
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaking the unconventional μ2-P bridging binding mode more conventional in phosphinine complexes†
Yuanfeng
Hou
a,
Zhongshu
Li
 *a,
Yaqi
Li
a,
Peng
Liu
b,
Cheng-Yong
Su
*a,
Florian
Puschmann
c and
Hansjörg
Grützmacher
*ac
*a,
Yaqi
Li
a,
Peng
Liu
b,
Cheng-Yong
Su
*a,
Florian
Puschmann
c and
Hansjörg
Grützmacher
*ac
aLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-Sen University, 510275 Guangzhou, China. E-mail: lizhsh6@mail.sysu.edu.cn
bGuangdong Engineering and Technology Research Center for Advanced Nanomaterials, School of Environment and Civil Engineering, Dongguan University of Technology, Dongguan 523808, China
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch
First published on 21st January 2019
Abstract
Phosphinines, as aromatic heterocycles, usually engage in coordination as η1-P σ-complexes or η6-phosphinine π-complexes. The μ2-P bridging coordination mode is rarely observed. With the aim to study the effect of different electronic configurations of phosphinines on the coordination modes, a series of anionic phosphinin-2-olates and neutral phosphinin-2-ols were prepared with moderate to high yield. Then the coordination chemistry of these two series was studied in detail towards coinage metals (Au(I) and Cu(I)). It is observed that the anionic phosphinin-2-olates possess a higher tendency to take a bridging position between two metal centers compared to the neutral phosphinin-2-ols. Based on these experimental findings bolstered by DFT calculations, some insight is gained on how the unconventional μ2-P phosphinine bridging coordination mode can be made more conventional and used for the synthesis of polynuclear complexes.
Introduction
Phosphinines have been suggested as useful ligands in homogeneously catalysed processes1 and valuable components for photoluminescent materials.2 As compared to their homologous pyridines, phosphinines are relatively weak σ-donor but strong π-acceptor ligands.3 Thus, the coordination mode of phosphinines through the phosphorus atom is the most common one and is frequently observed with late transition metals in low oxidation states (I, Scheme 1).1b–d,3e,4 It is rather challenging to prepare phosphinine complexes with metal centers in medium-to-high oxidation states because the smaller metal-to-ligand π-back donation leads to a reduced stability.5 But some of these complexes can be accessed with chelating polydentate phosphinines.6 Like other aromatic molecules with a delocalized 6π-electron system, phosphinines can also bind in a η6-fashion to metal centers (II, Scheme 1).7 | ||
| Scheme 1 Coordination modes of phosphinines. | ||
A relatively unusual coordination mode of phosphinines consists in bridging of two metal centers although a first example has been reported by Venanzi and co-workers more than 25 years ago (III, Scheme 1).8 It was suggested that the phosphorus atom of the phosphinine acts as two-electron donor only. Since then, few transition metal complexes with phosphinines as μ2-P bridging ligands have been reported (for a detailed listing of these complexes see Scheme 9 below).2c,9,10 Hartl et al. proposed that the μ2-bridging phosphorus atoms of a 2,2-biphosphinine as ligand in a triosmium cluster have a sp3-valence electron configuration and serve as four-electron donors (vide infra).9c,9d It is generally agreed that the metal centers donate significantly electron density back into π-type orbitals at the ligand.9,10
Recently, Mathey, et al. have reported phosphinin-2-ol, a functionalized phosphinine, which can adopt a μ2-P bridging coordination mode towards Cu(I) centers besides the common η1-P mode.11 The synthetic access to phosphinin-2-ol or phosphinin-2-olate was quite laborious and required multi-step syntheses.12 A simple and straight forward approach for the preparation of parent phosphinin-2-olate in high yield consists in the [2 + 4] cycloaddition of Na(O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P) to α-pyrones which proceeds under extrusion of CO2.10,13 After protonation with HCl, phosphinin-2-ol was isolated as colourless oil. We have now extended this approach and here we report the synthesis of a series of phosphinin-2-olates which were obtained with commercially available α-pyrones. The corresponding phosphinin-2-ols were prepared after simple protonation reactions. The coordination chemistry was investigated through the synthesis of various complexes with Cu(I) and Au(I) as metal centers with the aim to study the effect of the different electronic configurations of anionic phosphin-2-olates and neutral phosphinin-2-ols on the coordiantion modes.
P) to α-pyrones which proceeds under extrusion of CO2.10,13 After protonation with HCl, phosphinin-2-ol was isolated as colourless oil. We have now extended this approach and here we report the synthesis of a series of phosphinin-2-olates which were obtained with commercially available α-pyrones. The corresponding phosphinin-2-ols were prepared after simple protonation reactions. The coordination chemistry was investigated through the synthesis of various complexes with Cu(I) and Au(I) as metal centers with the aim to study the effect of the different electronic configurations of anionic phosphin-2-olates and neutral phosphinin-2-ols on the coordiantion modes.
Results
Density functional theory calculations (BP86/def2-TZVPP)14 were carried out for the parent phosphinine, PC5H5, the neutral phosphinin-2-ol, 2-(HO)PC5H4, and the anion phosphinin-2-olate, [2-O-PC4H4]−. The optimized structures were used for subsequent natural resonance theory (NRT) calculations.15 As expected, the structure with a delocalized 6π-electron configuration in the ring dominates the electronic ground state of PC5H5 with 70.9% while zwitterionic structures contribute with 9.1% and 8.5% [see (A) in Scheme 2]. Only the latter has two electron pairs at the phosphorus center. Also in 2-(HO)PC5H4 the structure with a delocalized 6π-electron configuration is the main-contributor, but two zwitterionic structures each with two lone pairs at the phosphorus center contribute with a total of 12.5% to the electronic ground state. The weight of individual resonance structures to the electronic ground state of the anionic phosphinin-2-olate is profoundly different. Here the structure with a 6π-electron configuration contributes only with 33.2% while the two structures with two lone pairs at phosphorus contribute almost equally with 30.4%. This weighting scheme does not change significantly when a [Na(18-crown-6)(H2O)]+ cation is included in the NRT calculation (33.5%, 18.6%, 10.0%, 19.6%; same order of resonance structures as given in Scheme 2). These results predict that the phosphinin-2-olate possesses a higher tendency to take a bridging position between metal centers and act as a four-electron donor when compared to neutral phosphinines. | ||
| Scheme 2 Resonance structures obtained by NRT computations on parent phosphinine (A), phosphinin-2-ol (B), and phosphinin-2-olate (C). Structures with circles or bows indicate an overlay of several similar resonance structures; the given percentages are the sum of the individual contributions of these electronic configurations. | ||
The positive charge determined by Natural Population Analyses (NPA) at the phosphorus atom decreases in the following order: phosphinine (+0.63 e) > phosphinin-2-ol (+0.49 e) > phosphinin-2-olate (+0.27 e), which agrees with the results from the NRT analysis.
We used a slightly modified procedure compared to our original one,13 to prepare a range of sodium phosphinin-2-olates 1a–1d (Scheme 3). These were prepared as yellowish solids with moderate to high yield in reactions between α-pyrones and Na(OCP).16 The 31P{1H} NMR spectra show a singlet at δ = 140.3, 133.2, 151.6, and 149.3 ppm, respectively, for the 31P nucleus of 1a–1d. In the 13C NMR spectra, a diagnostic doublet in the range of δ = 209.3–212.5 ppm with coupling constants 1JPC = 41.9–47.5 Hz are observed for the 13C nucleus C1 of 1a–1d. To reach completeness, the reaction mixtures need heating for several hours at 85 °C. Only for 1d, which is prepared from an α-pyrone with a carboxylate group, the reaction is complete within 10 min at room temperature. Electron-withdrawing groups activate α-pyrones in [4 + 2] cycloadditions.17
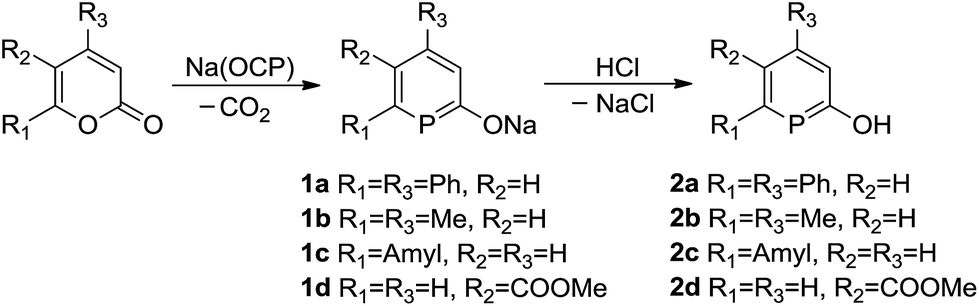 | ||
| Scheme 3 Synthesis of compounds phosphinin-2-olates 1a–1d and neutral phosphinin-2-ols 2a–2d. | ||
Subsequently the sodium phosphinin-2-olates 1a–1d were treated with anhydrous hydrochloric acid to give the phosphinin-2-ols 2a–2d in almost quantitative yield (Scheme 3). Singlets in the 31P{1H} NMR spectra at 131.4, 129.0, 146.2, and 147.9 ppm are observed for 2a–2d, respectively, which are slightly shifted to lower frequencies when compared to the 31P{1H} NMR spectra of the corresponding anions. In the 1H NMR spectra, a sharp singlet at 5.97 (2a), 5.68 (2b), 5.71 (2c), and 6.89 ppm (2d) in CDCl3 is assigned to the proton of the hydroxyl group and confirms the successful preparation of phosphinin-2-ols. In the 13C NMR spectra, a doublet in the range of 161.0–193.1 ppm with coupling constants 1JPC between 37.4 and 52.5 Hz is observed for the 13C nucleus C1 of the phosphinin-2-ols.
The solid-state structure of anionic phosphinin-2-olate 1a was determined as the 18-crown-6 adduct (Fig. 1a). But all attempts to obtain suitable single crystals for the neutral phosphinin-2-ols failed. With the aim to obtain a solid-state structure of a neutral phosphinine with a similar skeleton as seen in 1a, compound 3 was prepared by treating 1a with triphenyltin chloride as electrophile (Scheme 4). Single crystals of 3 suitable for X-ray diffraction studies were obtained and allowed to determine the structure of 3 and a comparison of the bonding parameters of an anionic and neutral phosphinine derivative (Fig. 1a and b). Notably, the P–C bond lengths, P1–C1 = 1.79 Å, P1–C5 = 1.76 Å, in 1a are different and slightly longer compared to neutral 3. There, both P–C bonds are almost equal in lengths, P1–C1 = 1.76 Å and P1–C5 = 1.75 Å. In both compounds, the P–C bond lengths are in between single and double P–C distances (Σrcov(P–C) = 1.86 Å, Σrcov(P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) = 1.69 Å)18 and are in the range of P–C bond distances of neutral phosphinines (1.73–1.77 Å).19 The C–O bond in 1a (1.28 Å) is significantly shorter than in 3 (1.33 Å) (Σrcov(C–O) = 1.38 Å; Σrcov(CO) = 1.24 Å).18 Furthermore, the C–C bonds in 1a show alternating bond lengths and vary between 1.39 Å and 1.43 Å while they are shorter and vary less in neutral 3. These observations indicate a stronger perturbation of the delocalized 6π-electron configuration of the PC5 ring in 1a compared to 3 and point to a stronger contribution of the resonance structures with two lone pairs at phosphorus center as discussed above.
