
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTranslocation of enzymes into a mesoporous MOF for enhanced catalytic activity under extreme conditions†
José
Navarro-Sánchez
 a,
Neyvis
Almora-Barrios
a,
Belén
Lerma-Berlanga
a,
J. Javier
Ruiz-Pernía
b,
Victor A.
Lorenz-Fonfria
a,
Iñaki
Tuñón
b and
Carlos
Martí-Gastaldo
*a
a,
Neyvis
Almora-Barrios
a,
Belén
Lerma-Berlanga
a,
J. Javier
Ruiz-Pernía
b,
Victor A.
Lorenz-Fonfria
a,
Iñaki
Tuñón
b and
Carlos
Martí-Gastaldo
*a
aInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Catedrático José Beltrán-2, Paterna, 46980, Spain. E-mail: carlos.marti@uv.es
bDepartamento de Química Física, Universidad de Valencia, Doctor Moliner-50, Burjassot, 46100, Spain
First published on 28th February 2019
Abstract
Enzymatic catalysis is of great importance to the chemical industry. However, we are still scratching the surface of the potential of biocatalysis due to the limited operating range of enzymes in harsh environments or their low recyclability. The role of Metal–Organic Frameworks (MOFs) as active supports to help overcome these limitations, mainly by immobilization and stabilization of enzymes, is rapidly expanding. Here we make use of mild heating and a non-polar medium during incubation to induce the translocation of a small enzyme like protease in the mesoporous MOF MIL-101(Al)-NH2. Our proteolytic tests demonstrate that protease@MIL-101(Al)-NH2 displays higher activity than the free enzyme under all the conditions explored and, more importantly, its usability can be extended to extreme conditions of pH and high temperatures. MOF immobilization is also effective in providing the biocomposite with long-term stability, recyclability and excellent compatibility with competing enzymes. This simple, one-step infiltration strategy might accelerate the discovery of new MOF-enzyme biocatalysts that meet the requirements for biotechnological applications.
Introduction
Metal–Organic Frameworks (MOFs) have proven to be promising alternatives for enzyme immobilization based on their high surface areas and unparalleled structural and chemical flexibility.1,2 Compared to other porous supports, MOFs also provide tailorable functionality to gain control over specific interactions with the enzyme that helps mitigating their poor stability at high temperatures or in organic solvents. Accordingly, MOF immobilization can improve the intrinsic properties of the enzyme for superior substrate specificity, catalytic stability under different pH conditions or its susceptibility to autolysis at higher temperatures, modulating and enlarging the range of conditions for which the enzyme can retain its activity.There are three main different strategies to produce enzyme-immobilized MOF composites depending on the chemical interaction between the enzyme and the solid support.1,3 Grafting of the enzyme to the external surface of the MOF by covalent linkage,4 biomimetic co-mineralization of the MOF from direct reaction of its precursors in the presence of the enzyme,5–7 and direct infiltration of the enzyme into the pores of pre-synthesized MOFs.8,9 Overall, all these strategies permit producing composites with better catalytic activities and environmental resistance than the free enzyme, but direct infiltration is arguably better fitted to prevent undesirable leaching and self-aggregation. Pore entrapment also renders superior stability due to the physical adsorption in a confined cavity. However, this approach is restricted to a handful of mesoporous materials with big pore apertures compatible with the diffusion of enzymes that typically feature large molecular dimensions.10–14 Finding alternative routes that permit overcoming this size limitation would permit enlarging the number of MOF-enzyme combinations that can be adapted to this route.
Previous reports confirm the ability of biomolecules to diffuse through comparatively narrow nanopores. By using fluorescence spectroscopy, Ma and co-workers demonstrated that the protein cytochrome c (2.6 × 3.3 × 3.3 nm) undergoes conformational changes due to the interaction with the surface of Tb-mesoMOF.15 Inspired by this work, we decided to explore if a similar protein translocation mechanism could also be extended to biologically active enzymes. This represents an additional challenge as the catalytic function of the enzyme is intimately linked to its native structure and its translocation into the MOF would need to be compatible with the regeneration of an active state after encapsulation. We presumed this could be demonstrated more easily for small enzymes formed using only one polypeptide chain, more likely to unfold partially and recover its native structure without irreversible changes that could destroy their function.16–18 Provided the enzyme could be reverted back to a biological active structure after infiltration, this would result in a very efficient immobilization as the enzyme would remain effectively trapped in the internal cavities of the MOF to avoid leaching.
Here we report the infiltration of a protease, an aspartic proteinase from Aspergillus saitoi (also known as Aspergillus phoenicis), into the mesoporous cavities of MIL-101(Al)-NH2 (ref. 19) (Fig. 1). Incubation of protease in an organic medium at mild temperatures facilitates partial unfolding of the enzyme, supported by molecular dynamics simulations and fluorescence spectroscopy, to enable its translocation across the pore windows of the host (50% smaller in size). Our tests confirm that the MOF-enzyme biocatalyst surpasses the proteolytic activity of the free enzyme in all cases and its operating range can be extended to extreme conditions of pH and temperature with excellent recyclability and tolerance to competing enzymes.
