
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect access to 2-aryl substituted pyrrolinium salts for carbon centre based radicals without pyrrolidine-2-ylidene alias cyclic(alkyl)(amino)carbene (CAAC) as a precursor†‡
Debdeep
Mandal
 a,
Sebastian
Sobottka
b,
Ramapada
Dolai
a,
Avijit
Maiti
a,
Debabrata
Dhara
a,
Pankaj
Kalita
c,
Ramakirushnan Suriya
Narayanan
a,
Vadapalli
Chandrasekhar
*ad,
Biprajit
Sarkar
*b and
Anukul
Jana
*a
a,
Sebastian
Sobottka
b,
Ramapada
Dolai
a,
Avijit
Maiti
a,
Debabrata
Dhara
a,
Pankaj
Kalita
c,
Ramakirushnan Suriya
Narayanan
a,
Vadapalli
Chandrasekhar
*ad,
Biprajit
Sarkar
*b and
Anukul
Jana
*a
aTata Institute of Fundamental Research Hyderabad, Gopanpally, Hyderabad-500107, Telangana, India. E-mail: ajana@tifrh.res.in
bInstitut für Chemie und Biochemie, Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34–36, 14195, Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de
cSchool of Chemical Sciences, National Institute of Science Education and Research, HBNI, Bhubaneswar-752050, India
dDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: vc@iitk.ac.in
First published on 28th February 2019
Abstract
The synthesis of organic radicals is challenging due to their inherent instability. In recent years, cyclic(alkyl)(amino)carbene (CAAC)-derived 2-substituted pyrrolinium salts have been used as synthons for the synthesis of isolable carbon-based radicals. Herein, we report a direct, easy and convenient method for the synthesis of 2-aryl substituted pyrrolinium salts without using CAAC as a precursor. These cations can be reduced to the corresponding radicals. The influence of the aryl substituent at the C-2 position on radical stabilization and dimerization has been investigated. Because of the large scope of our strategy (capability to modulate different substituents at all the C- and N-centres of the pyrrolinium salts), it has the merit to be an extremely effective and productive route for generating carbon-based radicals whose stability as well as reactivity can be varied.
Introduction
Radicals play crucial roles in various chemical and biological processes,1 as well as in industrial processes such as in polymer chemistry.2 Recent reports have also suggested their extensive use as catalysts and reactive intermediates to synthesize various types of molecules.3 However, because of their inherent instability and high reactivity most radicals exist as very short lived species. To understand the nature and reactivity of radicals formed in various reactions as intermediates their stabilization and isolation are very crucial. Efforts in this regard include trapping unstable radicals by coupling with other stable radicals4 or quenching them with suitable reagents.5 In recent years, cyclic(alkyl)(amino)carbenes (CAACs) have been used for the isolation of different kinds of radicals including those that are carbon based (Scheme 1),6 as well as hetero-atom based systems.7 In particular, Bertrand et al. have recently reported the syntheses of CAAC-derived mixed valence (radical and cation) organic compounds.8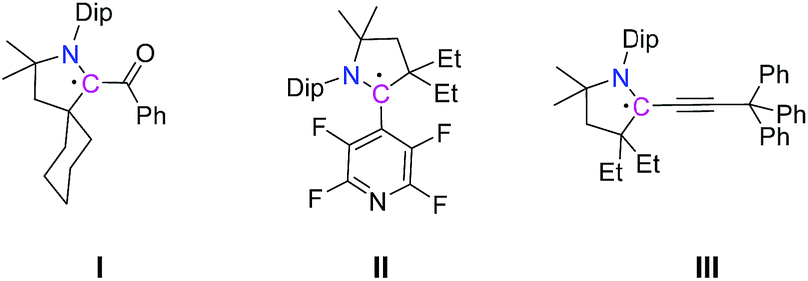 | ||
| Scheme 1 Selected chemical structures of CAAC-derived α-carbonyl radical I,6a tetrafluoropyridyl radical II,6c and propargyl radical III.6f | ||
So far all the reported CAAC-scaffold-based carbon radicals such as IV are generated from their corresponding pyrrolidinium salts, V, derived from CAAC, VI (Scheme 2). However, a serious limitation and disadvantage of using CAACs as precursors for generating radicals is their very limited number,9 which severely restricts the variation of the substituent scope in the products. To get CAAC-scaffold-based carbon centre radicals, the synthesis of 2-substituted pyrrolinium salts as immediate precursors is crucial (Scheme 2). In other words, if the synthesis of 2-substituted pyrrolinium salts can be achieved through alternate10 and more convenient routes, without using CAAC, this will enrich the library of 2-substituted pyrrolinium salts as well as the corresponding radicals. Therefore, a new methodology for synthesizing 2-substituted pyrrolinium salts, V, with a wide substrate scope is urgently required.