C) = 1.69 Å)18 and are in the range of P–C bond distances of neutral phosphinines (1.73–1.77 Å).19 The C–O bond in 1a (1.28 Å) is significantly shorter than in 3 (1.33 Å) (Σrcov(C–O) = 1.38 Å; Σrcov(CO) = 1.24 Å).18 Furthermore, the C–C bonds in 1a show alternating bond lengths and vary between 1.39 Å and 1.43 Å while they are shorter and vary less in neutral 3. These observations indicate a stronger perturbation of the delocalized 6π-electron configuration of the PC5 ring in 1a compared to 3 and point to a stronger contribution of the resonance structures with two lone pairs at phosphorus center as discussed above.
 | ||
| Fig. 1 Molecular structures of 1a and 3 in the solid state (H atoms and solvent were omitted for clarity; 50% probability thermal ellipsoids). Selected distances [Å] and angles [°]: (a) 1a: P1–C1 1.789(2), C1–C2 1.428(2), C2–C3 1.401(2), C3–C4 1.416(2), C4–C5 1.387(2), C5–P1 1.759(2), C1–O1 1.276(2), O1–Na1 2.260 (1), C1–P1–C5 102.2(7). (b) 3: Sn1–O1 2.028 (2), P1–C1 1.760(2), C1–C2 1.399(3), C2–C3 1.396(3), C3–C4 1.404(3), C4–C5 1.389(3), C5–P1 1.752(2), C1–O1 1.330(3), Sn1–O1–C1 134.1(1), P1–C1–O1 120.5(2), C1–P1–C5 100.8(1). | ||
 | ||
| Scheme 4 Synthesis of compound 3. | ||
All four phosphinin-2-ols 2a–2d were reacted with Cu(I)Cl (Scheme 5) to give complexes 4a–4d as yellowish powders in high yield (87–97%). Upon coordination to Cu(I), the 31P{1H} NMR resonances (4a: δ = 104.1, 4b: δ = 102.0, 4c: δ = 114.5, 4d: δ = 122.0 ppm) are shifted to lower frequencies compared to the free ligands 2a–2d.
 | ||
| Scheme 5 Synthesis of copper(I) complexes 4a–4d. | ||
The rather similar coordination shift, Δcoord = [δ(complex) − δ(2a–2d)], of about −30 ppm indicates similar electronic configurations at the phosphorus center in solution for all complexes including 4b (vide infra). The 1H NMR spectra show the proton of the OH groups as singlets at high frequencies in the range of 9.48–10.61 ppm which indicates the formation of hydrogen-bonds (see below).
For all complexes 4a–4d single crystals were grown and subjected to X-ray diffraction studies. Plots of the structures are shown in Fig. 2. The structure of 4a is best described as a coordination polymer which consists of four-membered Cu2(μ-Cl)2 rings as building blocks. In addition, each of the bridging μ-Cl centers binds to a third Cu(I) center of a neighbouring Cu2(μ-Cl)2 ring such that 1D polymer with a ladder structure is obtained in which the Cl centers take an overall μ3-binding mode. The Cu–Cl lengths vary between 2.33 Å and 2.50 Å. The transannular Cu–Cu distance is long (3.27 Å) and excludes any metal–metal interaction. The distorted tetrahedral coordination sphere at each Cu(I) center is completed by a terminal non-bridging phosphinine ligand at Cu–P1 = 2.18 Å. With the sterically less hindered phosphinin-2-ol 2b, a dinuclear Cu(I) complex is obtained in which the ratio of Cu to phosphinine is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. One of the phosphinine ligands takes an asymmetric bridging mode as shown for IV in Scheme 1. Significantly different Cu–P distances are observed. While the distances Cu1–P1 = 2.25 Å and Cu1–P2 = 2.24 Å are slightly shorter than in the other two known Cu(I) phosphinine complexes,2c,11 the Cu1′–P2 distance is significantly longer at 2.56 Å. Furthermore, the angles α(ct-P2–Cu1) = 169.2° and β(ct-P2–Cu1′) = 116.2° likewise differ by 53° (ct = center of the PC5 ring). A simple way to describe the structure of 4b is to assume that two CuCl (2b) units with a T-shape structure and the two phosphinines in trans-position to each other (P1–Cu–P2 = 133.4°) form a head-to-tail dimer.20 The Cu–Cu distance (2.70 Å) in 4b is much shorter than in 4a indicating a Cu⋯Cu interaction. The peculiar structure of 4b in combination with the 31P{1H} NMR spectrum, which shows only one signal with a chemical shift similar to the other complexes 4a, 4c, and 4d, strongly suggests that 4b either dissociates into monomers containing only mono-dentate phosphinine ligands in solution, or shows a dynamic phenomenon by which the phosphines rapidly change between a bridging and non-bridging position, or rearranges in solution into a structure like 4c.11 The structures of 4c and 4d contain again a Cu2(μ-Cl)2 core and either two phosphinines, as in 4c, or one, as in 4d, is bound in a non-bridging terminal fashion to the Cu center with Cu–P distances in the range of 2.18–2.23 Å. In both complexes the coordination sphere around Cu corresponds to a slightly distorted tetrahedron. Note, that the hydroxyl group of both, the bridging and non-bridging phosphinine, in all compounds isolated so far is involved in hydrogen-bonding to a Cl center (H⋯Cl ≈ 2.36 Å; cf. sum of van der Waals radii of 2.95 Å) in accord with the high frequency shift observed in the 1H NMR spectra which indicates that these O–H⋯Cl bridges persist in solution. This feature has also been observed by Mathey et al. in Cu(I) and Au(I) complexes with the unsubstituted parent phosphinin-2-ol.11 Importantly, the C–C bonds within the PC5 ring vary only very little from an average value of 1.39 Å indicating that in all solid state structures of 4a–4d the phosphinine ring maintains a delocalized 6π-electron system.
:2. One of the phosphinine ligands takes an asymmetric bridging mode as shown for IV in Scheme 1. Significantly different Cu–P distances are observed. While the distances Cu1–P1 = 2.25 Å and Cu1–P2 = 2.24 Å are slightly shorter than in the other two known Cu(I) phosphinine complexes,2c,11 the Cu1′–P2 distance is significantly longer at 2.56 Å. Furthermore, the angles α(ct-P2–Cu1) = 169.2° and β(ct-P2–Cu1′) = 116.2° likewise differ by 53° (ct = center of the PC5 ring). A simple way to describe the structure of 4b is to assume that two CuCl (2b) units with a T-shape structure and the two phosphinines in trans-position to each other (P1–Cu–P2 = 133.4°) form a head-to-tail dimer.20 The Cu–Cu distance (2.70 Å) in 4b is much shorter than in 4a indicating a Cu⋯Cu interaction. The peculiar structure of 4b in combination with the 31P{1H} NMR spectrum, which shows only one signal with a chemical shift similar to the other complexes 4a, 4c, and 4d, strongly suggests that 4b either dissociates into monomers containing only mono-dentate phosphinine ligands in solution, or shows a dynamic phenomenon by which the phosphines rapidly change between a bridging and non-bridging position, or rearranges in solution into a structure like 4c.11 The structures of 4c and 4d contain again a Cu2(μ-Cl)2 core and either two phosphinines, as in 4c, or one, as in 4d, is bound in a non-bridging terminal fashion to the Cu center with Cu–P distances in the range of 2.18–2.23 Å. In both complexes the coordination sphere around Cu corresponds to a slightly distorted tetrahedron. Note, that the hydroxyl group of both, the bridging and non-bridging phosphinine, in all compounds isolated so far is involved in hydrogen-bonding to a Cl center (H⋯Cl ≈ 2.36 Å; cf. sum of van der Waals radii of 2.95 Å) in accord with the high frequency shift observed in the 1H NMR spectra which indicates that these O–H⋯Cl bridges persist in solution. This feature has also been observed by Mathey et al. in Cu(I) and Au(I) complexes with the unsubstituted parent phosphinin-2-ol.11 Importantly, the C–C bonds within the PC5 ring vary only very little from an average value of 1.39 Å indicating that in all solid state structures of 4a–4d the phosphinine ring maintains a delocalized 6π-electron system.