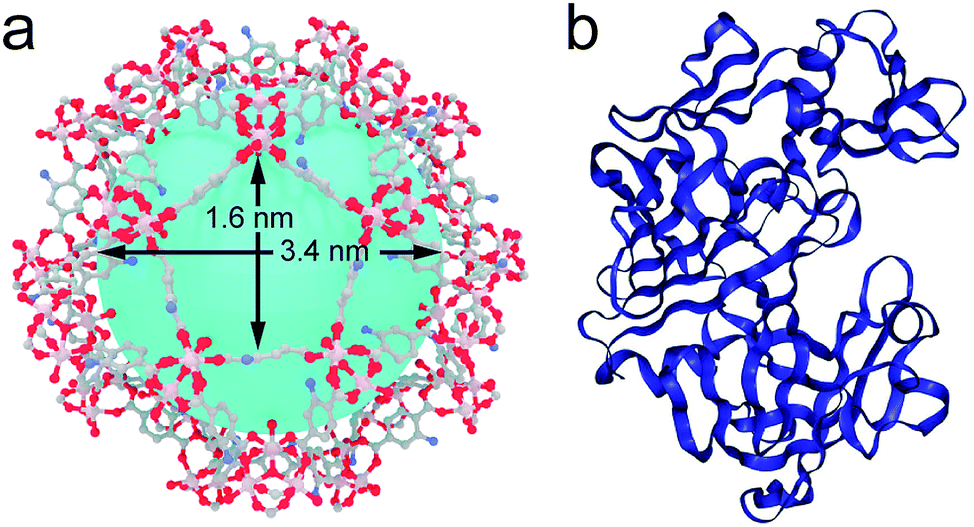 | ||
| Fig. 1 (a) Structure of MIL-101(Al)-NH2 highlighting the dimensions of the largest pore windows and mesoporous cages. (b) Crystal structure of the protease used in this work. Files available from CCDC (605510) and PDB (1IBQ). | ||
Results and discussion
Translocation of protease in MIL-101(Al)-NH2
Among the different candidates possible, we targeted a family of fungal proteases based on their relevance in biotechnological applications.20–22 Previous reports confirm that proteases can regain their native structure and function after denaturant or thermal-induced unfolding.23,24 Also important, compared to other common proteins like horseradish peroxidase (HPP, 44 kDa) or lipase (72 kDa), the smaller size of protease (17 kDa) would be compatible with the mesoporous cavities of MIL-101. As for the MOF, we selected MIL-101(Al)-NH2 due to several reasons. From a structural point of view, it displays a MTN zeotypic three-dimensional extended network that combines two types of mesoporous cavities of 3.6 and 2.9 nm that are interconnected by pentagonal and hexagonal windows of 1.6 and 1.2 nm (Fig. 1a). In principle, these mesoporous cages are large enough to accommodate small enzymes like Aspergillus saitoi protease. Dynamic light scattering (DLS) measurements of the protein in a TRIS buffer indicate an average distribution of sizes centered at 2.85 nm (Fig. S1†). The ratio between the diameters of the mesoporous cavity in MIL-101 and the protein is close to 1.2. Compared to other studies in which this value is close to one,13,25 this reduced environmental stress will result in a higher conformational freedom more likely to favor the regeneration of an active conformation after enzyme translocation. Previous reports confirm the biocompatibility of MIL-101 and its ability to endure the conditions used for the incubation of enzymes (physiological medium, pH, temperature, and prolonged time).26 Also important, we decided to use the amino functionalized MOF to favor the diffusion of the protease in the polar environment provided by the hydrophilic pores in MIL-101(Al)-NH2. This surface functionalization was also expected to render better dispersibility in aqueous medium and favor the stabilization of the encapsulated enzyme by formation of H-bond interactions.27Prior to attempting the infiltration experiments, we studied the effect of the buffer used for enzyme incubation on the structural integrity of the framework. MIL-101(Al)-NH2 was prepared by following a previously reported method,28 and immersed in TRIS, HEPES, carbonate and PBS buffers (0.1 M). As shown in Fig. S2,† PXRD of the solid recovered after 12 hours confirms that the structural integrity of MIL-101 is only maintained in TRIS whereas other buffers like HEPES and PBS accelerate its transformation into the MIL-53 phase. Accordingly, freshly made MIL-101(Al)-NH2 particles were immersed in 3 mL of a solution of the protease in TRIS buffer (0.1 M, pH = 7.4) followed by addition of 30 mL of anhydrous hexane with continuous stirring. The mixture was incubated under these conditions at 60 °C overnight. The addition of a non-polar solvent and a higher temperature were expected to favor the denaturation of the protease as result of reducing the hydrophobic effect and increasing kinetic energy of the system. The altered structure of the water in the aqueous polar medium by the presence of hexane, in combination with the increase in temperature, changes the landscape of energies associated with the array of supramolecular interactions (π–π stacking, H-bonds) with the solvent and within the protein that control its secondary structure. This process was analysed by means of MD simulations. As shown in Fig. 2a, the Aspergillus saitoi protease undergoes reversible unfolding under the conditions used in the experiment. Our 1.0 μs long MD simulation in a hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture at 60 °C shows a continuous drift from the native structure with time. The root-mean-square deviation (RMSD) allows a comparison between the structure of the native folded protein and the corresponding partially or fully unfolded structure. The RMSD measured in the presence of hexane increases continuously with time to reach larger values than those observed for the protein in a polar medium, aqueous solutions at 25 and 60 °C. This confirms that the combination of temperature and an organic solvent can favor partial denaturation of the enzyme. Comparison of the structure of the enzyme in the hexane:water mixture after 1.0 μs simulation with the reported X-ray structure shows that protein unfolding in the non-polar solution involves the opening of the cleft between the two protein lobes (Fig. 2a, insets). As shown in Fig. S3,† analysis of the secondary structure shows a gradual transformation of α-helices and β-sheets into unstructured regions, with a progressive increase of the number of residues belonging to regions without a defined secondary structure. Overall, our MD simulations confirm the potential ability of the enzyme to modify its conformation under the conditions used in the experiments required to translocate across the narrow pore windows in the MOF. For direct correlation with the experiment, we used fluorescence spectroscopy to compare the emission spectra of protease in TRIS buffer – native confirmation, in the incubation medium – partial unfolding – and after translocation into the MOF – encapsulated conformation. Protease presents 16 tyrosine (Tyr) and 3 tryptophan (Trp) residues that account for its emission spectrum. As shown in Fig. 2b incubating the free protease on a water–hexane dispersion under mild temperature conditions shifts the fluorescence emission maximum from 329 to 338 nm, corresponding to solvent-exposed Tyr and Trp.29 This red shift in fluorescence is a common situation when unfolding soluble proteins,29 and is fully reversed upon removal of the hexane and cooling down to room temperature (Fig. S4†). In contrast, refolding the protease in the presence of the MOF leads to a composite displaying a fluorescence emission maximum at 321 nm. This blue shift indicates that the protein located within MIL-101(Al)-NH2 cavities exists in a non-native folded conformation where Tyr/Trp residues experience an environment less polar than in the native folded state. The Raman spectra of free protease and protease@MIL-101(Al)-NH2 display the characteristics bands of disulphide bonds of cysteine (Fig. S5†). As reported for the encapsulation of microperoxidase in Tb-mesoMOF,30 the signals at 470 and 