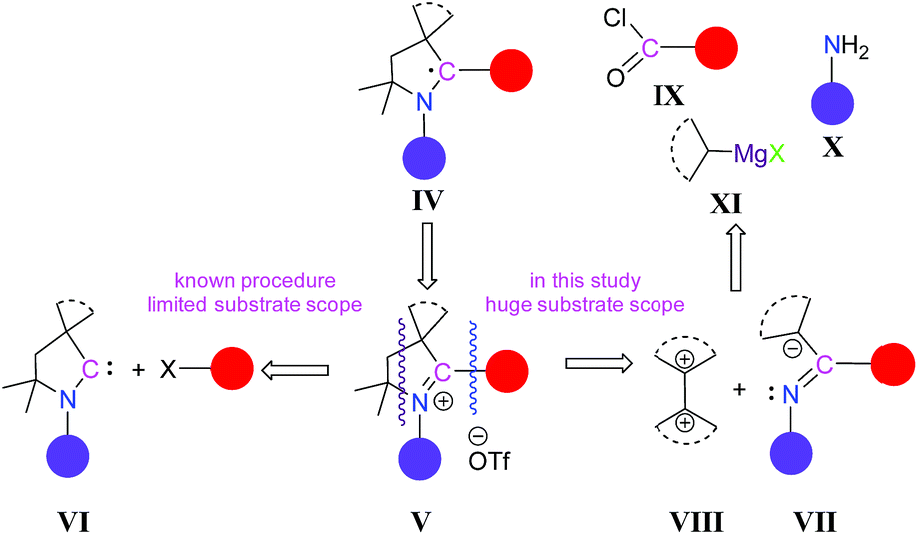 | ||
| Scheme 2 Reported strategy and our strategy for the synthesis of 2-aryl substituted pyrrolinium slats, V. | ||
Results and discussion
Herein, we have considered a new strategy where 2-substituted pyrrolinium salts, V, can be generated from a secondary ketimine, VII, which has only one α-hydrogen, and a compound having geminal electrophilic centres, VIII. The secondary ketimine, VII, can be obtained in three very convenient steps (Scheme 2), from commercially available reagents: acid chloride, IX; primary amine, X; and secondary Grignard reagents, R2CHMgX, XI.11 In this strategy, there is a huge scope for variation at all the C-centres as well as at the N-centre of the pyrrolinium ring. Moreover, the expansion of the five membered ring is also possible.To test our strategy, we considered 4-alkylbenzoyl chlorides, 1R, which are commercially available, as the starting precursors, keeping in mind that the 4-alkylphenyl substituent will be at the C-2 position of pyrrolinium salts which can influence the stability as well as the self-association of the radicals, which may be generated. Moreover, this choice will give access to the first non-fluorinated 2-aryl-substituted pyrrolinium salts and the subsequent radicals derived from them. For N-substitution we have considered the iPr-group.