 | ||
| Fig. 2 Molecular structures of 4a–4d in the solid state (H atoms except the ones from hydroxyl groups and solvents were omitted for clarity; 50% probability thermal ellipsoids). Selected distances [Å]: (a) 4a: Cu1–Cu1′ 3.268, Cu1–Cl1 2.3301(16), Cu1–Cl1′ 2.4550(16), Cu1–Cl1′′ 2.5019(19), Cu1–P1 2.1815, P1–C1 1.728(7), C1–C2 1.388(8), C2–C3 1.396(8), C3–C4 1.392(9), C4–C5 1.402(8), P1–C5 1.730(6), C1–O1 1.373(8), O1–Cl1′ 3.271; (b) 4b: Cu1–Cu1′ 2.6976(10), Cu1–P1 2.2534(10), Cu1–Cl1 2.3378(9), Cu1–P2 2.2424(9), Cu1–P2′ 2.5648(10), P1–C1 1.745(4), C1–C2 1.391(5), C2–C3 1.388(6), C3–C4 1.398(6), C4–C5 1.378(5), P1–C5 1.736(4), C1–O1 1.357(5), O1–Cl1 3.066; (c) 4c: Cu1–Cu1′ 3.0378(8), Cu1–Cl1 2.3946(8), Cu1–Cl1′ 2.4269(8), Cu1–P1 2.2276(7), Cu1–P2 2.2254(7), P1–C1 1.744(3), C1–C2 1.392(4), C2–C3 1.379(5), C3–C4 1.402(5), C4–C5 1.389(4), P1–C5 1.724(3), C1–O1 1.344(4), O1–Cl1 3.272; (d) 4d: Cu1–Cu1′ 3.261, Cu1–Cl1 2.4067(11), Cu1–Cl1′ 2.4347(11), Cu1–N1 1.974(4), Cu1–P1 2.1799(10), P1–C1 1.747(4), C1–C2 1.396(5), C2–C3 1.384(6), C3–C4 1.403(5), C4–C5 1.390(5), P1–C5 1.720(4), C1–O1 1.354(5), O1–Cl1′ 3.097. | ||
We investigated also the anionic phosphinin-2-olates 1a–1d in reactions with CuCl. However, only products which are NMR silent were obtained which we could not further characterize. But when 1f was reacted with one equivalent of CuCl in the presence of 1 equiv. of triphenylphosphane, an orange compound of composition [Cu4(1f)4(PPh3)4] (5) was obtained quantitatively (Scheme 6). In the 31P{1H} and 1H NMR spectra, only broad resonances are observed likely due to exchange phenomena. Suitable single crystals were grown by layering a dichloromethane solution with hexane and these were subjected to a X-ray diffraction study. A plot of the structure of 5 is shown in Fig. 3. The compound consists of a Cu4 tetrahedron (Cu⋯Cu = 3.63 Å, averaged) in which each Cu(I) adopts a distorted tetrahedral coordination sphere. One coordination site is occupied by the P atom of triphenylphosphane (Cu3–P7: 2.29 Å), the second of the O atom of the phosphinin-2-olate unit and the remaining two sites are taken by the P atoms of two phosphinine moieties each of which takes a bridging position between two Cu centers (Cu1–P1 = 2.32 Å; Cu2–P1 = 2.28 Å). The four sets of α or β angles (see Scheme 1) within this complex are within a narrow range between 126.2° to 129.5°. The phosphorus atom of the planar PC5 ring is likewise in a distorted tetrahedral environment. The structure of the phosphinine ring does change slightly upon coordination to both copper centers when compared to the free ligand 1a. The difference between both P–C bonds (P1–C1 = 1.784 Å; P1–C5 = 1.746 Å) is slightly larger as is the variance of the C–C bonds which show a more pronounced long-short-long-short alternation (C1–C2 1.424 Å, C2–C3 1.365 Å, 1.390 Å, 1.349 Å). The only related structure of a tetranuclear aggregate we could find is the one of a cesium salt of 6-methy-2-pyridonate in which N centers bridge each two Cs+ cations.21
 | ||
| Scheme 6 Synthesis of copper(I) complex 5. | ||
 | ||
| Fig. 3 Molecular structure of 5 in the solid state (H atoms and solvents were omitted for clarity; 50% probability thermal ellipsoids). Selected distances [Å] and angles [°]: Cu1–P1 2.322(2), Cu2–P1 2.285(2), Cu3–P7 2.295(2), P1–C1 1.784(6), C1–C2 1.424(8), C2–C3 1.365(9), C3–C4 1.390(9), C4–C5 1.349(9), C5–P1 1.746(6), C1–O1 1.296(7), O1–Cu3 2.003(4),; Cu1–P1–Cu2 101.32(7), C1–P1–C5 102.9(3). | ||
The coordination chemistry of the neutral 2-hyrdoxy-phosphinine 2a and anionic phosphinin-2-olate 1a towards [AuCl(SMe2)] was investigated. The results with 2a as ligand are summarized in Scheme 7. After 2 h the reaction was complete and gave complex 6 as yellow powder in 61% isolated yield. In the 1H NMR spectrum, a singlet at δ = 10.8 ppm is observed which is assigned to the hydroxyl group. In the 31P{1H} NMR spectrum, a shielded chemical shift of δ = 117.7 ppm is observed for 6 as compared to that of δ = 131.4 ppm of free ligand 2a. The molecular structure of 6, determined by single crystal X-ray diffraction analysis (Fig. 4a), shows a classical linear Au(I) complex (P–Au–Cl = 178°) in which the phosphinine binds as a mono-dentate two-electron donor ligand (Au1–P1 = 2.21 Å). The phosphorus center resides in a trigonal planar coordination environment [Σ(P1) = 360°], the P–C bonds (P1–C1 1.72 Å, C5–P1 1.74 Å) are slightly shorter and of similar length compared to the Ph3Sn-substituted neutral derivative 3. The C1–O1 bond is 1.35 Å long and the hydroxyl group forms a hydrogen bridge to the nitrogen center of an acetonitrile molecule with O1–N1 = 2.73 Å (Fig. 4a), which agrees with the high frequency shift of the hydroxyl proton in the 1H NMR spectrum. Note, that no further coordination occurs when 6 is treated with excess of [AuCl(SMe2)].
 | ||
| Scheme 7 Synthesis of complexes 6–8. | ||
 | ||
| Fig. 4 Molecular structures of 6 and 7 in the solid state (H atoms except the ones from hydroxyl groups and solvents except the one engaging in hydrogen-bond were omitted for clarity; 50% probability thermal ellipsoids). Selected distances [Å] and angles [°]: (a) 6: Au1–P1 2.210(2), Au1–Cl1 2.273(2), P1–C1 1.720(8), C1–C2 1.396(11), C2–C3 1.407(10), C3–C4 1.395(11), C4–C5 1.378(11), C5–P1 1.743(8), C1–O1 1.353(10), O1–N1 2.732, P1–Au1–Cl1 178.20(8); (b) 7: Au1–P1 2.2946(17), Au1–P2 2.2958(16), P1–C1 1.783(7), C1–C2 1.441(9), C2–C3 1.379(9), C3–C4 1.427(9), C4–C5 1.369(9), C5–P1 1.756(6), C1–O1 1.254(8), P1–Au1–P2 177.09(5), C1–P1–C5 107.2(3). | ||
The anionic heterocycle 1a was treated with an equimolar mixture of [AuCl(SMe2)] and triphenylphosphane in THF. This gives the red Au(I) complex [Au(η1-1a)PPh3] (7) in 67% yield (Scheme 7). In the 31P{1H} NMR spectrum, two singlets are observed located at δ = 89.6 ppm for the phosphinine ring and δ = 38.1 ppm for the triphenylphosphine. The rather strong coordination shift Δcoord = [δ(7) − δ(1a)] = −50.7 ppm indicates a significant change of the electronic configuration of the PC5 cycle upon coordination.
The result of an X-ray diffraction study with single crystals of 7 clearly shows these changes (Fig. 4b). Again, the gold(I) center resides in a two-fold linear coordination sphere (P1–Au–P2 = 177.1°; Au–P1 = 2.29 Å; Au–P2 = 2.30 Å). But the metrics of the coordinated phosphinine ring show significant changes when compared to the free ligand 1a and also to the Au(I) complex 6. The C1–O1 bond (1.25 Å) in 7 is shorter than in the free ligand and corresponds to a CO double bond (Σrcov(C–O) = 1.38 Å, Σrcov(CO) = 1.24 Å).18 The C–C bonds show a pronounced long-short-long-short alternation (C1–C2 1.441 Å, C2–C3 1.379 Å, 1.427 Å, 1.369 Å) exceeding slightly the one seen in the tetranuclear copper compound 5. Most remarkable is the significant deviation of the [Au(PPh3)]+ unit from co-planarity with the PC5 ring. This is reflected by the angle α(Au–P1-ct) = 156° and the sum of bond angles around P1, [Σ(P1) = 351.2°]. Nucleus independent chemical shift (NICS) values22 were calculated at 1 Å above the PC5 ring plane using simplified models 6H and 7H where the phenyl substituents in 6 and 7 were replaced by hydrogens. A comparison with parent phosphinin-2-ol, 2-HO-PC5H4 [NICS(1) = −8.5 ppm], shows that the aromaticity of the phosphinine ring is maintained in 6H [NICS(1) = −8.7 ppm]. However, the smaller NICS(1) values of −3.0 (opposite to the AuCl fragment) and −2.4 ppm (same side as the AuCl fragment) in 7H compared to the free anion phosphinin-2-olate (NICS(1) = −4.4 ppm) indicate loss of aromaticity {the difference between 7H and the one of the calculated structure of [Na(18-crown-6)(H2O)]+(O-PC5H4)−, NICS(1) = −6.0 ppm, is even larger}. The presence of a stereo-chemically active lone pair at P1 can be demonstrated by reacting 7 with a second equivalent of [AuCl(SMe2)] which cleanly leads to a new complex 8 in which the 31P nucleus shows a strongly low-frequency shifted (Δcoord = −145.5 ppm) resonance at δ31P{1H} = −5.2 ppm. Unfortunately, all attempts to obtain single crystals of 8 failed, but there is little doubt that the phosphinine takes a bridging position between two gold centers (see below). We calculated the structure of 8H which is shown in Fig. 5 together with 6H and 7H. The agreement between the experimental and calculated bond parameters of 6/6H and 7/7H is satisfactory. Specifically, the Au–P bond length and the planar coordination sphere around P is reproduced in 6H as well as the equilibrated C–C bond lengths in the PC5 ring. For 7H, a trigonal pyramidalized coordination sphere is calculated [Σ°(P) = 331.8°] and the C–C bond lengths alternate as observed experimentally. Also in 8H, the C–C bonds show a pronounced long-short-long-short alternation. Note, that the calculated P–C bonds in 8H (1.80 Å, 1.89 Å) are as long as P–C single bonds [rcov(P–C) = 1.86 Å].18 The NICS(1) values for 8H were calculated and are found to be even slightly positive, 0.7 [adjacent to Au(I)Cl] and 0.9 ppm {adjacent to [Au(I)(PPh3)]+}. In combination, these data indicate that the electronic structure of 2-oxy-phosphinines is heavily distorted when bound in anionic form in a bridging position between two metal centers.
 | ||
| Fig. 5 Optimized geometries for the simplified models 6H, 7H, and 8H. Bond lengths (at BP86/def2-TZVPP) are given in Å. | ||
Note in this context, that a dinuclear gold(I) complex [Au2(μ-N, O-pyridinone)(PPh3)2]BF4 of the nitrogen analogue of 1a, namely 2-pyridinonate, could be characterized by Schmidbaur and co-workers.23 In that complex, one Au(I) center binds to the N center and is almost coplanar with the C5N ring while the second Au center binds to the O atom.
