate should occur quickly before H· abstraction. Second, the oxidation potentials of Rf radical precursors should be much lower than that of alkenes. Otherwise, the undesired single C–O bond formation would be the predominant process instead of desired alkene difunctionalization. In this report, we establish that electrochemical difunctionalization of alkenes can be achieved using semi-aqueous conditions to afford unprecedented CF2H-containing lactones with CF2HSO2Na18 as the CF2H radical precursor under catalyst-free conditions. Moreover, this environmentally benign protocol could also be applicable for the access to CF3-containing lactones in the absence of a metal catalyst, chemical oxidant, and additional supporting electrolyte.
Results and discussion
Initially, we commenced the electrochemical carboxydifluoromethylation reaction by using 1c and CF2HSO2Na (2) as the model substrates in an undivided cell equipped with platinum electrodes (Table 1). When HOAc was employed as the additive with a mixture of CH3CN and H2O as the solvent, the isolated yield of the corresponding CF2H-containing lactone 3c was obtained to be 67% (entry 1). Interestingly, adding supporting electrolytes into this reaction mixture led to a decrease in the yields (entries 2 and 3).19 Changing the Pt electrodes to graphite failed to maintain the reaction yield (entries 4 and 5). When the reaction was carried out in the absence of HOAc, only a trace amount of the desired product 3c was detected (entry 6). This result suggested that the cathodic proton reduction may limit the overall reaction rate.20 Replacing HOAc with HCl only led to a trace amount of the product 3c (entry 7). Increasing or decreasing the current density failed to improve the yield (entries 8 and 9).|
|
||
|---|---|---|
| Entry | Changes from standard conditions | Yieldb (%) |
| a Reaction conditions: undivided cell, Pt plate (1.5 × 1.5 cm2, J = 6.7 mA cm−2), 1c (0.5 mmol), 2 (1.25 mmol), CH3CN/H2O (7/1 mL, v/v), rt, 3 h, and 3.4 F. b Isolated yield. | ||
| 1 | None | 67 |
| 2 | 0.1 M nBu4NPF6 was used as the electrolyte | 59 |
| 3 | 0.1 M LiClO4 was used as the electrolyte | 52 |
| 4 | Graphite(+) and Pt(−) were used as the electrodes | 47 |
| 5 | Pt(+) and graphite(−) were used as the electrodes | 39 |
| 6 | No HOAc | Trace |
| 7 | HCl was used instead of HOAc | Trace |
| 8 | J = 10 mA cm−2 | 61 |
| 9 | J = 5 mA cm−2 | 43 |

Having established the optimized reaction conditions, we then examined the substrate scope of electrochemical difluoromethylation triggered lactonization of alkenes. As shown in Table 2, the aromatic carboxylic acids were tolerated well to give the corresponding CF2H-containing lactones in moderate yields (3a–3o). For the substituents on the Ar2 ring, the para-substituents had little effect on the chemical yields (3b–3i). The ortho-substituted substrate 1j showed decreased reactivity to give the corresponding product 3j in 50% yield with TFA as the acidic additive instead of HOAc. When the fluoro group was placed at the meta position of the Ar2 ring, the corresponding lactone 3k was obtained in 79% yield. Replacing the phenyl group with the 1-naphthyl group decreased the yield of 3l to 57%.

When Ar2 was replaced with the methyl group, the corresponding lactone 3o was afforded in 46% yield. It is noteworthy that the challenging substrates of aliphatic carboxylic acids could also be tolerated to give the corresponding lactones 3p–3r in 38–42% yields.
To make this synthetic methodology more appealing, the electrochemical trifluoromethylation triggered lactonization of alkenes was then examined. As shown in Table 3, moderate to excellent yields of CF3-containing lactones were obtained regardless of the electronic nature of para-substitutions on the Ar2 ring (5a–5i). Changing the substitution on the Ar2 ring from the para-position to the ortho- or meta-position caused lower yields (5j–5l). The substrate containing a disubstituted Ar2 group was also tolerated well affording the product 5m in 64% yield. The fused ring substituted substrates also underwent the cyclizations smoothly to give the corresponding lactones 5n and 5p in 66% and 63% yields, respectively. Replacing the aromatic Ar1 or Ar2 group with aliphatic ones decreased the reaction efficiency, giving the corresponding lactones 5q and 5r in 53% and 69% yields, respectively. More importantly, the trisubstituted olefin was demonstrated to be a suitable substrate to give the lactone 5s in 42% yield.