2436 cm−1 in the enzyme are blue shifted close to 6 and 9 cm−1 after encapsulation as result of the interaction with the framework. After the insertion procedure, the resulting solid was thoroughly washed with the buffer and isolated by centrifugation to afford protease@MIL-101(Al)-NH2. UV-visible spectroscopy was used to ensure complete removal of the excess of non-encapsulated protease upon washing. As shown in Fig. 3a, there was no measurable protease in solution after two rounds. To further confirm that the protease was encapsulated rather than adsorbed on the external surface of the particles, we carried out two control experiments. We followed the same enzyme incubation protocol described above by using MIL-101(Cr). Immobilization is negligible in this case (Fig. S6 and S7†), likely due to the absence of –NH2 groups important in directing the translocation of the enzyme into the MOF. For the next test we made use of azocasein as a protease substrate (Fig. 3a, inset). Azocasein is hydrolysed non-specifically by proteolytic enzymes, releasing the azo dye into the media where it can be detected by its characteristic and intense absorbance at 440 nm. This permits rapid identification of the presence of the enzyme in MOFs incubated under different conditions. We ran five experiments by adding azocasein to five vials that contained: TRIS buffer as a blank sample (1) and dispersions in the same buffer of the MOF incubated with protease at 25 °C with hexane (2), the MOF incubated with protease at 60 °C without hexane (3), the protease@MIL-101(Al)-NH2 prepared as described above (4) and free protease (5). Samples 2, 3 and 4 where washed at least 3 times with fresh buffer before the test. After addition of azocasein, only samples 4 and 5 gave a positive result of proteolytic activity (yellow color), confirming the importance of temperature and the presence of a non-polar solvent in the incubation medium to facilitate the infiltration of the enzyme.
:water mixture at 60 °C shows a continuous drift from the native structure with time. The root-mean-square deviation (RMSD) allows a comparison between the structure of the native folded protein and the corresponding partially or fully unfolded structure. The RMSD measured in the presence of hexane increases continuously with time to reach larger values than those observed for the protein in a polar medium, aqueous solutions at 25 and 60 °C. This confirms that the combination of temperature and an organic solvent can favor partial denaturation of the enzyme. Comparison of the structure of the enzyme in the hexane:water mixture after 1.0 μs simulation with the reported X-ray structure shows that protein unfolding in the non-polar solution involves the opening of the cleft between the two protein lobes (Fig. 2a, insets). As shown in Fig. S3,† analysis of the secondary structure shows a gradual transformation of α-helices and β-sheets into unstructured regions, with a progressive increase of the number of residues belonging to regions without a defined secondary structure. Overall, our MD simulations confirm the potential ability of the enzyme to modify its conformation under the conditions used in the experiments required to translocate across the narrow pore windows in the MOF. For direct correlation with the experiment, we used fluorescence spectroscopy to compare the emission spectra of protease in TRIS buffer – native confirmation, in the incubation medium – partial unfolding – and after translocation into the MOF – encapsulated conformation. Protease presents 16 tyrosine (Tyr) and 3 tryptophan (Trp) residues that account for its emission spectrum. As shown in Fig. 2b incubating the free protease on a water–hexane dispersion under mild temperature conditions shifts the fluorescence emission maximum from 329 to 338 nm, corresponding to solvent-exposed Tyr and Trp.29 This red shift in fluorescence is a common situation when unfolding soluble proteins,29 and is fully reversed upon removal of the hexane and cooling down to room temperature (Fig. S4†). In contrast, refolding the protease in the presence of the MOF leads to a composite displaying a fluorescence emission maximum at 321 nm. This blue shift indicates that the protein located within MIL-101(Al)-NH2 cavities exists in a non-native folded conformation where Tyr/Trp residues experience an environment less polar than in the native folded state. The Raman spectra of free protease and protease@MIL-101(Al)-NH2 display the characteristics bands of disulphide bonds of cysteine (Fig. S5†). As reported for the encapsulation of microperoxidase in Tb-mesoMOF,30 the signals at 470 and 2436 cm−1 in the enzyme are blue shifted close to 6 and 9 cm−1 after encapsulation as result of the interaction with the framework. After the insertion procedure, the resulting solid was thoroughly washed with the buffer and isolated by centrifugation to afford protease@MIL-101(Al)-NH2. UV-visible spectroscopy was used to ensure complete removal of the excess of non-encapsulated protease upon washing. As shown in Fig. 3a, there was no measurable protease in solution after two rounds. To further confirm that the protease was encapsulated rather than adsorbed on the external surface of the particles, we carried out two control experiments. We followed the same enzyme incubation protocol described above by using MIL-101(Cr). Immobilization is negligible in this case (Fig. S6 and S7†), likely due to the absence of –NH2 groups important in directing the translocation of the enzyme into the MOF. For the next test we made use of azocasein as a protease substrate (Fig. 3a, inset). Azocasein is hydrolysed non-specifically by proteolytic enzymes, releasing the azo dye into the media where it can be detected by its characteristic and intense absorbance at 440 nm. This permits rapid identification of the presence of the enzyme in MOFs incubated under different conditions. We ran five experiments by adding azocasein to five vials that contained: TRIS buffer as a blank sample (1) and dispersions in the same buffer of the MOF incubated with protease at 25 °C with hexane (2), the MOF incubated with protease at 60 °C without hexane (3), the protease@MIL-101(Al)-NH2 prepared as described above (4) and free protease (5). Samples 2, 3 and 4 where washed at least 3 times with fresh buffer before the test. After addition of azocasein, only samples 4 and 5 gave a positive result of proteolytic activity (yellow color), confirming the importance of temperature and the presence of a non-polar solvent in the incubation medium to facilitate the infiltration of the enzyme.