Finally, [AuCl(SMe2)] was reacted with phosphinin-2-olate 1a in the absence of a co-ligand like PPh3. In this case and under the assumption that 1a strongly prefers a μ2-bridging position, oligomers can be expected which have structures similar to those of very well studied gold thiolates.24 The reaction between 1a and [AuCl(SMe2)] in THF as solvent is complete within 2 h at room temperature. After precipitation with hexane, the product 9 [Au5(μ2-1a)5 × (AuCl)(SMe2)] was isolated as orange powder in 70% yield (Scheme 8). The 31P{1H} NMR spectrum shows a singlet at −3.2 ppm for the phosphorus nucleus. As for 9, a very large coordination shift Δcoord = −143.5 ppm is observed. Note that in a related structurally characterized dinuclear Au(I) phosphinine complex, in which the phosphinine bridges the two Au centers, a comparable large Δcoord of −81.2 ppm has been observed.25
 | ||
| Scheme 8 Synthesis of gold cluster 9. | ||
A single crystal X-ray diffraction analysis was used to unambiguously identify the structure of complex 9 (Fig. 6). A flat pentagon is obtained which consists of five Au(I) centers symmetrically bridged by five μ2-1a ligands arranged around a [AuCl(SMe2)] moiety in the center. The Au–P distances vary in a very narrow range from 2.316(3) Å to 2.324(4) Å. The Au(I) centers are linearly surrounded by two phosphorus centers (P–Au–P 177°), the Au–P–Au angles are on the average 111° (108° for ideal pentagon). The five sets of α and β angles vary between 108.1° and 142.1°. The averaged P–C bond lengths are 1.82 Å in agreement with the predicted calculated value observed in 8H (Fig. 5) indicating P–C single bonds.18 Furthermore, the C–C bonds show the most pronounced long–short–long–short alternation of all structurally characterized complexes (C1–C2 1.483 Å, C2–C3 1.336 Å, C3–C4 1.459 Å, C4–C5 1.353 Å) in full agreement with the calculated data for 8H. The average C1–O1 bond distance of 1.23 Å indicates a CO double bond. This clearly shows that the resonance structures with a formal negative charge and two lone pairs at the phosphorus center prevails in the electronic ground state as shown in Scheme 2C. That is, the electronic structure of 1a as bridging ligand is best described as the anion of 2-phospininone. A further remarkable feature of complex 9 is the inclusion of an AuCl(SMe2) unit in the center of the Au5 pentagon. This central Au(I) center is fixed by five “aurophilic” interactions with “magic” Au–Au distances of about 3.2 Å.26 While there are many reports on gold clusters with capping monodentate phosphines as ligands,27 polynuclear gold complexes with bridging phosphorus centers are still very rare and have only been reported very recently.28
 | ||
| Fig. 6 Molecular structure of 9 in the solid state (H atoms and solvents were omitted for clarity; 50% probability thermal ellipsoids). Selected distances [Å] and angles [°]: Au1–Au2 3.1424(7), Au1–Au3 3.2960(7), Au1–Au4 3.1301(7), Au1–Au5 3.2908(7), Au1–Au6 3.2381(7), Au1–Cl1 2.298(3), Au1–S1 2.290(4), Au2–P1 2.316(3), Au2–P2 2.305(3), Au3–P2 2.324(4), Au3–P3 2.314(3), Au4–P3 2.321(3), Au4–P4 2.322(3), Au5–P4 2.319(3), Au5–P5 2.319(3), Au6–P5 2.314(3), Au6–P1 2.313(3), P1–C1 1.827(14), C1–C2 1.483(18), C2–C3 1.336(18), C3–C4 1.459(17), C4–C5 1.353(17), P1–C5 1.810(13), C1–O1 1.204(16), Au6–P1–Au2 112.89(12), P1–Au2–P2 177.21(12), C1–P1–C5 105.7(6). | ||
Discussion and conclusions
In summary, a straight forward synthesis of a series of phosphinin-2-olates and the corresponding phosphinin-2-ol derivatives has been developed by using readily accessible Na(OCP) and commercially available α-pyrones. Yields are high to moderate. DFT calculations suggest that especially the anionic phosphinin-2-olates serve as flat bridging ligands while neutral 2-alkoxy and hydroxy-substituted phosphinines should prefer the classical terminal binding mode. This prediction was tested through reactions with Cu(I) and Au(I) halides as precursor complexes. In reactions with neutral 2-hydroxy-phosphinines, mostly complexes with a terminal bound phosphinine are observed.These are characterized by a modest coordination shift, Δcoord, of about −30 ppm when δ31P{1H} of the complex and free ligand are compared. Furthermore, the P–C and C–C bonds in the PC5 ring show little variation in free and coordinated form. We therefore assume that in these complexes the phosphinine serves as classical two-electron donor. In complex 4b and related Cu(I)2c,11 and Ag(I)10 complexes reported in the literature (A–C, Scheme 9), a neutral phosphinine takes a bridging position. Again Δcoord is modestly negative. However, in none of these Cu(I) or Ag(I) complexes, the NMR data support the observed solid state structures in solution. Note that this two-electron μ2-binding mode is established for classical phosphanes (as well as arsanes and stibanes)29 and therefore in principle is possible for phosphinines as well. There are two examples in the literature where this is likely to be the case. One is a Pd3 cluster in which each Pd(0) center is coordinated by a terminal PEt3 ligand and two μ2-bridging 2,4,6-triphenyl phosphinine ligands (D, Scheme 9). In this complex, which retains its structure in solution, little variation of the C–C bond lengths is observed and the 31P resonance is shifted to even higher frequencies (Δcoord = +10 ppm). Another is a dinuclear Ru complex with 2,2′-biphosphinine as ligand in which one PC5 ring takes a bridging position and the other not (E, Scheme 9). No NMR data are available but the structure shows that for both PC5 rings, the P–C bond lengths are beyond 1.75 Å and all C–C bonds are almost equally long (av. 1.39 Å). In reactions with the anionic, and hence more polarizable, phosphinin-2-olates 1a–1d exclusively structures with μ2-bridging ligands are obtained. These are characterized by strong coordination shifts to much lower frequencies (negative Δcoord). Furthermore, the structure of the μ2-bridging PC5 rings is severely perturbed: the P–C distances approach P–C single bond lengths and the four C–C bonds show a pronounced long–short–long–short alternation. This is not only observed in the Cu(I) complex 5 and the Au(I) complexes 7–9 reported here but also in the AuCl complex G (Scheme 9).23 Furthermore, Le Floch et al. synthesized and fully characterized triosium complexes in which the 2,2′-biphosphinine ligand is assumed to be reduced by two electrons (F, Scheme 9).9c,9d As a consequence, very strong coordination shifts were observed and a strong localisation of the conjugated 6π-electron system into a long–short–long–short C–C bonding sequence. It is proposed, that complexes which show these structural features combined with strongly negative Δcoord values contain μ2-bridging phosphinines which act as four-electron donors. Venanzi et al. noted first the possibility that 2-(2′pyridyl)-4,5-dimethylphosphinine may act as four-electron donor but discarded this idea on the basis of the structural data which they deemed to be of insufficient evidence (H, Scheme 9).8 However, in view of the Δcoord = −77 ppm and the C–C bond length alternation they observed, this complex clearly belongs into the category of complexes with bridging four-electron donor phosphinine ligands. Although the experimental results presented here must be further bolstered by theoretical methods, it becomes evident that the binding mode of the P atom of phosphinines can be divided into three classes: one in which they act as classical terminal two-electron donors (2e); a second one in which they act as μ2-bridging two-electron donors (μ2-2e), and a third one in which they act as four-electron donors (μ2-4e). The latter binding mode is especially interesting for the synthesis of stable new metallooligo- and polymers. The syntheses of a tetra-nuclear Cu cluster and a “golden crown” composed of five Au(I) centers, five phosphinine units, and a sixth Au(I) center in the center serve as proof-of-principle. In combination with previously reported results, the data reported here indicate that the μ2-4e-binding mode can be reached by employing either an easily polarizable anionic phosphinine or by binding an easily reducible (highly π-conjugated) phosphinine to an electron rich metal core.
 | ||
| Scheme 9 Reported examples of metal complexes with μ2-P bridging phosphinines. The coordination shifts, Δcoord = [δ(complex) − δ(ligand)], are given for the relevant 31P nuclei. | ||
Experimental
General considerations
All manipulations were performed under an inert atmosphere of dry nitrogen, using standard Schlenk techniques and a glovebox. Dry, oxygen-free solvents were employed unless otherwise mentioned. The sodium phosphaethynolate,16b sodium 1,3-diphenyl phosphaphenolate (1a)30 and sodium phosphinin-2-olate (1f)13 were prepared by following literature procedures while all other starting materials were purchased from commercial sources. NMR spectra were recorded on Bruker Avance 400 MHz spectrometers (1H: 400.1 MHz; 13C: 100.5 MHz; 31P: 161.9 MHz). All spectra were obtained in the solvent indicated at 25 °C. The chemical shifts (δ) were measured according to IUPAC and expressed in ppm relative to SiMe4 (1H, 13C), and 85% H3PO4 (31P). Coupling constants J are reported in hertz [Hz] as absolute values. The elemental analyses were acquired on Vario EL cube elemental analyser. UV/vis spectra were measured on Shimadzu UV/vis/NIR UV-3600-spectrometer. IR spectra were obtained on a Perkin-Elmer Spectrum ATR 2000 FT-IR-Raman spectrometer with KBr beam splitter (range 500–4000 cm−1). The ATR technique was used for the analysis of solid compounds. Melting points (M. P.) were measured on Jiahang JH-30 apparatus.Synthesis of compound 1b