In order to provide a rationale for the reaction pathway proposed in Scheme 1c, cyclic voltammetric (CV) experiments were carried out. As shown in Fig. 1, CF2HSO2Na and CF3SO2Na have the oxidation potentials of 0.72 V and 1.06 V, respectively. However, the oxidation potential of alkenes is 1.58 V. These results indicated that CF2HSO2Na and CF3SO2Na are much easier to be electrochemically oxidized to generate fluoromethyl radicals than the alkene moiety. The CV experiments which were carried out in CH3CN/HOAc or CH3CN/H2O also indicated that CF2HSO2Na and CF3SO2Na are much easier to be electrochemically oxidized than the alkene moiety (see the ESI† for details). The much lower oxidation potentials of CF2H and CF3 radical precursors than that of alkenes are the key to electrochemical carboxyfluoromethylation reactions.
 | ||
| Fig. 1 Cyclic voltammograms of substrates in 0.1 M LiClO4/CH3CN, using a Pt wire working electrode and glassy carbon and Ag/AgNO3 (0.1 M in CH3CN) as counter and reference electrodes at a 100 mV s−1 scan rate: (a) background (0.1 M LiClO4 in CH3CN), (b) CF3SO2Na (5 mmol L−1), (c) CF2HSO2Na (5 mmol L−1), and (d) 1a (5 mmol L−1). | ||
Experimental
An undivided cell was equipped with a magnet stirrer and platinum plate (1.5 × 1.5 cm2) electrodes. The substrate 2-(1-phenylvinyl)benzoic acid 1a (112 mg, 0.5 mmol), CF3SO2Na 4 (195 mg, 1.25 mmol) and additive HOAc (86 μL, 1.5 mmol) were added to a mixed solvent of CH3CN/H2O (7/1 v/v). The resulting mixture was allowed to stir and electrolyze under constant current conditions (J = 6.7 mA cm−2) at room temperature for 3 hours. Then the volatile solvent was removed with a rotary evaporator and then water (10 mL) was added. The resulting mixture was extracted with ethyl acetate (10 × 3 mL). The combined organic layer was dried over Na2SO4 and concentrated under vacuum. The residue was purified by column chromatography (ethyl acetate/petroleum ether = 1/15–1/10) on silica gel to afford the desired product 5a in 93% yield.Conclusions
We have developed the first example of electrochemical difluoromethylation triggered lactonization of alkenes. Under additional supporting electrolyte- and catalyst-free conditions, a wide array of CF2H-containing lactones were obtained in moderate yields. Moreover, this environmentally benign method is also applicable to access pharmaceutically important CF3-containing lactones in the absence of a metal catalyst, chemical oxidant, and additional supporting electrolyte.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21702113, U1504208 and 21602119), project funded by the China Postdoctoral Science Foundation, and Program for Science and Technology Innovation Talents in Universities of Henan Province (19HASTIT033).Notes and references
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity Applications, Wiley-VCH, Weinheim, 2004 CrossRef; (b) T. Yamazaki, T. Taguchi and I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, Great Britain, 2009 Search PubMed.
- For recent reviews on trifluoromethylations, see: (a) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (c) X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163 RSC; (d) H.-X. Song, Q.-Y. Han, C.-L. Zhao and C.-P. Zhang, Green Chem., 2018, 20, 1662 RSC; (e) S. Barata-Vallejo, M.-V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287 CrossRef CAS.