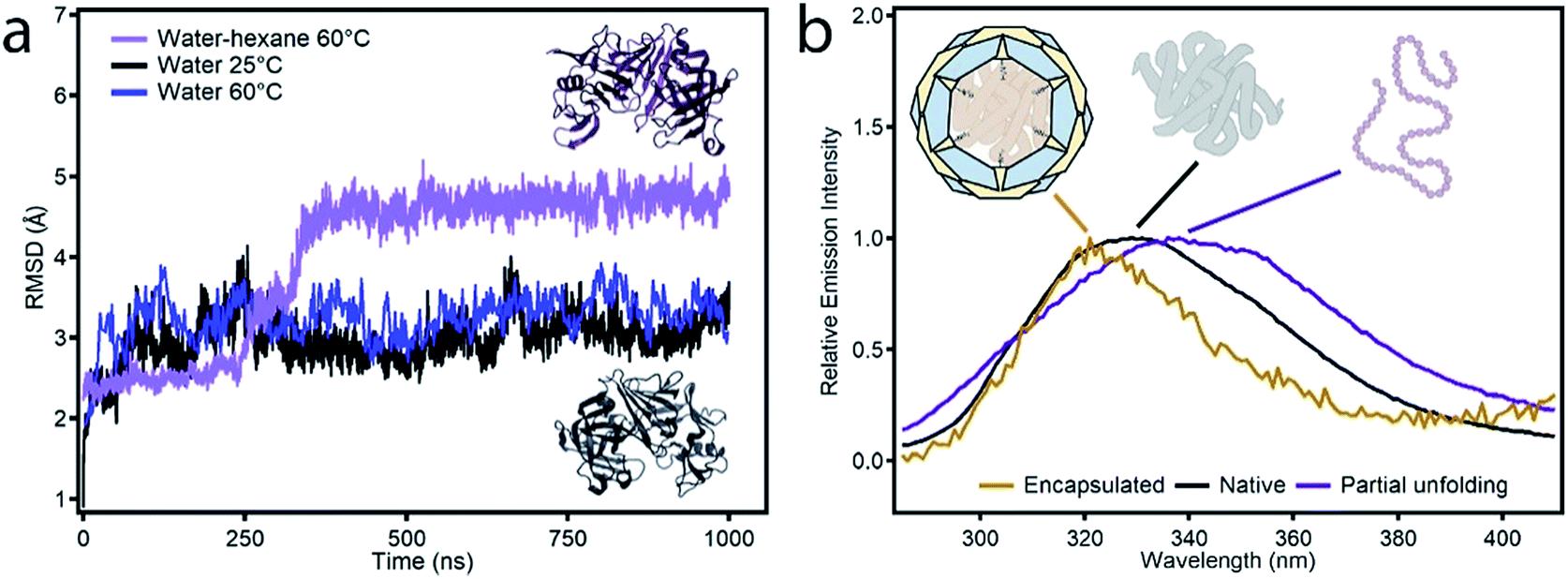 | ||
| Fig. 2 (a) Representation of the RMSD of the configurations obtained during MD simulations in different media with respect to the reported X-ray structure of protease (PDB code 1IBQ): hexane:water at 60 °C (purple), aqueous solution at 60 °C (blue) and 25 °C (black). The inset highlights the changes in the structure obtained after 1.0 μs of simulation in hexane:water (dark purple) compared to the original structure (black). (b) Fluorescence spectra of free protease (black), protease in the non-polar incubation medium at 60 °C (purple) and protease@MIL-101(Al)-NH2 (yellow). | ||
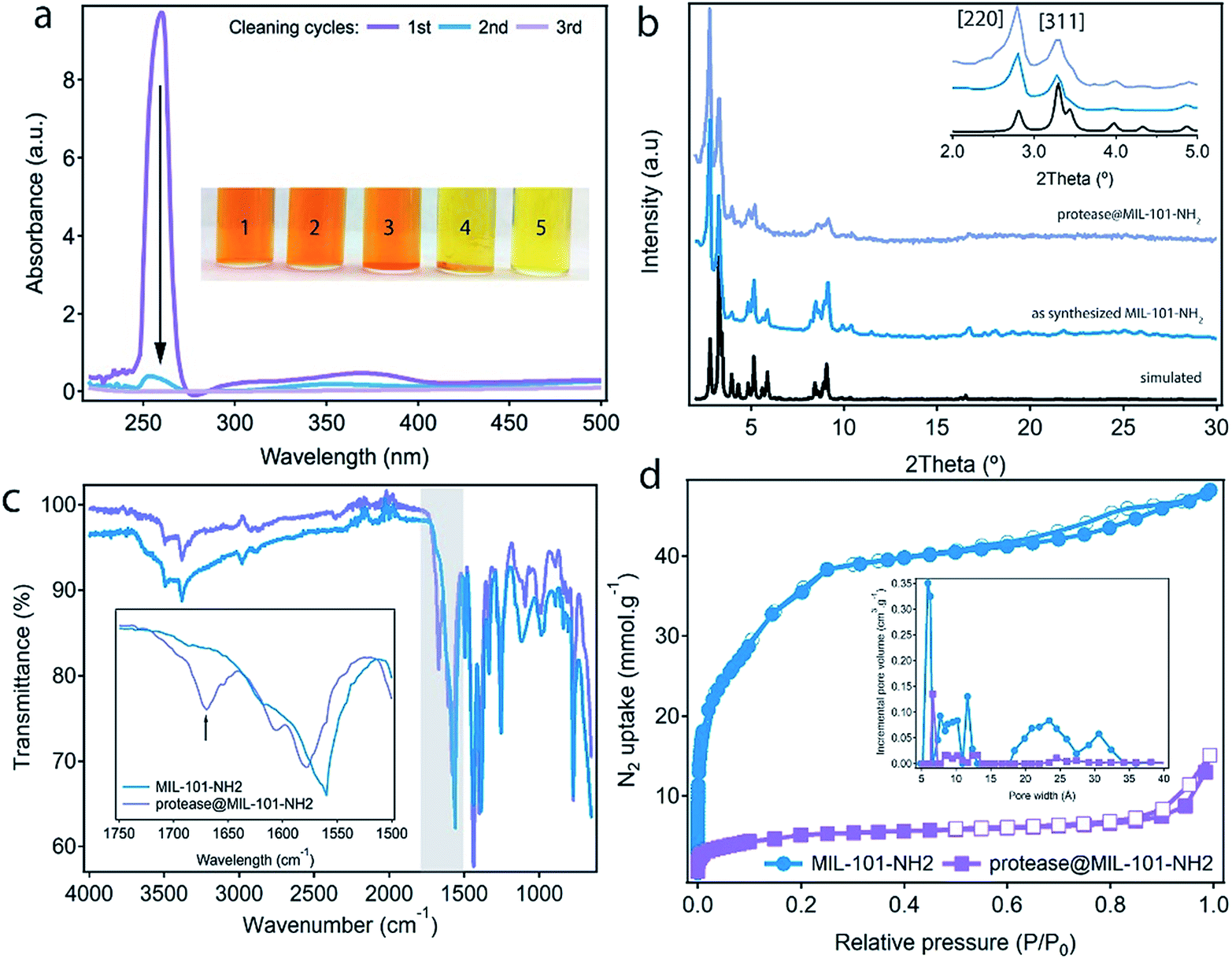 | ||
| Fig. 3 (a) UV-vis spectra of the supernatant solution, confirming the complete removal of non-encapsulated protease after the second cleaning of protease@MIL-101(Al)-NH2. Insets show control experiments with addition of azocasein to a TRIS buffer (1; reference), dispersions in the same buffer of the MOF incubated with protease at 25 °C with hexane (2), 60 °C without hexane (3), 60 °C with hexane (4) and the free enzyme (5). (b) PXRDs of MIL-101(Al)-NH2: simulated (black), the as-synthesized material (blue) and after infiltration of protease (purple). The inset shows most representative Bragg peaks at low-theta values. (c) FT-IR spectra showing the appearance of stretching band characteristic of the enzyme (marked with an arrow) in protease@MIL-101(Al)-NH2. (d) N2 isotherms confirming the drop in porosity after encapsulation. The inset shows the PSD of both materials analysed by density functional theory. | ||
Enzyme uptake and physicochemical characterization of protease@MIL-101(Al)-NH2
Comparison of the thermogravimetric (TGA) profile of the MOF before and after encapsulation confirms that protease@MIL-101(Al)-NH2 displays an additional weight loss at 230 °C that corresponds to the decomposition of the enzyme (Fig. S8†). The amount of protease was calculated to be 5%. The uptake of protease was also evaluated by using UV-vis spectroscopy, by correlating the drop in the absorption of the band intrinsic to protease, at 260 nm, with the concentrations of non-encapsulated enzyme in the supernatants after washing the material (Fig. S9†). The uptake calculated by this experiment is 0.05 g g−1. This value is very close to the content estimated from TGA, and quite similar to the values reported for the encapsulation of other enzymes in mesoporous hosts like Tb-mesoMOF31 or NTU-1000.9,12Protease@MIL-101(Al)-NH2 was fully characterized by PXRD, scanning electron microscopy (SEM), FT-IR spectroscopy and adsorption/desorption experiments. As shown in Fig. 3b, the infiltration of the enzyme proceeds without significant changes to the crystallinity of the MOF. After incubation in TRIS, there are no additional diffraction lines that could account for a structural transformation during encapsulation. Only the change in the relative intensity of the Bragg peaks seems to account for the filling of the pores. Phase purity was also confirmed by Le Bail refinement of the PXRDs of the material before and after infiltration (Fig. S10, Table S1†). SEM pictures confirm that the infiltration of protease