4,6-Dimethyl-2-pyrone (1 g, 8.0 mmol), sodium phosphaethynolate ([Na(OCP)·(dioxane)2.5]), (2.4 g, 8.0 mmol) and THF (50 mL) were successively added into a Teflon sealed flask. After stirring for 10 h at 85 °C, the precipitate was filtered off and the filtrate was dried under reduced pressure. Then, the remaining solid was extracted with ethyl ether and washed with hexane. Drying the residue in vacuo afforded 1b as yellowish powder (0.79 g, 4.9 mmol, 61% yield). 1H NMR (CD3CN, 400 MHz): δ = 6.67 (d, 2JPH = 8.0 Hz, 1H, CarH, PH), 6.43 (d, 2JPH = 4.0 Hz, 1H, CarH, PH), 2.44 (d, 3H, CH3), 2.21 (d, 3H, CH3); 13C{1H} NMR (CD3CN, 100.5 MHz): δ = 209.5 (d, 1JPC = 42.2 Hz, OCP), 167.2 (d, 2JPC = 50.2 Hz, PCC), 141.7 (d, 3JPC = 7.0 Hz, PCCC), 122.6 (t, 1JPC = 4.0 Hz, 1JPC = 4.0 Hz, CPC), 122.5 (s, Car), 23.9 (d, JPC = 36.1 Hz, CH3), 22.9 (d, JPC = 2.1 Hz, CH3). 31P{1H} NMR (CD3CN, 161.9 MHz): δ = 133.2 (s). M. P. = 248 °C. IR (ATR, [cm−1]): 3592 (w, CH stretch), 2909 (w, CH stretch), 1659 (w), 1562 (m), 1372 (m), 1335 (w), 1262 (s, C–O), 1141 (m), 1124 (w), 838 (w), 562 (m), 456 (w). Elemental analysis (%): calcd for C7H8OPNa; C 51.87, H 4.97; found: C 51.73, H 5.02. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 323.8 (1089).Synthesis of compound 1c
6-Amyl-2-pyrone (0.5 g, 3.0 mmol), sodium phosphaethynolate ([Na(OCP)·(dioxane)2.5]) (0.9 g, 3.0 mmol) and THF (50 mL) were successively added into a Teflon sealed flask. After stirring for 4 h at 85 °C, the precipitate was filtered off and the filtrate was dried under reduced pressure. Then, the remaining solid was extracted with ethyl ether and washed with hexane. Drying the residue in vacuo afforded 1c as yellowish powder (0.33 g, 1.70 mmol, 57% yield). 1H NMR (CD3CN, 400 MHz): δ = 7.07 (m, 1H, CarH), 6.75 (t, 3JPH = 8.0 Hz, 3JPH = 4.0 Hz, 1H, CarH, PH), 6.52 (m, 1H, CarH), 2.69 (m, 2H, CH2), 1.65 (m, 2H, CH2), 1.37 (m, 4H, 2CH2), 0.93 (t, 3H, CH3); 13C{1H} NMR (CD3CN, 100.5 MHz): δ = 209.1 (d, 1JPC = 43.2 Hz, OCP), 173.9 (d, 1JPC = 54.3 Hz, CPC), 131.5 (d, 3JPC = 8.0 Hz, PCCC), 121.5 (d, 2JPC = 8.0 Hz, PCC), 119.5 (d, 2JPC = 13.0 Hz, PCC), 39.0 (d, JPC = 31.3 Hz, CH2), 33.6 (d, JPC = 7.1 Hz, CH2), 31.4 (s, CH2), 22.3 (s, CH2), 13.4 (s, CH3). 31P{1H} NMR (CD3CN, 161.9 MHz): δ = 151.6 (s). M. P. = 182 °C. IR (ATR, [cm−1]): 3524 (w, CH stretch), 2921 (w, CH stretch), 1657 (w), 1544 (w), 1392 (m), 1278 (s, C–O), 1183 (w), 1172 (w), 1129 (w), 810 (w), 775 (m), 626 (w). Elemental analysis (%): calcd for C10H14OPNa; C 58.82, H 6.91; found: C 58.61, H 7.02. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 329.3 (1383), 386.1 (3258).Synthesis of compound 1d
Methyl coumalate (0.5 g, 3.2 mmol), sodium phosphaethynolate ([Na(OCP)·(dioxane)2.5]), 1.0 g, 3.2 mmol and THF (50 mL) were successively added into a Teflon sealed flask. After stirring at room temperature for 10 min, the precipitate was filtered off and the filtrate was dried under reduced pressure. Then, the remaining solid was washed with ethyl ether. Drying the residue in vacuo afforded 1d as yellowish powder (0.33 g, 1.70 mmol, 53% yield). 1H NMR (THF-D8, 400 MHz): δ = 9.21 (d, 2JPH = 36.0 Hz, 1H, CarH, PH), 7.81 (d, 3JPH = 12.0 Hz, 1H, CarH, PH), 6.46 (d, 4JPH = 12.0 Hz, 1H, CarH, PH), 3.75 (s, 3H, CH3); 13C{1H} NMR (THF-D8, 100.5 MHz): δ = 212.2 (d, 1JPC = 47.2 Hz, OCP), 165.7 (d, 3JPC = 3.0 Hz, PCCC), 160.0 (d, 1JPC = 55.3 Hz, PCC), 133.5 (d, 2JPC = 4.2 Hz, PCC), 122.4 (d, 3JPC = 12.1 Hz, PCCC), 118.1 (d, 2JPC = 13.1 Hz, PCC), 50.1 (s, CH3); 31P{1H} NMR (THF-D8, 161.9 MHz): δ = 149.3 (s, JPH = 32.4 Hz). M. P. = 247 °C. IR (ATR, [cm−1]): 2982 (w, CH stretch), 2947 (w, CH stretch), 1671 (m), 1550 (w), 1440 (w), 1327 (s, C–O), 1260 (s, C–O), 1215 (w), 1093 (w), 840 (w), 759 (m), 589 (w), 452 (w). Elemental analysis (%): calcd for C7H6O3PNa; C 43.77, H 3.15; found: C 43.45, H 3.31. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 328.3 (15395), 372.0 (5119).
Synthesis of compound 2a
Anhydrous hydrochloric acid (0.6 mL, 1.20 mmol) was added in portions to a stirred solution of 1a (308 mg, 1.07 mmol) in Et2O (5 mL). After stirring for 1 h, the reaction mixture was filtered. The filtrate was dried under reduced pressure to afford 2a as yellow powder (260 mg, 0.98 mmol, 93% yield). 1H NMR (CDCl3, 400 MHz): δ = 7.90 (d, 3JPH = 8.0 Hz, 1H, CarH, PH), 7.68 (t, 4H, CarH), 7.51 (m, 7H, CarH), 5.97 (s, 1H, OH); 13C{1H} NMR (CDCl3, 100.5 MHz): δ = 188.9 (d, 1JPC = 49.2 Hz, OCP), 170.8 (d, 1JPC = 46.2 Hz, CPC), 146.4 (d, 3JPC = 12.1 Hz, PCCC), 143.1 (d, 3JPC = 26.1 Hz, PCCC), 141.6 (d, 2JPC = 3.0 Hz, PCC), 129.0 (Car), 128.9 (Car), 128.2 (Car), 128.1 (d, 1JPC = 2.0 Hz, Car), 127.8 (Car), 127.6 (t, 1JPC = 2.0 Hz, 2JPC = 2.0 Hz, Car), 127.5 (Car), 119.5 (d, 1JPC = 8.1 Hz, Car); 31P{1H} NMR (CDCl3, 161.9 MHz): δ = 131.4 (s). M. P. = 123 °C. IR (ATR, [cm−1]): 3393 (w, CH stretch), 3028 (w, CH stretch), 1577 (w), 1490 (w), 1478 (w), 1361 (w), 1294 (s, C–O), 1153 (m), 951 (w), 877 (w), 756 (m), 690 (m), 648 (w). Elemental analysis (%): calcd for C17H13OP; C 77.27, H 4.96; found: C 77.15, H 5.61. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 352.2 (6865).Synthesis of compound 2b
Anhydrous hydrochloric acid (0.7 mL, 1.4 mmol) was added in portions to a stirred solution of 1b (200 mg, 1.2 mmol) in Et2O (5 mL). After stirring for 1 h, the reaction mixture was filtered. The filtrate was dried under reduced pressure to afford 2b as colorless oil (150 mg, 1.1 mmol, 87% yield). 1H NMR (CDCl3, 400 MHz): δ = 7.23 (d, 2JPH = 4.0 Hz, 1H, CarH, PH), 7.02 (d, 2JPH = 4.0 Hz, 1H, CarH, PH), 5.68 (s, 1H, OH), 2.64 (d, 3H, CH3), 2.40 (d, 3H, CH3); 13C{1H} NMR (CDCl3, 100.5 MHz): δ = 188.7 (d, 1JPC = 47.2 Hz, OCP), 167.0 (d, 2JPC = 45.2 Hz, PCC), 143.4 (d, 3JPC = 12.1 Hz, PCCC), 130.5 (d, 1JPC = 53.3 Hz, CPC), 120.3 (d, 2JPC = 10.1 Hz, PCC), 24.7 (d, JPC = 39.2 Hz, CH3), 23.5 (d, JPC = 2.7 Hz, CH3); 31P{1H} NMR (CDCl3, 161.9 MHz): δ = 129.0 (s). M. P. = 123 °C. IR (ATR, [cm−1]): 3232 (w, CH stretch), 2911 (w, CH stretch), 2854 (w, CH stretch), 1739 (w), 1579 (m), 1435 (w), 1368 (m), 1304 (w), 1233 (s, C–O), 1198 (w), 853 (w), 607 (m). UV/vis (THF, λ (nm) ε (M−1 cm−1)): 328.5 (3494).Synthesis of compound 2c
Anhydrous hydrochloric acid (0.3 mL, 0.60 mmol) was added in portions to a stirred solution of 1c (113 mg, 0.55 mmol) in Et2O (5 mL). After stirring for 1 h, the reaction mixture was filtered. The filtrate was dried under reduced pressure to afford 2c as colorless oil (89 mg, 0.49 mmol, 89% yield). 1H NMR (CDCl3, 400 MHz): δ = 7.40 (m, 2H, CarH), 7.16 (m, 1H, CarH), 5.71 (s, 1H, OH), 2.89 (m, 2H, CH2), 1.70 (m, 2H, CH2), 1.38 (m, 4H, 2CH2), 0.92 (t, 3H, CH3); 13C{1H} NMR (CDCl3, 100.5 MHz): δ = 160.8 (d, 1JPC = 37.2 Hz, OCP), 149.6 (d, 1JPC = 36.2 Hz, CPC), 118.5 (d, 3JPC = 9.0 Hz, PCCC), 115.4 (d, 2JPC = 10.1 Hz, PCC), 108.7 (d, 2JPC = 6.0 Hz, PCC), 48.7 (d, JPC = 25.3 Hz, CH2), 44.5 (d, JPC = 7.1 Hz, CH2), 42.9 (s, CH2), 36.3 (s, CH2), 29.9 (s, CH3); 31P{1H} NMR (CDCl3, 161.9 MHz): δ = 146.2 (s). IR (ATR, [cm−1]): 3349 (w, CH stretch), 2955 (w, CH stretch), 2925 (w, CH stretch), 2854 (w, CH stretch), 1566 (w), 1465 (w), 1394 (m), 1160 (s, C–O), 1120 (m), 1067 (w), 772 (m), 693 (w), 610 (m). UV/vis (THF, λ (nm) ε (M−1 cm−1)): 329.6 (3370).Synthesis of compound 2d
Anhydrous hydrochloric acid (0.5 mL, 1.0 mmol) was added in portions to a stirred solution of 1d (170 mg, 0.89 mmol) in Et2O (5 mL). After stirring for 1 h, the reaction mixture was filtered. The filtrate was dried under reduced pressure to afford 2d as white powder (146 mg, 0.86 mmol, 97% yield). 1H NMR (CDCl3, 400 MHz): δ = 9.40 (d, 2JPH = 44.0 Hz, 1H, CarH, PH), 8.22 (d, 3JPH = 8.0 Hz, 1H, CarH, PH), 7.36 (dd, 4JPH = 12.0 Hz, 1H, CarH, PH), 6.89 (s, 1H, OH), 3.96 (s, 3H, CH3); 13C{1H} NMR (CDCl3, 100.5 MHz): δ = 192.8 (d, 1JPC = 52.3 Hz, OCP), 166.2 (d, 3JPC = 4.0 Hz, PCCC), 155.9 (d, 1JPC = 51.3 Hz, PCC), 134.6 (d, 2JPC = 12.1 Hz, PCC), 128.3 (d, 3JPC = 17.1 Hz, PCCC), 121.5 (d, 2JPC = 7.0 Hz, PCC), 52.6 (s, CH3); 31P{1H} NMR (CDCl3, 161.9 MHz): δ = 147.9 (s). M. P. = 88 °C. IR (ATR, [cm−1]): 3210 (w, CH stretch), 2947 (w, CH stretch), 1676 (m), 1422 (w), 1311 (s, C–O), 1204 (s, C–O), 1194 (m), 1133 (w), 1090 (m), 871 (w), 754 (m), 671 (w), 596 (w), 490 (w). Elemental analysis (%): calcd for C7H7O3P; C 49.43, H 4.15; found: C 49.26, H 4.25. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 328.5 (5843).Synthesis of compound 3