The reaction of 1R with iPrNH2/Et3N, PCl5, and iPrMgCl sequentially leads to the imine, 2R, as a colorless liquid (Scheme 2).12 Subsequently, the reaction of 2R with nBuLi, isobutylene oxide and triflic anhydride sequentially in one pot leads to the formation of 2-aryl substituted pyrrolinium triflates, 3R, as white solids (Scheme 3).12
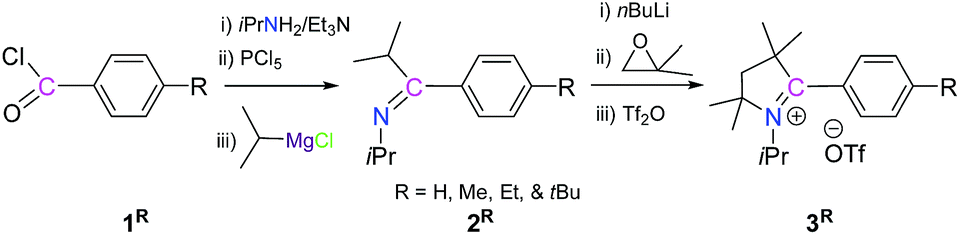 | ||
| Scheme 3 Synthesis of 2-aryl substituted pyrrolinium triflate, 3R. | ||
All the 2-aryl substituted pyrrolinium triflates, 3R, were characterized by NMR spectroscopy as well as by single-crystal X-ray measurements (see Fig. 1 for the molecular structure of 3H and Fig. S1–S3 for the molecular structures of 3Me, 3Et, and 3tBu in the ESI‡). An examination of the molecular structures reveals that the pyrrolinium- and the phenyl rings are almost perpendicular to each other (torsion angle: 86.93° for 3H, 89.60° for 3Me, 87.55° for 3Et, and 90° for 3tBu).12
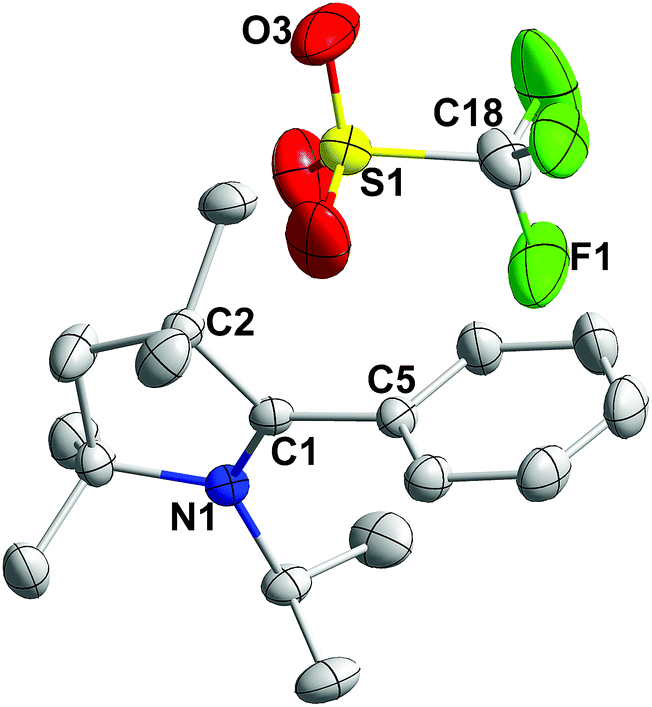 | ||
| Fig. 1 Solid state molecular structure of 3H, with thermal ellipsoids at 30%. All hydrogens are omitted for clarity. Selected bond lengths (Å) and angles (°): N1–C1 1.275(3), C1–C2 1.508(3), C1–C5 1.486(3), and N1–C1–C2 113.34(19). | ||
The cyclovoltammograms of 2-aryl substituted pyrrolinium triflates, 3R, reveal two reduction steps, the first event at around E1/2 = −1.8 V (vs. FcH/FcH+) which is completely reversible on the cyclic voltammetry timescale and indicates the formation of a neutral radicals, 3R˙ (Fig. 2). The substituents on the phenyl rings do not have any significant influence on the redox potentials of these compounds. The second reduction step is irreversible and is seen around E1/2 = −2.53 to −2.75 V indicating the formation of a 2-e reduced anionic compounds, 3R−, which could not be generated chemically (Fig. 2).6c Furthermore, the second reduction step has a profound influence on the reversibility of the first reduction step even on the timescale of the cyclic voltammetry experiments (see Fig. S17–S21 in the ESI‡).