does not cause changes to the size or morphology of the particles or induce the formation of an amorphous, contaminant phase (Fig. S11†). Infrared (IR) spectroscopy was used to study vibrational differences between the bare MOF and the protease@MIL-101(Al)-NH2. Fig. 3c shows that some spectral differences exist between them, in particular the presence of a new vibrational band at around 1670 cm−1 in protease@MIL-101(Al)-NH2. This band can be tentatively assigned to the amide I vibration ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the peptide bond of the protease. Encapsulation seems to shift the maximum of this band outside the typical range for folded proteins and peptides in solution (between 1660 and 1630 cm−1).32,33 We also observe a shift from 1560 to 1580 cm−1 in the position of the peak characteristic of δ(N–H) vibrations from the amino groups in the MOF after infiltration of protease, suggesting the presence of weak interactions between the enzyme and the host. However, changes in this frequency range could also be caused by overlapping contributions from the amide II vibration of the peptide bond (∼1550 cm−1).33 N2 adsorption isotherms were used to confirm the effect of encapsulation on the porosity of the material. MIL-101(Al)-NH2 displays the reported behaviour with a high uptake capacity for a Brunauer–Emmett–Teller (BET) surface area of 3000 m2 g−1, that is reduced to 245 m2 g−1 after encapsulation (Fig. 3d). The analysis of the pore size distribution in MIL-101(Al)-NH2 and protease@MIL-101(Al)-NH2 by using density functional theory (DFT) shows that the incremental pore volume corresponding to the mesoporous cages, centered at 2.0–2.5 and 3.0–3.4 nm, drops from 0.08 to 0.005 cm3 g−1 and 0.06 to 0.001 cm3 g−1 after the encapsulation of protease. This suggests that the majority of the mesoporous space in MIL-101(Al)-NH2 is occupied by the protease with a small fraction of microporosity (0.6 nm), still accessible for substrate diffusion. This behaviour is very similar to the reduction of porosity reported for COFs after encapsulation of lipase.34
O) of the peptide bond of the protease. Encapsulation seems to shift the maximum of this band outside the typical range for folded proteins and peptides in solution (between 1660 and 1630 cm−1).32,33 We also observe a shift from 1560 to 1580 cm−1 in the position of the peak characteristic of δ(N–H) vibrations from the amino groups in the MOF after infiltration of protease, suggesting the presence of weak interactions between the enzyme and the host. However, changes in this frequency range could also be caused by overlapping contributions from the amide II vibration of the peptide bond (∼1550 cm−1).33 N2 adsorption isotherms were used to confirm the effect of encapsulation on the porosity of the material. MIL-101(Al)-NH2 displays the reported behaviour with a high uptake capacity for a Brunauer–Emmett–Teller (BET) surface area of 3000 m2 g−1, that is reduced to 245 m2 g−1 after encapsulation (Fig. 3d). The analysis of the pore size distribution in MIL-101(Al)-NH2 and protease@MIL-101(Al)-NH2 by using density functional theory (DFT) shows that the incremental pore volume corresponding to the mesoporous cages, centered at 2.0–2.5 and 3.0–3.4 nm, drops from 0.08 to 0.005 cm3 g−1 and 0.06 to 0.001 cm3 g−1 after the encapsulation of protease. This suggests that the majority of the mesoporous space in MIL-101(Al)-NH2 is occupied by the protease with a small fraction of microporosity (0.6 nm), still accessible for substrate diffusion. This behaviour is very similar to the reduction of porosity reported for COFs after encapsulation of lipase.34