Triphenyltin chloride (192 mg, 0.5 mmol) was added in portions to a stirred solution of 1a (144 mg, 0.5 mmol) in THF (5 mL). After stirring for 2 h, the reaction mixture was filtered. The filtrate was dried under reduced pressure and washed with hexane. Drying the residue in vacuo afforded 3 as white powder (155 mg, 0.26 mmol, 51% yield). Colorless crystals of 3 were obtained from a dichloromethane solution layered with hexane on top and stored at −30 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 7.87 (m, 6H, CarH), 7.73 (m, 1H, CarH), 7.56 (m, 11H, CarH), 7.45 (m, 9H, CarH); 13C{1H} NMR (CD2Cl2, 100.5 MHz): δ = 197.6 (d, 1JPC = 43.2 Hz, OCP), 170.8 (d, 1JPC = 50.3 Hz, CPC), 145.6 (d, 3JPC = 10.1 Hz, PCCC), 143.7 (d, 2JPC = 24.1 Hz, PCC), 142.1 (d, 2JPC = 3.0 Hz, PCC), 137.5 (Car), 137.1 (Car), 136.9 (Car), 136.3 (Car), 136.1 (Car), 130.6 (Car), 129.5 (Car), 129.2 (Car), 128.9 (Car), 128.7 (Car), 127.7 (Car), 127.6 (Car), 127.5 (Car), 127.4 (Car), 125.5 (Car), 125.3 (Car), 123.4 (Car); 31P{1H} NMR (CD2Cl2, 161.9 MHz): δ = 146.4 (s). M. P. = 158 °C. IR (ATR, [cm−1]): 3064 (w, CH stretch), 1565 (w), 1473 (w), 1427 (w), 1364 (w), 1284 (w), 1203 (m), 1203 (s, C–O), 1186 (w), 1074 (m), 954 (m), 875 (w), 773 (m), 728 (w), 694 (s), 659 (w), 518 (w). Elemental analysis (%): calcd for C35H27SnOP; C 68.55, H 4.44; found: C 68.41, H 4.59. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 421.9 (6750).Synthesis of complex 4a
CuCl (38 mg, 0.38 mmol) was added in portions to a stirred solution of 2a (100 mg, 0.38 mmol) in THF (10 mL). After stirring for 10 h, the solid was removed and the filtrate was dried in vacuo. The remaining solid was washed with ethyl ether and dried in vacuo to afford 4a as yellow powder (118 mg, 0.33 mmol, 87% yield). Yellow crystals of 4a were obtained from an acetonitrile solution layered with ether on top and stored at −30 °C. 1H NMR (CD3CN, 400 MHz): δ = 10.33 (s, 1H, OH), 7.90 (d, 3JPH = 12.0 Hz, 1H, CarH, PH), 7.73 (m, 4H, CarH), 7.51 (m, 7H, CarH); 13C{1H} NMR (CD3CN, 100.5 MHz): δ = 186.3 (d, 1JPC = 14.1 Hz, OCP), 161.7 (d, 1JPC = 15.1 Hz, CPC), 145.3 (d, 3JPC = 15.1 Hz, PCCC), 141.4 (d, 2JPC = 10.1 Hz, PCC), 141.3 (d, 2JPC = 6.0 Hz, PCC), 141.2 (Car), 129.0 (Car), 128.8 (Car), 128.3 (Car), 128.2 (d, JPC = 13.1 Hz, Car), 127.6 (d, JPC = 2.0 Hz, Car), 127.4 (d, JPC = 13.1 Hz, Car), 120.9 (d, JPC = 5.1 Hz, Car); 31P{1H} NMR (CD3CN, 161.9 MHz): δ = 104.1 (s). M. P. = 247 °C. IR (ATR, [cm−1]): 3382 (w, CH stretch), 3028 (w, CH stretch), 1576 (w), 1482 (m), 1411 (w), 1364 (w), 1229 (m), 1192 (s, C–O), 1181 (w), 957 (w), 873 (w), 759 (m), 691 (m), 546 (w). Elemental analysis (%): calcd for [C17H13OPClCu]n; C 56.21, H 3.61; found: C 55.92, H 3.72. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 352.2 (4328).Synthesis of complex 4b
CuCl (106 mg, 1.07 mmol) was added in portions to a stirred solution of 2b (150 mg, 1.07 mmol) in THF (10 mL). After stirring for 10 h, the solid was removed and the filtrate was dried in vacuo. The remaining solid was washed with diethyl ether and dried in vacuo to afford 4b as yellow powder (116 mg, 0.31 mmol, 57% yield). Yellow crystals of 4b were obtained from an acetonitrile solution layered with ether on top and stored at −30 °C. 1H NMR (CD3CN, 400 MHz): δ = 9.48 (s, 1H, OH), 7.29 (d, 3JPH = 12.0 Hz, 1H, CarH, PH), 7.03 (d, 3JPH = 12.0 Hz, 1H, CarH, PH), 2.52 (s, 3H, CH3), 2.36 (s, 3H, CH3); 13C{1H} NMR (100.5 MHz, CD3CN): δ = 185.5 (d, 1JPC = 10.5 Hz, OCP), 158.8 (d, 2JPC = 11.1 Hz, PCC), 143.7 (d, 3JPC = 17.1 Hz, PCCC), 131.0 (d, 1JPC = 13.1 Hz, CPC), 121.9 (d, 2JPC = 7.0 Hz, PCC), 22.9 (d, JPC = 3.5 Hz, CH3), 22.3 (d, JPC = 31.6 Hz, CH3); 31P{1H} NMR (CD3CN, 161.9 MHz): δ = 102.0 (s). M. P. = 170 °C. IR (ATR, [cm−1]): 3239 (w, CH stretch), 3177 (m, CH stretch), 1729 (w), 1578 (m), 1415 (m), 1369 (w), 1311 (w), 1258 (s, C–O), 1206 (s, C–O), 1154 (w), 859 (w), 851 (w), 606 (m). Elemental analysis (%): calcd for [C14H18O2P2ClCu]2; C 44.34, H 4.78; found: C 44.42, H 4.62. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 328.8 (16325).
Synthesis of complex 4c
CuCl (73 mg, 0.74 mmol) was added in portions to a stirred solution of 2c (135 mg, 0.74 mmol) in THF (10 mL). After stirring for 10 h, the solid was removed and the filtrate was dried in vacuo. The remaining solid was washed with hexane and dried in vacuo to afford 4c as yellow powder (128 mg, 0.28 mmol, 75% yield). Yellow crystals of 4c were obtained from an acetonitrile solution layered with ether on top and stored at −30 °C. 1H NMR (CD3CN, 400 MHz): δ = 10.13 (s, 1H, OH), 7.39 (m, 2H, CarH), 7.14 (m, 1H, CarH), 2.85 (m, 2H, CH2), 1.69 (m, 2H, CH2), 1.36 (m, 4H, 2CH2), 0.91 (t, 3H, CH3); 13C{1H} NMR (CD3CN, 100.5 MHz): δ = 185.2 (d, 1JPC = 9.0 Hz, OCP), 163.5 (d, 1JPC = 11.1 Hz, CPC), 131.5 (d, 3JPC = 17.1 Hz, PCCC), 128.3 (d, 2JPC = 14.1 Hz, PCC), 120.9 (d, 2JPC = 5.0 Hz, PCC), 37.3 (d, JPC = 27.3 Hz, CH2), 32.9 (d, JPC = 8.1 Hz, CH2), 31.0 (s, CH2), 22.2 (s, CH2), 13.3 (s, CH3); 31P{1H} NMR (CD3CN, 161.9 MHz): δ = 114.5 (s). M. P. = 162 °C. IR (ATR, [cm−1]): 3376 (w, CH stretch), 2955 (w, CH stretch), 2927 (w, CH stretch), 2855 (w, CH stretch), 1962 (w), 1564 (w), 1414 (s, C–O), 1380 (w), 1252 (m), 1186 (m), 784 (m), 677 (w), 599 (m). Elemental analysis (%): calcd for [C20H30O2P2ClCu]2; C 51.84, H 6.53; found: C 51.45, H 6.72. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 331.1 (6348).Synthesis of complex 4d
CuCl (32 mg, 0.32 mmol) was added in portions to a stirred solution of 2d (55 mg, 0.32 mmol) in CH3CN (5 mL). After stirring for 10 h, the solid was removed and the filtrate was dried in vacuo. The remaining solid was washed with hexane and dried in vacuo to afford 4d as yellow powder (80 mg, 0.26 mmol, 80% yield). Yellow crystals of 4d were obtained from an acetonitrile solution layered with ether on top and stored at −30 °C. 1H NMR (THF-D8, 400 MHz): δ = 10.35 (s, 1H, OH), 9.18 (dd, CarH, PH), 8.17 (m, 1H, CarH), 7.30 (m, 1H, CarH), 3.89 (s, 3H, CH3); 13C{1H} NMR (CD3CN, 100.5 MHz): δ = 190.0 (d, 1JPC = 20.1 Hz, OCP), 165.2 (d, 3JPC = 8.0 Hz, PCCC), 147.5 (d, 1JPC = 21.1 Hz, PCC), 133.5 (d, 2JPC = 14.1 Hz, PCC), 128.6 (d, 3JPC = 17.1 Hz, PCCC), 123.0 (d, 2JPC = 4.0 Hz, PCC), 52.6 (s, CH3); 31P{1H} NMR (CD3CN, 161.9 MHz): δ = 122.0 (s, JPH = 32.4 Hz). M. P. = 225 °C. IR (ATR, [cm−1]): 3394 (w, CH stretch), 2967 (w, CH stretch), 1706 (m), 1428 (w), 1408 (w), 1296 (m), 1242 (s, C–O), 1180 (s, C–O), 992 (w), 862 (w), 750 (m), 687 (w), 614 (w), 479 (w). Elemental analysis (%): calcd for [C9H10O3PNClCu]2; C 34.85, H 3.25; found: C 34.99, H 3.41. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 328.5 (3449).Synthesis of complex 5
A solution of PPh3 (705 mg, 2.69 mmol) in 10 mL of THF was added dropwise to a mixture of CuCl (133 mg, 1.35 mmol) dissolved in 10 mL of acetonitrile. After stirring for 1 h, 1f (180 mg, 1.34 mmol) was suspended in 10 mL of THF and added dropwise to the reaction mixture. The reaction mixture was stirred for another 1 h. Subsequently the solvent was removed under reduced pressure, and the product was extracted with dichloromethane (10 mL). The filtrate was concentrated under reduced pressure and the remaining solid was washed with hexane and dried in vacuo affording 5 as an orange powder (574 mg, 0.33 mmol, 98% yield). Orange crystals of 5 were obtained from a dichloromethane solution layered with hexane on top and stored at −30 °C. M. P. = 175 °C. IR (ATR, [cm−1]): 3047 (w, CH stretch), 3013 (w), 1736 (s), 1716 (w), 1584 (w), 1563 (w), 1480 (w), 1479 (w), 1433 (m), 1415 (w), 1381 (m), 1349 (m), 1261 (s, C–O), 1201 (w), 1092 (m), 1027 (m), 995 (w), 856 (w), 799 (w), 740 (s), 682 (s), 614 (m). Elemental analysis (%): calcd for [C23H19OP2Cu]4; C 63.23, H 4.38; found: C 63.02, H 4.42. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 445 nm, 252 nm.Synthesis of complex 6
2a (53 mg, 0.20 mmol) in THF (2 mL) was added dropwise to a stirred solution of AuCl(SMe2) (59 mg, 0.20 mmol) in THF (2 mL). After stirring for 2 h. The solvent was removed under reduced pressure. The remaining solid was washed with hexane and dried in vacuo affording 6 as a light yellow powder (70 mg, 0.13 mmol, 61% yield). Yellow crystals of 6 were obtained from a acetonitrile solution layered with hexane on top and stored at −30 °C. 1H NMR (THF-D8, 400 MHz): δ = 10.77 (s, 1H, OH), 8.13 (d, 3JPH = 20.0 Hz, 1H, CarH, PH), 7.80 (m, 5H, CarH), 7.52 (m, 6H, CarH); 13C{1H} NMR (THF-D8, 100.5 MHz): δ = 182.0 (d, 1JPC = 25.1 Hz, OCP), 160.7 (d, 1JPC = 27.1 Hz, CPC), 146.0 (d, 3JPC = 19.1 Hz, PCCC), 140.8 (d, 3JPC = 5.0 Hz, PCCC), 139.8 (d, 2JPC = 15.1 Hz, PCC), 129.5 (Car), 128.9 (Car), 128.8 (Car), 128.5 (Car), 128.2 (d, JPC = 10.1 Hz, Car), 127.5 (d, JPC = 10.1 Hz, Car), 123.0 (Car); 31P{1H} NMR (THF-D8, 161.9 MHz): δ = 117.7 (s). M. P. = 148 °C. IR (ATR, [cm−1]): 3209 (w, CH stretch), 3026 (w, CH stretch), 1570 (w), 1480 (m), 1413 (w), 1364 (w), 1308 (m), 1202 (s, C–O), 1077 (w), 962 (w), 869 (w), 760 (m), 694 (m), 575 (w). Elemental analysis (%): calcd for C17H13OPClAu; C 41.11, H 2.64; found: C 42.32, H 2.60. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 354 (16000), 313 (21935).