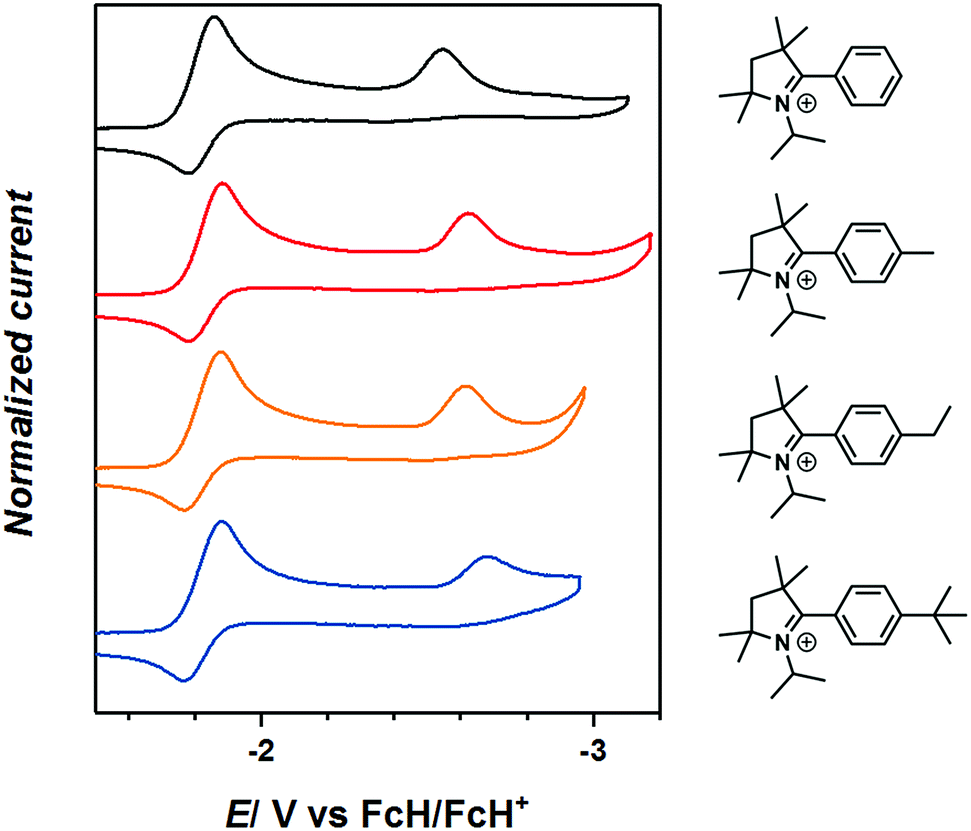 | ||
| Fig. 2 CV of 3R in THF/0.1 NBu4PF6 measured at a GC working electrode at 100 mV s−1. | ||
In order to chemically access the species that are generated electrochemically, we carried out the reduction of 3H with KC8 in THF (Scheme 4), which leads to a dark red solution from which pale yellow crystals could be harvested after keeping the n-hexane extraction at −35 °C, overnight. Analysis through a single crystal X-ray diffraction study showed the formation of a dimer, 3H2 (Fig. 3), which must have been formed by a head-to-tail dimerization of the initially formed radical, 3H˙ as in the case of the Gomberg triphenylmethyl radical, Ph3C˙.13 The formation of the radical, 3H˙ in solution was confirmed by ESR, which reveals a signal centered at g = 2.003 (Fig. 4). Compound, 3H2 exists in equilibrium, in solution, with the monomer, 3H˙. Also there is a possibility of formation of 3H2 by a coupling between the starting cation, 3H, and the 2-electron reduced compound, 3H−, which has been observed in a spectroelectrochemistry study.12 This is analogous to the reactivity of the trityl cation which forms adducts with neutral bases.14 Both mechanisms were considered for simulating the cyclic voltammograms and this provided the best fits with the experimental voltammograms (see Fig. S33–S37 in the ESI‡).