Enzymatic activity of encapsulated protease
In order to assess the protective effect of the MOF host on the chemical and thermal stability of the enzyme, we studied the proteolytic ability of free and immobilized protease to cleave the peptide bond of glycyl-L-tyrosine (Gly–Tyr) under extreme conditions of pH and temperature (see S5† for more details). Activity data were collected before 3 hours to discard the effect or protease autolysis. Previous studies confirm that this enzyme half-life is retained for at least 14 hours.35Fig. 4a shows the evolution of the proteolytic activity maintained after 3 hours at 25 °C with changes to the pH between 1 and 12. Compared to the free enzyme, protease@MIL-101(Al)-NH2 is more active in all cases even under the optimal conditions of this alkali protease at pH 2–3. Moreover, immobilization almost doubles the proteolytic activity of the enzyme at pH 1 and permits retaining the enzymatic reactivity in a basic medium above pH 8, for which the activity of the free enzyme is completely lost (Fig. S13†). The effect of temperature was studied with equivalent experiments by varying the bath temperature between 25 and 125 °C whilst fixing the pH at 7.4. We hypothesized that temperature would have a positive effect on reactivity due to thermodynamic reasons, provided the integrity of the enzyme was preserved. As shown in Fig. 4b, the free enzyme displays a slight decrease in activity at 55 °C to become inactive at higher temperatures. Under the same conditions, encapsulation permits the enzyme to retain its function in a broader range of temperatures, with a maximum of 78% at 95 °C. Heating above this temperature has a detrimental effect leading to complete loss of activity above 105 °C (Fig. S14†). These assays confirm the beneficial effect of encapsulation on the stability of the enzyme, which can retain enzymatic activity under extreme conditions of pH and temperature during which the free enzyme is denatured.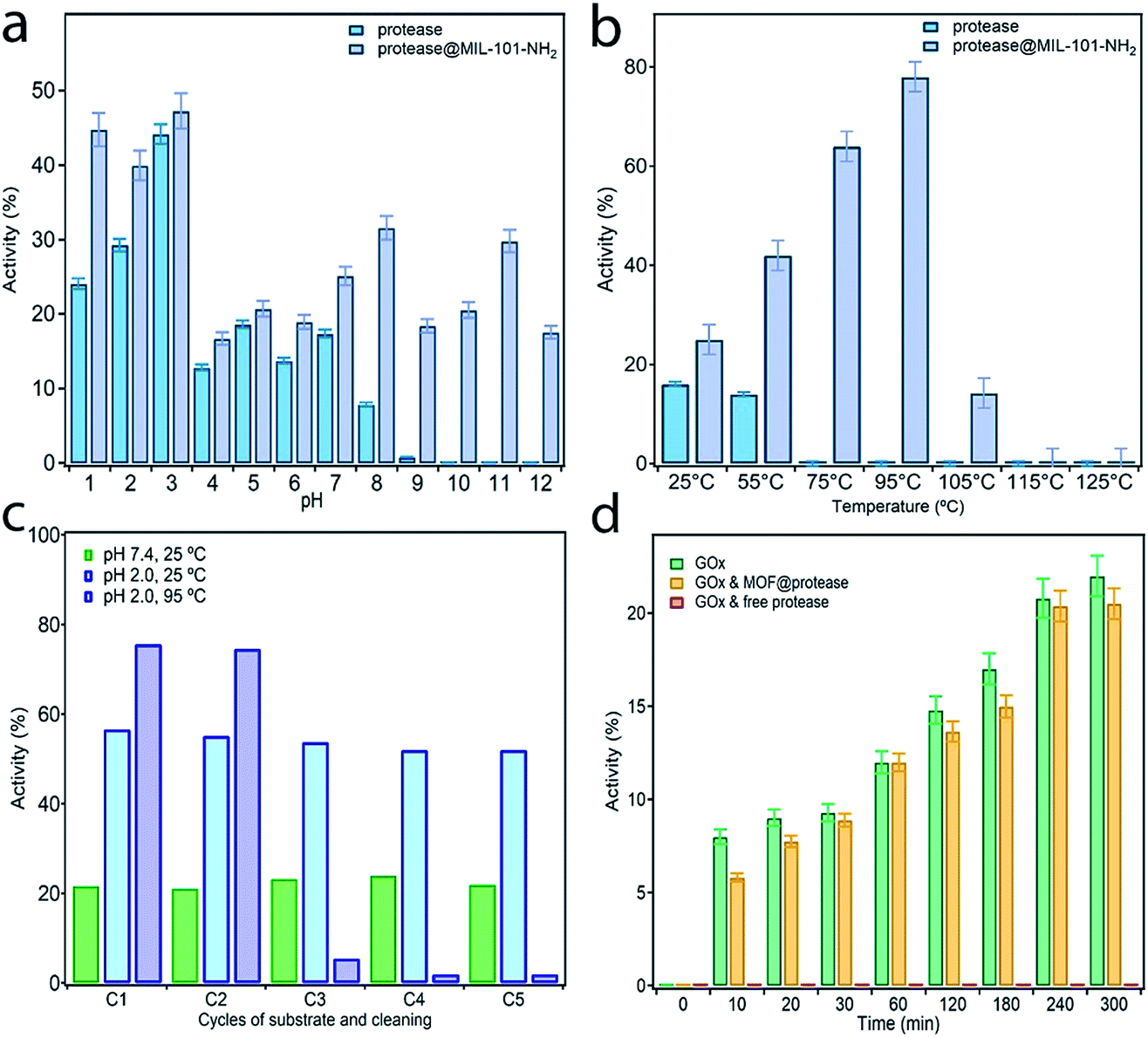 | ||
| Fig. 4 Comparison of the activity of protease before and after encapsulation tested at variable (a) pH from 1 to 12, temperature fixed at 25 °C and (b) temperature from 25 to 125 °C, pH = 7.4. (c) Activity and recyclability of the dried biocomposite, stored at room temperature for two weeks, at 25 °C and pH 7 (green) and at pH 2 and 25 °C (blue) or 95 °C (purple), confirming long-term stability and excellent recyclability at room temperature. (d) Activity at room temperature in a buffered medium of GOx alone (red), in the presence of protease@MIL-101(Al)-NH2 (yellow) and free protease (blue). MOF encapsulation prevents GOx digestion and retains its activity very similar to that of glucose oxidase alone. | ||