- For recent examples on trifluoromethylations, see: (a) W. Yang, D. Ma, Y. Zhou, X. Dong, Z. Lin and J.-W. Sun, Angew. Chem., Int. Ed., 2018, 57, 12097 CrossRef CAS PubMed; (b) E. Valverde, S. Kawamura, D. Sekine and M. Sodeoka, Chem. Sci., 2018, 9, 7115 RSC; (c) H. Wang, Q. Xu and S.-Y. Yu, Org. Chem. Front., 2018, 5, 2224 RSC; (d) B.-K. Mai, K.-J. Szabó and F. Himo, ACS Catal., 2018, 8, 4483 CrossRef CAS; (e) A.-J. Borah and Z.-Z. Shi, Chem. Commun., 2017, 53, 3945 RSC; (f) H.-S. Han, E.-Y. Oh, Y.-S. Jung and S.-B. Han, Org. Lett., 2018, 20, 1698 CrossRef CAS PubMed; (g) B. Yang, D. Yu, X.-H. Xu and F.-L. Qing, ACS Catal., 2018, 8, 2839 CrossRef CAS; (h) M. Imiołek, G. Karunanithy, W.-L. Ng, A.-J. Baldwin, V. Gouverneur and B.-G. Davis, J. Am. Chem. Soc., 2018, 140, 1568 CrossRef PubMed.
- For recent reviews, see: (a) J.-B. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (b) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2015, 21, 12836 CrossRef CAS PubMed; (c) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676 CrossRef CAS PubMed; (d) A. D. Dilman and V. V. Levin, Acc. Chem. Res., 2018, 51, 1272 CrossRef CAS PubMed. For selected recent works, see: (e) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed; (f) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494 CrossRef CAS PubMed; (g) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed; (h) T. Iida, R. Hashimoto, K. Aikawa, S. Ito and K. Mikami, Angew. Chem., Int. Ed., 2012, 51, 9535 CrossRef CAS PubMed; (i) P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2015, 54, 6558 CrossRef CAS PubMed; (j) N.-B. Heine and A. Studer, Org. Lett., 2017, 19, 4150 CrossRef CAS PubMed; (k) T. T. Tung, S. B. Christensen and J. Nielsen, Chem.–Eur. J., 2017, 23, 18125 CrossRef CAS PubMed.
- (a) M. A. Chowdhury, K. R. A. Abdellatif, Y. Dong, D. Das, M. R. Suresh and E. E. Knaus, J. Med. Chem., 2009, 52, 1525 CrossRef CAS PubMed; (b) N.-A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed.
- T. Koike and M. Akita, Chem, 2018, 4, 409 CAS and references cited therein..
- (a) P. Xu, S. Guo, L. Wang and P. Tang, Synlett, 2014, 26, 36 CrossRef PubMed; (b) R. Sakamoto, H. Kashiwagi and K. Maruoka, Org. Lett., 2017, 19, 5126 CrossRef CAS PubMed; (c) S.-Q. Zhu, Y.-L. Liu, H. Li, X.-H. Xu and F.-L. Qing, J. Am. Chem. Soc., 2018, 140, 11613 CrossRef CAS PubMed.
- (a) X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246 CrossRef CAS PubMed; (b) Y. Xiang, Y. Li, Y. Kuang and J. Wu, Chem.–Eur. J., 2016, 23, 1032 CrossRef PubMed; (c) J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J.-B. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743 CrossRef CAS PubMed; (d) Q.-Y. Lin, X.-H. Xu, K. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2016, 55, 1479 CrossRef CAS PubMed; (e) J.-S. Lin, F.-L. Wang, X.-Y. Dong, W.-W. He, Y. Yuan, S. Chen and X.-Y. Liu, Nat. Commun., 2017, 8, 14841 CrossRef PubMed; (f) N. Noto, T. Koike and M. Akita, Chem. Sci., 2017, 8, 6375 RSC.
- For recent reviews, see: (a) R. Feng, J. A. Smith and K. D. Moeller, Acc. Chem. Res., 2017, 50, 2346 CrossRef CAS PubMed; (b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (c) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS PubMed; (d) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS; (e) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS; (f) J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS PubMed; (g) Y. Okada and K. Chiba, Chem. Rev., 2018, 118, 4592 CrossRef CAS PubMed; (h) Y. Jiang, K. Xu and C.-C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS PubMed; (i) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC.