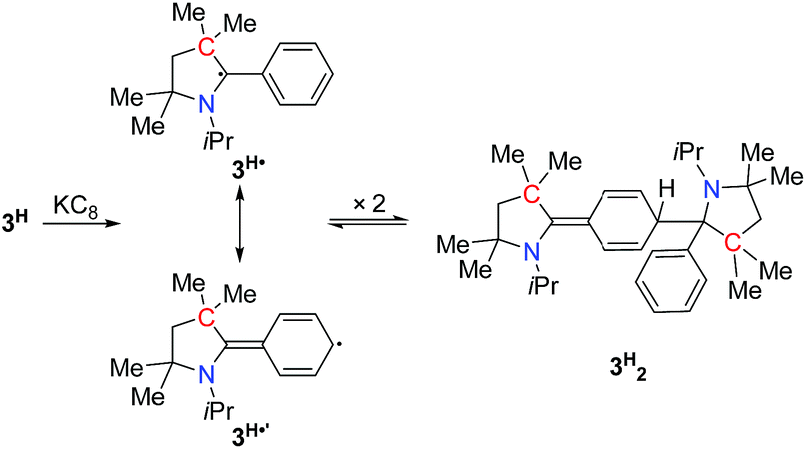 | ||
| Scheme 4 Synthesis of 3H2. | ||
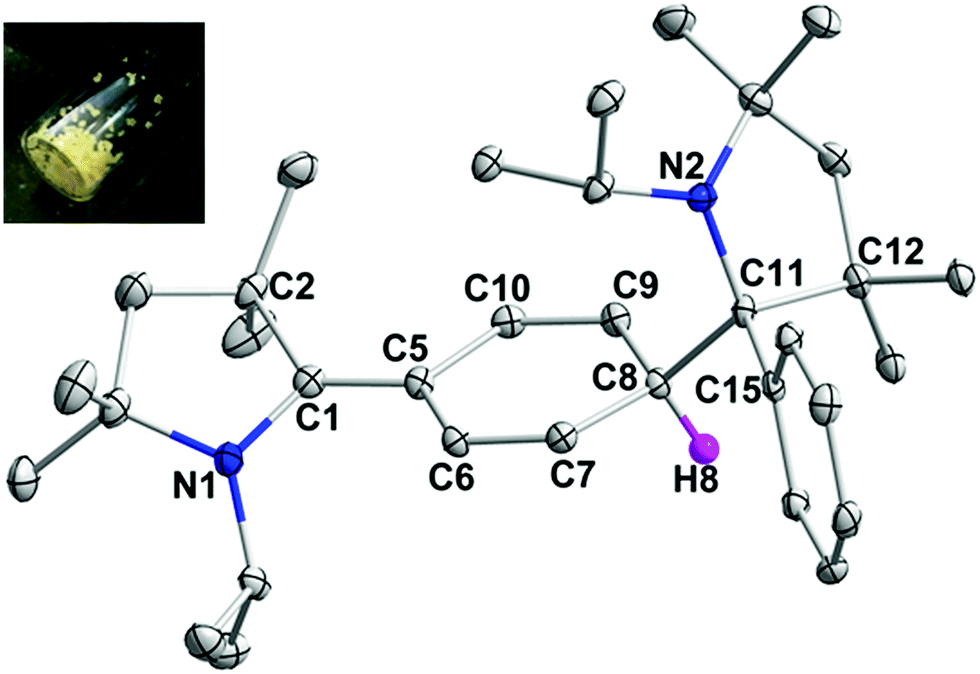 | ||
| Fig. 3 Molecular structure of 3H2, with thermal ellipsoids at 30% and all hydrogens except C8–H are omitted for clarity. Selected bond lengths (Å) and angles (°): C1–C5 1.370(4), C5–C10 1.450(5), C10–C9 1.331(4), C9–C8 1.513(4), C8–C11 1.603(4), and C11–C15 1.545(4); N1–C1–C2 108.4(3), C5–C1–N1 124.2(3), C5–C1–C2 127.2(3), C9–C8–C11 114.8(2), C7–C8–C11 114.9(2), and N2–C11–C12 102.9(2) (inset: yellow crystalline compounds of 3H2). | ||
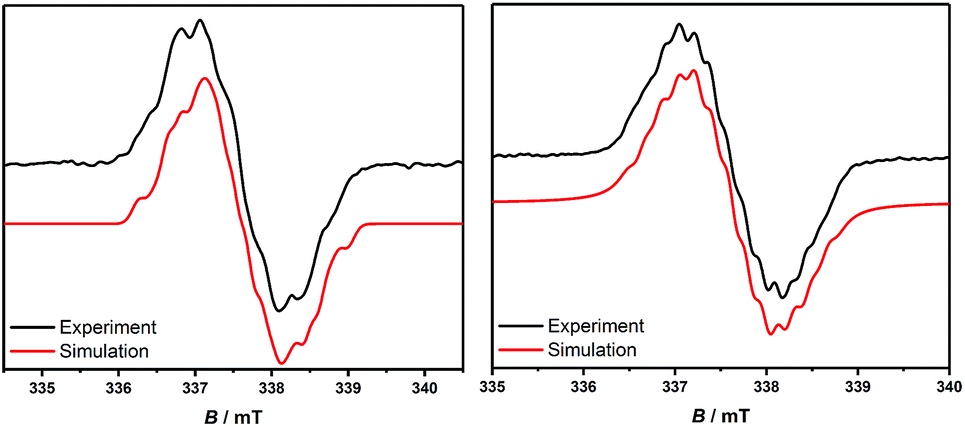 | ||
| Fig. 4 EPR spectra of 3H˙ and 3tBu˙ in 0.1 M NBu4PF6 in THF. | ||
Apart from the head-to-tail dimerization of 3H˙, there is a possibility of tail-to-tail and head-to-head coupling, the latter being very unlikely because of steric reasons. Theoretical calculations reveal that head-to-tail dimerization is the energetically favourable process. On the other hand, the formation of the tail-to-tail dimer is an endothermic process and is destabilized compared to the head-to-tail dimer (see Table S15 and Fig. S52 in the ESI‡).
Subsequently, we carried out the reduction of other 2-aryl substituted pyrrolinium salts with KC8 (Scheme 5), where the substitution at the 4-position of the aryl group can block the dimerization.15 Both 3Me and 3Et afford a mixture of decomposition products at room temperature although the reduction of 3Et immediately results in a red solution at room temperature. In the case of 3tBu, KC8 reduction produced a deep red color solution at about −50 °C which remains sufficiently intense until its work up at room temperature. Thus, after removing the THF from the reaction mixture, we extracted the residue with dry n-hexane. Keeping this extract at −80 °C afforded deep red crystals (Scheme 5). Unfortunately, these crystals were polycrystalline in nature. Several attempts to grow single crystals in solvents of various polarities like THF and toluene failed to give suitable single crystals for X-ray diffraction study.