Encouraged by the chemical and thermal stability of the biocomposite we decided to test its long-term stability and recyclability for recovery after use. One of the key advantages of enzyme encapsulation is the possibility of simplifying the handling, storage or transportation of the biocatalyst whilst retaining its biological function intact.
To investigate this possibility, we dried protease@MIL-101(Al)-NH2 in a vacuum and stored it at room temperature for two weeks. This sample was used to perform recyclability tests by recovering the sample by centrifugation after each catalytic round. Activity was measured by following the same protocol described above at pH 7.4 and 25 °C after 3 hours. After two weeks the proteolytic activity remained unchanged, with only small deviations with respect to the freshly prepared material (Fig. 4c). The activity remains constant after 5 cycles, ruling out enzyme leaching or partial denaturation. The PXRD of the material after recycling also confirms that its structure remains unchanged (Fig. S15†). Besides the protective shield offered by the MOF, we argue this excellent stability might be also linked to the surface chemistry of amino groups for stronger immobilization of the enzyme by supramolecular interactions. We decided to push the limits of the material at higher temperatures and performed, at pH 2, two equivalent tests at 25 °C and 95 °C. Although the initial boost in activity is consistent with the value recorded for the fresh material, we observe an abrupt decrease in performance after two catalytic cycles until the material becomes inactive after five rounds at high temperatures. This behaviour is not caused by ineffective encapsulation but the structural transformation of the MOF under these conditions. As shown in Fig. S15,† heating at 95 °C in an acidic medium favors the transformation of the MIL-101 phase into the MIL-53 one and thus the leaching of the enzyme. We argue the use of alternative MOFs, more stable at acidic pH, will allow for the preparation of biocomposites by using this same methodology with excellent recyclability also under extreme conditions.
Finally, to also prove the ability of MOF encapsulation to neglect the effect of inhibitors on the reactivity of the enzyme we tested the activity of protease@MIL-101(Al)-NH2 in the presence of a second enzyme. The compatibility of Glucose Oxidase (GOx) with proteases is an important issue for biotechnological applications in food and health industries.36 The small pore openings provided by MIL-101-NH2 would be a promising alternative in this regard as they shall only enable the diffusion of small substrates, thus preventing the digestion of GOx. To confirm this possibility, we measured the activity of GOx in the presence of free protease and protease@MIL-101(Al)-NH2 under buffered conditions at room temperature (Fig. 4d). However free protease cancels the activity of GOx immediately, and the activity of GOx in the presence of the encapsulated enzyme is very similar to that of glucose oxidase alone (Fig. S16†). This suggests that our encapsulation method also provides excellent compatibility with competing enzymes due to size exclusion.