- For selected recent examples, see: (a) Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448 CrossRef CAS PubMed; (b) T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317 CrossRef CAS PubMed; (c) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575 CrossRef CAS PubMed; (d) Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487 CrossRef CAS PubMed; (e) S. Tang, L. Zeng and A. Lei, J. Am. Chem. Soc., 2018, 140, 13128 CrossRef CAS PubMed; (f) R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913 CrossRef CAS PubMed; (g) S. Zhang, L. Li, M. Xue, R. Zhang, K. Xu and C. Zeng, Org. Lett., 2018, 20, 3443 CrossRef CAS PubMed; (h) K.-S. Du and J.-M. Huang, Org. Lett., 2018, 20, 2911 CrossRef CAS PubMed; (i) H. Wang, J. Zhang, J. Tan, L. Xin, Y. Li, S. Zhang and K. Xu, Org. Lett., 2018, 20, 2505 CrossRef CAS; (j) Q. Liu, B. Sun, Z. Liu, Y. Kao, B.-W. Dong, S.-D. Jiang, F. Li, G. Liu, Y. Yang and F.-Y. Mo, Chem. Sci., 2018, 9, 8731 RSC; (k) J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2018, 57, 5695 CrossRef CAS PubMed; (l) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22 CrossRef CAS PubMed; (m) B. Schille, N. O. Giltzau and R. Francke, Angew. Chem., Int. Ed., 2018, 57, 422 CrossRef CAS PubMed. For some pioneering electrochemical fluorination studies, see: (n) A. Konno, K. Nakagawa and T. Fuchigami, J. Chem. Soc., Chem. Commun., 1991, 1027 RSC; (o) K. Uneyama, Tetrahedron, 1991, 47, 555 CrossRef CAS.
- A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868 CrossRef PubMed.
- P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460 CrossRef CAS PubMed.
- (a) Y.-J. Zhang, T. Abe, T. Tanaka, C.-R. Yang and I. Kouno, J. Nat. Prod., 2001, 64, 1527 CrossRef CAS PubMed; (b) J. J. Beck and S.-C. Chou, J. Nat. Prod., 2007, 70, 891 CrossRef CAS PubMed.
- The use of α-bromodifluoromethyl substituted esters and amides to stabilize the in situ generated CF2H· to access to CF2H-containing lactones has been described, see: Y. Da, S. Han, X. Du, S. Liu, L. Liu and J. Li, Org. Lett., 2018, 20, 5149 CrossRef CAS PubMed.
- (a) S. Zhang, F. Lian, M. Xue, T. Qin, L. Li, X. Zhang and K. Xu, Org. Lett., 2017, 19, 6622 CrossRef CAS PubMed; (b) S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C.-C. Zeng, Org. Lett., 2018, 20, 252 CrossRef CAS PubMed.
- A previous work on the construction of two C–O bonds during lactonization of alkenes was reported by Moeller and co-workers, see: R. J. Perkins, H.-C. Xu, J. M. Campbell and K. D. Moeller, Beilstein J. Org. Chem., 2013, 9, 1630 CrossRef PubMed.
- For selected recent reviews, see: (a) G. Yin, X. Mu and G. Liu, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed; (b) J. R. Coombs and J. P. Morken, Angew. Chem., Int. Ed., 2016, 55, 2636 CrossRef CAS PubMed. For recent examples of electrochemical alkene difunctionalizations, see: (c) Y. Yuan, Y. Gao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871 CrossRef CAS; (d) K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438 CrossRef CAS PubMed; (e) Y. Wang, L. Deng, H. Mei, B. Du, J. Han and Y. Pan, Green Chem., 2018, 20, 3444 RSC; (f) C.-Y. Cai and H.-C. Xu, Nat. Commun., 2018, 9, 3551 CrossRef PubMed; (g) S. Zhang, L. Li, P. Wu, P. Gong, R. Liu and K. Xu, Adv. Synth. Catal., 2019, 361, 485 Search PubMed. For recent example of electrochemical alkyne difunctionalizations, see: (h) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383 CrossRef CAS PubMed.
- Z. He, P. Tan, C. Ni and J.-B. Hu, Org. Lett., 2015, 17, 1838 CrossRef CAS PubMed.
- The addition of additional electrolyte may be favourable for the formation of a more defined double layer, which could slow the bimolecular reactions.
- The adding of acidic additives also lower the pH of the solvent. The low pH of the solvent is unfavourable for the unexpected Kolbe reactions.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00100j |
| This journal is © The Royal Society of Chemistry 2019 |