 | ||
| Scheme 5 Reduction of 3Me, 3Et, and 3tBu (left) and picture of 3tBu˙ (right). | ||
Theoretical calculations reveal 3Me˙, 3Et˙, and 3tBu˙ to be more stable than their corresponding higher energy dimers.12 This is the reason why 3Me˙ and 3Et˙ afford decomposed products rather than the dimer. Due to the steric bulk 3tBu˙ is relatively more stable. The formation of 3tBu˙in situ in solution has been confirmed by ESR spectroscopy by the observation of a signal at g = 2.003 similar to what was seen for 3H˙ (Fig. 4). The hyperfine couplings to the relevant nuclei are likely not resolved due to unfavorable hyperfine coupling/line-width ratios. However, for 3tBu˙ a partial resolution of the hyperfine couplings is observed in the experimental spectrum. The calculated spin densities at the PBE0/IGLO-III level of theory reveal a localization of the spin densities at the C-2 and the N-1 atoms of the CAAC-scaffold with only minor contributions at the phenyl substituent at the C2 position (Fig. 5).
 | ||
| Fig. 5 Spin densities of 3H˙ (left) and 3tBu˙ (right) at PBE0/IGLO-III, iso-value 0.01. | ||
In order to gain insight into the redox properties and the electronic structures of the various redox states (spectro)electrochemical and (TD-)DFT calculations were performed on the 2-pyrrolinium salts, 3R. In the UV/vis spectra all the four 2-pyrrolinium salts are transparent in the visible region, as expected for such colorless compounds. However, the in situ generated one-electron-reduced neutral forms (measured exemplarily for two compounds) 3H˙ and 3tBu˙ each display one absorption at around 350 nm and a second broad absorption at around 485 nm (see Fig. S58, S59 and Table S19 in the ESI‡). The UV/vis spectroelectrochemical measurements clearly display the reversible nature of the first reduction step, as seen by the re-generation of the starting spectrum with quantitative intensities on reversing the potential back to the starting potential (see Fig. S57–S60 in the ESI‡). The first reduction step is thus also seen to be reversible on the spectroelectrochemical time scale. A full TD-DFT calculation using B3LYP reproduces the bands of both the cationic and the neutral radical forms with reasonable accuracy (see Fig. S66 in the ESI‡).
On performing a second reduction on the compounds via in situ UV/vis spectroelectrochemistry the bands at around 350 and 485 nm that belong to the radical form disappear, and the spectra become more or less transparent in the visible region except for a weak shoulder at 330 or 380 nm (see Table S19 in the ESI‡). As discussed above for the cyclic voltammetry measurements, this second reduction step is not reversible, and switching the potential back to the starting potential does not lead to the regeneration of the starting spectra. Furthermore, on scanning the potential back, the spectra corresponding to the one-electron reduced compounds are not regenerated either. ESR spectroelectrochemistry at the second reduction potential shows a disappearance of the signal corresponding to the radical species, possibly indicating the generation of a diamagnetic compound. These measurements thus point to a drastic change in the structure of the compounds on reducing them with two electrons.
Conclusions
In conclusion, we have disclosed a new strategy for the synthesis of 1(N)- and 2-substituted pyrrolinium salts which are immediate precursors for the synthesis of CAAC-scaffold based carbon centre radicals. This strategy allows a large variation of substrate scope at all the C- and N-centres of the pyrrolinium moieties. As a proof of principle, we demonstrated the synthesis of 1(N)-isopropyl-2-(4-R-phenyl)-pyrrolinium triflates (R = H, Me, Et, and tBu). These compounds can be converted to the corresponding neutral radicals which were characterized by electrochemical, in situ UV/vis, and spectroelectrochemical (ESR) methods as well as with DFT calculations. In the case of the 2-phenyl substituted radical, the formation of a head-to-tail dimer was observed as in the case of the Gomberg triphenylmethyl radical. In view of the above results, we believe that our strategy will be useful for the synthesis of different CAAC-scaffold based carbon centre radicals. Currently, we are actively pursuing this strategy to realize new carbon-centered multi-radical centres including diradicaloids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the TIFR Hyderabad. V. C. is thankful to the DST for a National J. C. Bose fellowship. The high-performance computing facilities at ZEDAT, FU-Berlin, are acknowledged for access to computing resources.Notes and references
- (a) T. L. Dormandy, Ann. R. Coll. Surg. Engl., 1980, 62, 188–194 CAS; (b) B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, 2015 CrossRef; (c) M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- P. Nesvadba, Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, 2012, vol. 4, pp. 1701–1736 Search PubMed.