Synthesis of complex 7
1a (72 mg, 0.25 mmol) in THF (2 mL) was added dropwise to the stirred reaction mixture of PPh3 (65 mg, 0.25 mmol) and AuCl(SMe2) (74 mg, 0.25 mmol) in THF (5 mL). After stirring for 1 h, the solvent was removed under reduced pressure and the product was extracted with dichloromethane (10 mL). The filtrate was concentrated under reduced pressure, the remaining solid was washed with hexane and dried in vacuo affording 7 as red powder (125 mg, 0.17 mmol, 67% yield). Red crystals of 7 were obtained from a dichloromethane solution layered with hexane on top and stored at −30 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 7.65 (m, 2H, CarH), 7.55 (m, 4H, CarH), 7.46 (m, 17H, CarH), 7.34 (m, 3H, CarH), 6.93 (m, 1H, CarH); 13C{1H} NMR (CD2Cl2, 100.5 MHz): δ = 206.2 (Car), 164.0 (d, 1JPC = 16.1 Hz, CPC), 149.1 (Car), 142.9 (d, 2JPC = 14.1 Hz, Car), 142.6 (d, 2JPC = 3.0 Hz, Car), 134.1 (Car), 134.0 (Car), 131.6 (Car), 129.9 (Car), 129.4 (Car), 129.2 (Car), 129.1 (Car), 128.6 (Car), 128.0 (Car), 127.8 (Car), 127.5 (Car), 127.4 (Car), 125.5 (Car), 125.2 (Car), 123.0 (Car); 31P{1H} NMR (161.9 MHz, CD2Cl2): δ = 89.6 (s), 38.1 (s). M. P. = 141 °C. IR (ATR, [cm−1]): 3051 (w, CH stretch), 1560 (w, CH stretch), 1466 (w), 1433 (m), 1307 (w), 1098 (s, C–O), 1026 (w), 997 (w), 849 (w), 748 (s), 690 (m), 500 (s). Elemental analysis (%): calcd for C35H27OP2Au; C 58.18, H 3.77; found: C 57.96, H 3.84. UV/vis (THF, λ (nm) ε (M−1 cm−1)): 470 (6121).Synthesis of complex 8
AuCl(SMe2) (50 mg, 0.17 mmol) in THF (2 mL) was added dropwise to the stirred reaction of 7 (125 mg, 0.17 mmol) in THF (5 mL). After stirring for 1 h, the solvent was removed and the remaining solid was washed with hexane and dried in vacuo affording 8 as red powder (132 mg, 0.13 mmol, 80% yield). 31P{1H} NMR (161.9 MHz, CD2Cl2): δ = 33.2 (s), −5.4 (s).Synthesis of complex 9
1a (72 mg, 0.25 mmol) was added in portions to a stirred solution of AuCl(SMe2) (88 mg, 0.30 mmol) in THF (10 mL). After stirring for 2 h, the solid was filtrated and then washed by hexane, and dried in vacuo to afford 9 as orange powder (100 mg, 0.03 mmol, 70% yield). Orange crystals of 9 were obtained from a dichloromethane solution layered with hexane on top and stored at −30 °C. 1H NMR (CD2Cl2, 400 MHz): δ = 7.83 (m, 9H, CarH), 7.55 (m, 11H, CarH), 7.46 (m, 21H, CarH), 7.34 (m, 14H, CarH), 6.64 (m, 5H, CarH), 3.65 (s, 6H, 2CH3); 31P {1H} NMR (161.9 MHz, CD2Cl2): δ = −3.2 (s). M. P. = 120 °C. IR (ATR, [cm−1]): 3515 (w, CH stretch), 3051 (w, CH stretch), 1602 (w), 1586 (w), 1497 (w), 1352 (w), 1309 (m), 1245 (w), 1171 (s, C–O), 1029 (w), 998 (w), 857 (w), 751 (m), 686 (w), 621 (w). Elemental analysis (%): calcd for C87H66O5P5SAu6Cl; C 40.26, H 2.56; found: C 40.40, H 2.34. UV/vis (CH2Cl2, λ (nm) ε (M−1 cm−1)): 327.5 (69200).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21603280, 21720102007, 21821003), the Fundamental Research Funds for the Central Universities (171gpy78), the Guangdong Provincial Key Platform and Major Scientific Research Projects for Colleges and Universities (2016KTSCX135), and ETH Zürich (0-20406-18).Notes and references
- (a) B. Breit, R. Winde, T. Mackewitz, R. Paciello and K. Harms, Chem.–Eur. J., 2001, 7, 3106–3121 CrossRef CAS PubMed; (b) L. Weber, Angew. Chem. Int. Ed., 2002, 41, 563–572 ( Angew. Chem. , 2002 , 114 , 583–592 ) CrossRef CAS; (c) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef CAS; (d) C. Müller and D. Vogt, Dalton Trans., 2007, 5505–5523 RSC; (e) L. Kollár, Chem. Rev., 2010, 110, 4275–4302 CrossRef PubMed; (f) R. J. Newland, M. F. Wyatt, R. L. Wingad and S. M. Mansell, Dalton Trans., 2017, 46, 6172–6176 RSC; (g) M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2017, 46, 86–95 RSC.
- (a) J. Moussa, T. Cheminel, G. R. Freeman, L. M. Chamoreau, J. A. G. Williams and H. Amouri, Dalton Trans., 2014, 43, 8162–8165 RSC; (b) P. Roesch, J. Nitsh, M. Lutz, J. Wiecko, A. Steffen and C. Müller, Inorg. Chem., 2014, 53, 9855–9859 CrossRef CAS PubMed; (c) X. Chen, Z. Li, Y. Fan and H. Grützmacher, Eur. J. Inorg. Chem., 2016, 633–638 CrossRef.
- (a) P. D. Burrow, A. J. Ashe III, D. J. Bellville and K. D. Jordan, J. Am. Chem. Soc., 1982, 104, 425–429 CrossRef CAS; (b) K. K. Baldridge and M. S. Gordon, J. Am. Chem. Soc., 1988, 110, 4204–4208 CrossRef CAS; (c) L. Nyulászi, Chem. Rev., 2001, 101, 1229–1246 CrossRef; (d) A. Modelli, B. Hajató, J. F. Nixon and L. Nyulászi, J. Phys. Chem. A, 2004, 108, 7440–7447 CrossRef CAS; (e) C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187–202 CrossRef.
- Selected examples see: (a) H. Vahrenkamp and H. Nöth, Chem. Ber., 1973, 106, 2227–2235 CrossRef CAS; (b) P. le Floch, L. Ricard and F. Mathey, Polyhedron, 1990, 9, 991–997 CrossRef CAS; (c) C. Elschenbroich, M. Novotny, J. Kroker, A. Behrendt, W. Massa and S. Wocadlo, J. Organomet. Chem., 1993, 459, 157–167 CrossRef CAS; (d) C. Elschenbroich, M. Nowotny, A. Behrendt, K. Harms, S. Wocadlo and J. Pebler, J. Am. Chem. Soc., 1994, 116, 6217–6219 CrossRef CAS; (e) B. Breit, R. Winde and K. Harms, J. Chem. Soc., Perkin Trans. 1, 1997, 2681–2682 RSC; (f) P. Le Floch and F. Mathey, Coord. Chem. Rev., 1998, 179–180, 771–791 CrossRef; (g) C. Elschenbroich, S. Voss, O. Schiemann, A. Lippek and K. harms, Organometallics, 1998, 17, 4417–4424 CrossRef CAS; (h) C. Elschenbroich, J. Six and K. Harms, Chem. Commun., 2006, 3429–3431 RSC.
- (a) C. Elschenbroich, S. Voss and K. Harms, Z. Naturforsch. B Chem. Sci., 1999, 54, 209–213 CAS; (b) C. Elschenbroich, J. Six, K. Harms, G. Frenking and G. Heydenrych, Eur. J. Inorg. Chem., 2008, 3303–3309 CrossRef CAS; (c) S. B. Clendenning, P. b. Hitchcock, G. A. Lawless, J. F. Nixon and C. W. Tate, J. Organomet. Chem., 2010, 695, 717–720 CrossRef CAS; (d) L. E. E. Broeckx, M. Lutz, D. Vogt and C. Müller, Chem. Commun., 2011, 47, 2003–2005 RSC; (e) I. de Krom, L. E. E. Broeckx, M. Lutz and C. Müller, Chem.–Eur. J., 2013, 19, 3676–3684 CrossRef CAS PubMed.