Conclusions
In summary, we have demonstrated how the conformational flexibility of simple enzymes can be exploited to chemically induce the structural changes required for their infiltration into mesoporous hosts featuring comparatively smaller pore windows. Translocation into the host is enabled by using a non-polar medium and mild temperatures during enzyme incubation. Our proteolytic tests demonstrate that protease@MIL-101(Al)-NH2 displays higher activity than the free enzyme in all cases but, more importantly, extends its usability to extreme conditions of pH (1–12) and temperatures up to 95 °C. MOF shielding is also effective in providing the biocomposite with long-term stability, recyclability and excellent compatibility with competing enzymes. The role of MOFs in the development of biocatalysts of interest for biotechnological applications is rapidly expanding. In this context, our methodology offers a simple approach for the preparation of new biocatalysts by encapsulation and stabilization of other enzymes for applications that demand ease of storage, good recyclability and tolerance to a broad range of operating conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EU (ERC Stg Chem-fs-MOF 714122) and Spanish MINECO (Project CTQ2017-83486-P, RYC-2012-10894, RYC-2013-13114 and FPU16/04162), MINECO/FEDER (CTQ2015-74523-JIN(AEI/FEDER), Unit of Excellence Marí de Maeztu (MDM-2015-0538) and Generalitat Valenciana AICO/2018/238.References
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H. C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC
.
- R. Riccò, W. Liang, S. Li, J. J. Gassensmith, F. Caruso, C. Doonan and P. Falcaro, ACS Nano, 2018, 12, 13–23 CrossRef PubMed
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed
- S. Jung, Y. Kim, S. J. Kim, T. H. Kwon, S. Huh and S. Park, Chem. Commun., 2011, 47, 2904–2906 RSC
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed
- F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu, K. C.-W. Wu and C.-K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed
- M. V. de Ruiter, R. Mejia-Ariza, J. J. L. M. Cornelissen and J. Huskens, Chem, 2016, 1, 29–31 CAS
- P. Li, J. A. Modica, A. J. Howarth, L. E. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Chem, 2016, 1, 154–169 CAS
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed
- X. Lian, Y. P. Chen, T. F. Liu and H. C. Zhou, Chem. Sci., 2016, 7, 6969–6973 RSC
- P. Li, S. Y. Moon, M. A. Guelta, S. P. Harvey, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 8052–8055 CrossRef CAS PubMed
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson, D. A. Gómez-Gualdrón, F. M. Cetin, J. Jagiello, A. M. Asiri, J. F. Stoddart and O. K. Farha, Chem, 2018, 4, 1022–1034 CAS
- D. Feng, T. F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y. P. Chen, X. Wang, K. Wang, X. Lian, Z. Y. Gu, J. Park, X. Zou and H. C. Zhou, Nat. Commun., 2015, 6, 1–8 Search PubMed
- Y. Chen, V. Lykourinou, C. Vetromile, T. Hoang, L. J. Ming, R. W. Larsen and S. Ma, J. Am. Chem. Soc., 2012, 134, 13188–13191 CrossRef CAS PubMed
- V. Daggett, Acc. Chem. Res., 2002, 35, 422–429 CrossRef CAS PubMed
- S. E. Jackson and A. R. Fersht, Biochemistry, 1991, 30, 10428–10435 CrossRef CAS PubMed
- F. N. Zaidi, U. Nath and J. B. Udgaonkar, Nat. Struct. Biol., 1997, 4, 1016–1024 CrossRef CAS PubMed
- P. Serra-Crespo, E. V. Ramos-Fernandez, J. Gascon and F. Kapteijn, Chem. Mater., 2011, 23, 2565–2572 CrossRef CAS
- T. A. S. e Silva, A. Knob, C. R. Tremacoldi, M. R. Brochetto-Braga and E. C. Carmona, World J. Microbiol. Biotechnol., 2011, 27, 2491–2497 CrossRef
- R. J. S. de Castro and H. H. Sato, J. Food Process. Technol., 2014, 2014, 372352 Search PubMed
- K. Liburdi, I. Benucci and M. Esti, Food Biotechnol., 2010, 24, 282–292 CrossRef CAS
- S. W. Englander and L. Mayne, Proc. Natl. Acad. Sci., 2014, 111, 15873–15880 CrossRef CAS PubMed
- K. A. Dill and H. S. Chan, Nat. Struct. Biol., 1997, 4, 10–19 CrossRef CAS PubMed
- X. Lian, Y. Huang, Y. Zhu, Y. Fang, R. Zhao, E. Joseph, J. Li, J. P. Pellois and H. C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5725–5730 CrossRef CAS PubMed
- F. X. Qin, S. Y. Jia, F. F. Wang, S. H. Wu, J. Song and Y. Liu, Catal. Sci. Technol., 2013, 3, 2761–2768 RSC
- J. L. England and G. Haran, Annu. Rev. Phys. Chem., 2011, 62, 257–277 CrossRef CAS PubMed
- C. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed
-
Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006, pp. 29–575 Search PubMed
- Y. Chen, S. Han, X. Li, Z. Zhang and S. Ma, Inorg. Chem., 2014, 53, 10006–10008 CrossRef CAS PubMed
- V. Lykourinou, Y. Chen, X. S. Wang, L. Meng, T. Hoang, L. J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed
- S. Y. Venyaminov and N. N. Kalnin, Biopolymers, 1990, 30, 1259–1271 CrossRef CAS PubMed
- E. Goormaghtigh, V. Cabiaux and J. M. Ruysschaert, Subcell. Biochem., 1994, 23, 405–450 CAS
- Q. Sun, C. W. Fu, B. Aguila, J. Perman, S. Wang, H. Y. Huang, F. S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef CAS PubMed
- N. Bombara, A. M. R. Pilosof and M. C. Añón, J. Food Biochem., 1994, 18, 31–41 CrossRef CAS
- S. Patra, T. Hidalgo Crespo, A. Permyakova, C. Sicard, C. Serre, A. Chaussé, N. Steunou and L. Legrand, J. Mater. Chem. B, 2015, 3, 8983–8992 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00082h |
| This journal is © The Royal Society of Chemistry 2019 |