- (a) C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237–1286 CrossRef CAS; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- (a) D. H. R. Barton, V. N. L. Gloahec and J. Smith, Tetrahedron Lett., 1998, 39, 7483–7486 CrossRef CAS; (b) E. G. Janzen, Acc. Chem. Res., 1971, 4, 31–40 CrossRef CAS.
- I. H. Leave and G. C. Ramsay, Tetrahedron, 1969, 25, 5669–5675 CrossRef.
- Selected references are: (a) J. K. Mahoney, D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef CAS PubMed; (b) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS PubMed; (c) J. K. Mahoney, D. Martin, F. Thomas, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (d) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem.–Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (e) D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed; (f) D. Mandal, R. Dolai, N. Chrysochos, P. Kalita, R. Kumar, D. Dhara, A. Maiti, R. S. Narayanan, G. Rajaraman, C. Schulzke, V. Chandrasekhar and A. Jana, Org. Lett., 2017, 19, 5605–5608 CrossRef CAS PubMed; (g) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed.
- Selected references are: (a) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed; (b) B. Li, S. Kundu, C. Stückl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed; (c) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed; (d) O. Back, M. Ali Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed.
- M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- U. S. D. Paul and U. Radius, Chem.–Eur. J., 2017, 23, 3993–4009 CrossRef CAS PubMed.
- H. G. Richey Jr, K. Narasaka, M. Yamane, Y. Chen and W. Tang, Encyclopedia of Reagents for Organic Synthesis, 2017 Search PubMed.
- See the ESI‡ for the details.
- (a) M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef; (b) H. Lankamp, W. T. Nauta and C. MacLean, Tetrahedron Lett., 1968, 9, 249–254 CrossRef; (c) C. Lichtenberg, L. Viciu, M. Vogt, R. E. Rodríguez-Lugo, M. Adelhardt, J. Sutter, M. M. Khusniyarov, K. Meyer, B. de Bruin, E. Billd and H. Grützmacher, Chem. Commun., 2015, 51, 13890–13893 RSC; (d) S. Rösel, J. Becker, W. D. Allen and P. R. Schreiner, J. Am. Chem. Soc., 2018, 140, 14421–14432 CrossRef PubMed; (e) S. Rösel, C. Balestrieri and P. R. Schreiner, Chem. Sci., 2017, 8, 405–410 RSC.
- (a) P. A. Chase, A. L. Gille, T. M. Gilbert and D. W. Stephan, Dalton Trans., 2009, 7179–7188 RSC; (b) L. Cabrera, G. C. Welch, J. D. Masuda, P. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef CAS; (c) E. Follet, P. Mayer, D. S. Stephenson, A. R. Ofial and G. Berionni, Chem.–Eur. J., 2017, 23, 7422–7427 CrossRef CAS PubMed.
- (a) T. H. Colie and E. S. Lewis, J. Am. Chem. Soc., 1979, 101, 1810–1814 CrossRef; (b) D. Dünnebacke, W. P. Neumann, A. Penenory and U. Stewen, Chem. Ber., 1989, 122, 533–535 CrossRef.
Footnotes |
| † Dedicated to Professor Vinod K. Singh on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, NMR spectra, X-ray crystallographic data and theoretical details. CCDC 1868053 (3H), 1868056 (3Me), 1868057 (3Et), 1868055 (3tBu) and 1868054 (3H2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05477k |
| This journal is © The Royal Society of Chemistry 2019 |