- For selected examples see: (a) D. Carmichael, P. Le Floch, L. Ricard and F. Mathey, Inorg. Chim. Acta, 1992, 198–200, 437–441 CrossRef CAS; (b) P. Le Floch, S. Mansuy, L. Ricard and F. Mathey, Organometallics, 1996, 15, 3267–3274 CrossRef CAS; (c) J. J. M. Weemers, J. Wiecho, E. A. Pidko, M. Weber, M. Lutz and C. Müller, Chem.–Eur. J., 2013, 19, 14458–14469 CrossRef CAS PubMed; (d) L. E. E. Broeckx, S. Güven, F. J. L. Heutz, M. Lutz, D. Vogt and C. Müller, Chem.–Eur. J., 2013, 19, 13087–13098 CrossRef CAS PubMed; (e) I. de Krom, M. Lutz and C. Müller, Dalton Trans., 2015, 44, 10304–10314 RSC; (f) X. Chen, Z. Li and H. Grützmacher, Chem.–Eur. J., 2018, 24, 8432–8437 CrossRef CAS PubMed.
- For selected examples see: (a) J. Deberitz and H. Nöth, Chem. Ber., 1970, 103, 2541–2547 CrossRef CAS; (b) H. Vahrenkamp and H. Nöth, Chem. Ber., 1972, 105, 1148–1157 CrossRef CAS; (c) J. Deberitz and H. Nöth, Chem. Ber., 1973, 106, 2222–2226 CrossRef CAS; (d) C. Elschenbroich, M. Nowotny, B. Metz, W. Massa, J. Graulich, K. Biehler and W. Sauer, Angew. Chem., Int. Ed. Engl., 1991, 30, 547–550 CrossRef; (e) F. Knoch, F. Kremer, U. Schmidt and U. Zenneck, Organometallics, 1996, 15, 2713–2719 CrossRef CAS; (f) P. Le Floch, F. Knoch, F. Kremer, F. Mathey, J. Scholz, W. Scholz, K. H. Thiele and U. Zenneck, Eur. J. Inorg. Chem., 1998, 119–126 CrossRef CAS; (g) N. Mézailles, L. Ricard, F. Mathey and P. Le Floch, Organometallics, 2001, 20, 3304–3307 CrossRef; (h) M. Doux, L. Ricard, F. Mathey, P. Le Floch and N. Mézailles, Eur. J. Inorg. Chem., 2003, 687–698 CrossRef CAS.
- B. Schmid, L. M. Venanzi, T. Gerfin, V. Gramlich and F. Mathey, Inorg. Chem., 1992, 31, 5117–5122 CrossRef CAS.
- (a) M. T. Reetz, E. Bohres, R. Goddard, M. C. Holthausen and W. Thiel, Chem.–Eur. J., 1999, 5, 2101–2108 CrossRef CAS; (b) P. Rosa, L. Ricard, F. Mathey and P. Le Floch, Organometallics, 2000, 19, 5247–5250 CrossRef CAS; (c) M. J. Bakker, F. W. Vergeer, F. Hartl, K. Goubitz, J. Fraanje, P. Rosa and P. Le Floch, Eur. J. Inorg. Chem., 2000, 843–845 CrossRef CAS; (d) M. J. Bakker, F. W. Vergeer, F. Hartl, P. Rosa, L. Ricard, P. Le Floch and M. J. Calhorda, Chem.–Eur. J., 2002, 8, 1741–1752 CrossRef CAS PubMed.
- M. H. Habicht, F. Wossidlo, T. Bens, E. A. Pidko and C. Müller, Chem.–Eur. J., 2018, 24, 944–952 CrossRef CAS PubMed.
- Y. Mao, K. M. H. Lim, Y. Li, R. Ganguly and F. Mathey, Organometallics, 2013, 32, 3562–3565 CrossRef CAS.
- (a) G. Märkl and A. Kallmünzer, Tetrahedron Lett., 1989, 30, 5245–5248 CrossRef; (b) Y. Mao and F. Mathey, Chem.–Eur. J., 2011, 17, 10745–10751 CrossRef CAS PubMed; (c) K. Nakajima, W. Liang and Y. Nishibayashi, Org. Lett., 2016, 18, 5006–5009 CrossRef CAS PubMed; (d) Y. Mei, D.-J. Wu, J. E. Borger and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 5512–5515 ( Angew. Chem. , 2018 , 130 , 5610–5613 ) CrossRef CAS PubMed.
- X. D. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkö, Z. S. Li, G. Becker, H. F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 ( Angew. Chem. , 2014 , 126 , 1667–1671 ) CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2009, full reference see ESI†.
- J. K. B. e. D. Glendening, A. E. Reed, J. E. Carpenter, J. A. Bohman, C. M. Morales, C. R. Landis, F. Weinhold, Vol. NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- (a) G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS; (b) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H. F. Grützmacher and H. Grützmacher, Angew. Chem. Int. Ed., 2011, 50, 8420–8423 ( Angew. Chem. , 2011 , 123 , 8570–8574 ) CrossRef CAS PubMed; (c) I. Krummenacher and C. C. Cummins, Polyhedron, 2012, 32, 10–13 CrossRef CAS; (d) A. R. Jupp and J. M. Goicoechea, Angew. Chem. Int. Ed., 2013, 52, 10064–10067 ( Angew. Chem. , 2013 , 125 , 10248–10251 ) CrossRef CAS PubMed; (e) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 831–840 RSC; (f) R. Stuter, Z. Benkö, M. Bispinghoff and H. Grützmacher, Angew. Chem. Int. Ed., 2017, 56, 11226–11231 ( Angew. Chem. , 2017 , 129 , 11378–11383 ) CrossRef PubMed.
- L. Liu, J. Zhu and Y. Zhao, Chem. Commun., 2014, 50, 11347–11349 RSC.
- (a) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC; (b) P. Pyykko and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- For selected examples see: (a) G. Maas, J. Fink, H. Wingert, K. Blatter and M. Regitz, Chem. Ber., 1987, 120, 819–824 CrossRef CAS; (b) P. Rosa, L. Ricard, F. Mathey and P. Le Floch, Chem. Commun., 1999, 537–538 RSC; (c) N. Mézailles, N. Maigrot, S. Hamon, L. Ricard, F. Mathey and P. Le Floch, J. Org. Chem., 2001, 66, 1054–1056 CrossRef; (d) S. Choua, C. Dutan, L. Cataldo, T. Berclaz, M. Geoffroy, N. Mézailles, A. Moores, L. Ricard and P. Le Floch, Chem.–Eur. J., 2004, 10, 4080–4090 CrossRef CAS PubMed; (e) M. Dochnahl, M. Doux, E. Faillard, L. Ricard and P. Le Floch, Eur. J. Inorg. Chem., 2005, 125–134 CrossRef CAS; (f) A. Moores, T. Cantat, L. Ricard, N. Mézailles and P. Le Floch, New J. Chem., 2007, 31, 1493–1498 RSC; (g) C. Müller, D. Wasserberg, J. J. M. Weemers, E. A. Pidko, S. Hoffmann, M. Lutz, A. L. Spek, S. C. J. Meskers, R. A. J. janssen, R. A. van Santen and D. Vogt, Chem.–Eur. J., 2007, 13, 4548–4559 CrossRef PubMed.
- Selected recent examples for T-shaped Cu complexes: (a) S. Kumar, G. Mani, D. Dutta and S. Mishra, Inorg. Chem., 2014, 53, 700–709 CrossRef CAS PubMed; (b) J. I. van der Vlugt, E. A. Pidko, D. Vogt, M. Lutz and A. L. Spek, Inorg. Chem., 2009, 48, 7513–7515 CrossRef CAS PubMed.
- W. Clegg and D. M. Tooke, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 603–612 CrossRef CAS PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- S. E. Thwaite, A. Schier and H. Schmidbaur, Inorg. Chim. Acta, 2004, 357, 1549–1557 CrossRef CAS.
- For selected examples see: (a) C. F. Shaw III, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS PubMed; (b) D. Jiang, R. L. Whetten, W. Luo and S. Dai, J. Phys. Chem. C, 2009, 113, 17291–17295 CrossRef CAS; (c) H. Qian, W. T. Eckenhoff, M. E. Bier, T. Pintauer and R. Jin, Inorg. Chem., 2011, 50, 10735–10739 CrossRef CAS PubMed; (d) B. M. Barngrover and C. M. Aikens, J. Phys. Chem. A, 2011, 115, 11818–11823 CrossRef CAS PubMed; (e) Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed; (f) B. M. Barngrover and C. M. Aikens, J. Am. Chem. Soc., 2012, 134, 12590–12595 CrossRef CAS PubMed.
- H. Huang, G. Tao, Z. Wei, J. Hou, M. Wang, Z. Duan and F. Mathey, Organometallics, 2018, 37, 464–468 CrossRef CAS.
- For selected examples see: (a) R. E. Bachman, M. S. Fioritto, S. K. Fetics and T. M. Cocker, J. Am. Chem. Soc., 2001, 123, 5376–5377 CrossRef CAS PubMed; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC; (c) A. Codina, E. J. Fernández, P. G. Jones, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, J. Pérez and M. A. Rodríguez, J. Am. Chem. Soc., 2012, 124, 6781–6786 CrossRef; (d) W. L. Li, H. T. Liu, T. Jian, G. V. Lopez, Z. A. Piazza, D. L. Huang, T. T. Chen, J. Su, P. Yang, X. Chen, L. S. Wang and J. Li, Chem. Sci., 2016, 7, 475–481 RSC.
- (a) J. J. B. W. P. Bosman and J. H. Noordik, J. Chem. Soc., Chem. Commun., 1981, 1218–1219 Search PubMed; (b) K. P. Hall, B. R. C. Theobald, D. I. Gilmour, D. M. P. Mingos, A. J. Welch and J. W. A. V. D. Velden, J. Chem. Soc., Chem. Commun., 1982, 528–530 RSC; (c) M. Schulz-Dobrick and M. Jansen, Eur. J. Inorg. Chem., 2006, 4498–4502 CrossRef CAS; (d) Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed; (e) Y. Shichibu, M. Zhang, Y. Kamei and K. Konishi, J. Am. Chem. Soc., 2014, 136, 12892–12895 CrossRef CAS PubMed; (f) S. Yamazoe, S. Mastsuo, S. Muramatsu, S. Takano, K. Nitta and T. Tsukuda, Inorg. Chem., 2017, 56, 8319–8325 CrossRef CAS PubMed.
- (a) A. Hinz, A. Schulz and A. Villinger, Chem. Commun., 2016, 52, 6328–6331 RSC; (b) Z. Li, Y. Hou, Y. Li, A. Hinz and X. Chen, Chem.–Eur. J., 2018, 24, 4849–4855 CrossRef CAS PubMed.
- H. Werner, Angew. Chem. Int. Ed., 2004, 43, 938–954 ( Angew. Chem. , 2004 , 116 , 956–972 ) CrossRef CAS PubMed.
- Y. Q. Li, Z. S. Li, Y. F. Hou and H. Grützmacher, Inorg. Chem., 2018, 57, 13235–13245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, crystallographic data, and description of the computational protocol. CCDC 1872113–1872122 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05281f |
| This journal is © The Royal Society of Chemistry 2019 |